
Insight into the Inhibitory Mechanism of Embryonic Ectoderm Development Subunit by Triazolopyrimidine Derivatives as Inhibitors