
A Computational Workflow to Predict Biological Target Mutations: The Spike Glycoprotein Case Study

Abstract
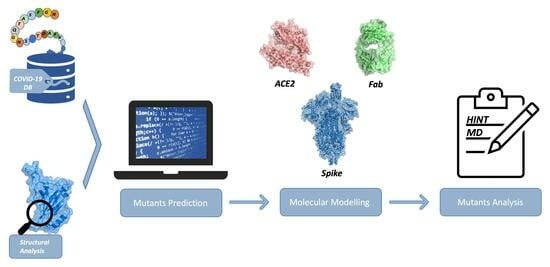
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Constraint-Based Modeling
3.2. Protein Structures Selection
3.3. RBD VOC Mutations
3.4. Protein Preparation
3.5. HINT Calculation
3.6. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, Y.-F.; Zhao, X.-M. A Survey on the Computational Approaches to Identify Drug Targets in the Postgenomic Era. BioMed Res. Int. 2015, 2015, 239654. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.-A.; Hochreiter, S. Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, V.; Dhankhar, P.; Dalal, V. Promising antivirals for PLpro of SARS-CoV-2 using virtual screening, molecular docking, dynamics, and MMPBSA. J. Biomol. Struct. Dyn. 2023, 41, 4650–4666. [Google Scholar] [CrossRef] [PubMed]
- Dhankhar, P.; Dalal, V.; Kumar, V. Screening of Severe Acute Respiratory Syndrome Coronavirus 2 RNA-Dependent RNA Polymerase Inhibitors Using Computational Approach. J. Comput. Biol. 2021, 28, 1228–1247. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.A.; Sharma, M.; Dalal, V.; Singh, V.; Tomar, S.; Kumar, P. Multifunctional inhibitors of SARS-CoV-2 by MM/PBSA, essential dynamics, and molecular dynamic investigations. J. Mol. Graph. Model. 2021, 107, 107969. [Google Scholar] [CrossRef]
- Dhankhar, P.; Dalal, V.; Singh, V.; Tomar, S.; Kumar, P. Computational guided identification of novel potent inhibitors of N-terminal domain of nucleocapsid protein of severe acute respiratory syndrome coronavirus 2. J. Biomol. Struct. Dyn. 2022, 40, 4084–4099. [Google Scholar] [CrossRef]
- Eweas, A.F.; Osman, H.-E.H.; Naguib, I.A.; Abourehab, M.A.S.; Abdel-Moneim, A.S. Virtual Screening of Repurposed Drugs as Potential Spike Protein Inhibitors of Different SARS-CoV-2 Variants: Molecular Docking Study. Curr. Issues Mol. Biol. 2022, 44, 3018–3029. [Google Scholar] [CrossRef]
- Lynch, M.; Ackerman, M.S.; Gout, J.F.; Long, H.; Sung, W.; Thomas, W.K.; Foster, P.L. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016, 17, 704–714. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef]
- Saifi, S.; Ravi, V.; Sharma, S.; Swaminathan, A.; Chauhan, N.S.; Pandey, R. SARS-CoV-2 VOCs, Mutational diversity and clinical outcome: Are they modulating drug efficacy by altered binding strength? Genomics 2022, 114, 110466. [Google Scholar] [CrossRef]
- Akkiz, H. Implications of the Novel Mutations in the SARS-CoV-2 Genome for Transmission, Disease Severity, and the Vaccine Development. Front. Med. 2021, 8, 636532. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Cozzini, P.; Agosta, F.; Dolcetti, G.; Righi, G. How a Blockchain Approach Can Improve Data Reliability in the COVID-19 Pandemic. ACS Med. Chem. Lett. 2022, 13, 517–519. [Google Scholar] [CrossRef]
- Han, W.; Chen, N.; Xu, X.; Sahil, A.; Zhou, J.; Li, Z.; Zhong, H.; Gao, E.; Zhang, R.; Wang, Y.; et al. Predicting the antigenic evolution of SARS-COV-2 with deep learning. Nat. Commun. 2023, 14, 3478. [Google Scholar] [CrossRef]
- Saldivar-Espinoza, B.; Garcia-Segura, P.; Novau-Ferré, N.; Macip, G.; Martínez, R.; Puigbò, P.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallve, S. The Mutational Landscape of SARS-CoV-2. Int. J. Mol. Sci. 2023, 24, 9072. [Google Scholar] [CrossRef] [PubMed]
- Palu, A.D.; Torroni, P. 25 years of applications of logic programming in Italy. In A 25-Year Perspective on Logic Programming; Springer: Berlin/Heidelberg, Germany, 2010; pp. 300–328. [Google Scholar]
- Falkner, A.; Friedrich, G.; Schekotihin, K.; Taupe, R.; Teppan, E.C. Industrial Applications of Answer Set Programming. Künstliche Intell. 2018, 32, 165–176. [Google Scholar] [CrossRef]
- Erdem, E.; Gelfond, M.; Leone, N. Applications of Answer Set Programming. AI Mag. 2016, 37, 53–68. [Google Scholar] [CrossRef]
- Apt, K.R.; Marek, V.W.; Truszczynski, M.; Warren, D.S. The Logic Programming Paradigm: A 25-Year Perspective; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Edwards, D.; Forster, J.W.; Cogan, N.O.I.; Batley, J.; Chagné, D. Single Nucleotide Polymorphism Discovery. In Association Mapping in Plants; Springer: New York, NY, USA, 2007; pp. 53–76. [Google Scholar] [CrossRef]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On Human Disease-Causing Amino Acid Variants: Statistical Study of Sequence and Structural Patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Rees-Spear, C.; Muir, L.; Griffith, S.A.; Heaney, J.; Aldon, Y.; Snitselaar, J.L.; Thomas, P.; Graham, C.; Seow, J.; Lee, N.; et al. The effect of spike mutations on SARS-CoV-2 neutralization. Cell Rep. 2021, 34, 108890. [Google Scholar] [CrossRef]
- Gómez, S.A.; Rojas-Valencia, N.; Gómez, S.; Cappelli, C.; Restrepo, A. The Role of Spike Protein Mutations in the Infectious Power of SARS-COV-2 Variants: A Molecular Interaction Perspective. ChemBioChem 2022, 23, e202100393. [Google Scholar] [CrossRef]
- Lin, H.-X.; Feng, Y.; Wong, G.; Wang, L.; Li, B.; Zhao, X.; Li, Y.; Smaill, F.; Zhang, C. Identification of residues in the receptor-binding domain (RBD) of the spike protein of human coronavirus NL63 that are critical for the RBD–ACE2 receptor interaction. J. Gen. Virol. 2008, 89, 1015–1024. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Agosta, F.; Kellogg, G.E.; Cozzini, P. From oncoproteins to spike proteins: The evaluation of intramolecular stability using hydropathic force field. J. Comput. Aided Mol. Des. 2022, 36, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Sobitan, A.; Rhoades, R.; Liu, D.; Tang, Q. Systemic effects of missense mutations on SARS-CoV-2 spike glycoprotein stability and receptor-binding affinity. Brief. Bioinform. 2021, 22, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Laha, S.; Chakraborty, J.; Das, S.; Manna, S.K.; Biswas, S.; Chatterjee, R. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect. Genet. Evol. 2020, 85, 104445. [Google Scholar] [CrossRef]
- Salleh, M.Z.; Derrick, J.P.; Deris, Z.Z. Structural Evaluation of the Spike Glycoprotein Variants on SARS-CoV-2 Transmission and Immune Evasion. Int. J. Mol. Sci. 2021, 22, 7425. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.F.; Rezaei, R.; Jamieson, T.R.; Dave, J.; Martin, N.T.; Singaravelu, R.; Crupi, M.J.F.; Boulton, S.; Tucker, S.; Duong, J.; et al. Characterization of Critical Determinants of ACE2–SARS CoV-2 RBD Interaction. Int. J. Mol. Sci. 2021, 22, 2268. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; de Masi, L.; Argenio, M.A.; Facchiano, A. Structural Dissection of Viral Spike-Protein Binding of SARS-CoV-2 and SARS-CoV-1 to the Human Angiotensin-Converting Enzyme 2 (ACE2) as Cellular Receptor. Biomedicines 2021, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, G.E.; Semus, S.F.; Abraham, D.J. HINT: A new method of empirical hydrophobic field calculation for CoMFA. J. Comput. Aided Mol. Des. 1991, 5, 545–552. [Google Scholar] [CrossRef]
- Kellogg, G.E.; Abraham, D.J. Hydrophobicity: Is LogPo/w more than the sum of its parts? Eur. J. Med. Chem. 2000, 35, 651–661. [Google Scholar] [CrossRef]
- Nethercote, N.; Stuckey, P.J.; Becket, R.; Brand, S.; Duck, G.J.; Tack, G. MiniZinc: Towards a standard CP modelling language. In Proceedings of the International Conference on Principles and Practice of Constraint Programming, Providence, RI, USA, 23–27 September 2007; Volume 4741 of LNCS. [Google Scholar]
- Agosta, F.; Cozzini, P. Hint approach on Transthyretin folding/unfolding mechanism comprehension. Comput. Biol. Med. 2023, 155, 106667. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.H.; Catalano, C.; Portillo, S.C.; Safo, M.K.; Scarsdale, N.; Kellogg, G.E. 3D interaction homology: The hydropathic interaction environments of even alanine are diverse and provide novel structural insight. J. Struct. Biol. 2019, 207, 183–198. [Google Scholar] [CrossRef]
- Mughram, M.H.A.L.; Catalano, C.; Bowry, J.P.; Safo, M.K.; Scarsdale, J.N.; Kellogg, G.E. 3D Interaction Homology: Hydropathic Analyses of the ‘π–Cation’ and ‘π–π’ Interaction Motifs in Phenylalanine, Tyrosine, and Tryptophan Residues. J. Chem. Inf. Model. 2021, 61, 2937–2956. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 Add. Mutation | 2 Add. Mutations | 3 Add. Mutations | 4 Add. Mutations | |
|---|---|---|---|---|
| Group A | 130 structures | 8178 structures | = | = |
| Group B | 10 structures | 33 structures | 36 structures | = |
| Group C | 33 structures | 461 structures | 3531 structures | 16,002 structures |
| Additional Mutations | HINTscore | ∆HINTscore | Variant |
|---|---|---|---|
| R408S | 10,972.15 | 223.76 | BA.2, BA.4, BA.5 |
| L452R | 11,552.56 | 804.17 | BA.4, BA.5, XE |
| D405N | 10,978.56 | 229.72 | BA.4, BA.5, XE |
| G476S | 11,384.25 | 635.86 | BA.1 |
| D405N, L452R | 10,860.45 | 112.06 | BA.4, BA.5, XE |
| D405N, R408S | 11,047.57 | 299.18 | BA.2, BA.4, BA.5, XE |
| R408S, L452R | 10,818.82 | 70.43 | BA.4, BA.5, XE |
| S371F | 10,926.9 | 178.51 | BA.2, BA.4, BA.5, XE |
| T376A | 11,025.84 | 277.45 | BA.2, BA.4, BA.5, XE |
| S371F, T376A | 11,216.69 | 468.3 | BA.2, BA.4, BA.5, XE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozzini, P.; Agosta, F.; Dolcetti, G.; Dal Palù, A. A Computational Workflow to Predict Biological Target Mutations: The Spike Glycoprotein Case Study. Molecules 2023, 28, 7082. https://doi.org/10.3390/molecules28207082
Cozzini P, Agosta F, Dolcetti G, Dal Palù A. A Computational Workflow to Predict Biological Target Mutations: The Spike Glycoprotein Case Study. Molecules. 2023; 28(20):7082. https://doi.org/10.3390/molecules28207082
Chicago/Turabian StyleCozzini, Pietro, Federica Agosta, Greta Dolcetti, and Alessandro Dal Palù. 2023. "A Computational Workflow to Predict Biological Target Mutations: The Spike Glycoprotein Case Study" Molecules 28, no. 20: 7082. https://doi.org/10.3390/molecules28207082
APA StyleCozzini, P., Agosta, F., Dolcetti, G., & Dal Palù, A. (2023). A Computational Workflow to Predict Biological Target Mutations: The Spike Glycoprotein Case Study. Molecules, 28(20), 7082. https://doi.org/10.3390/molecules28207082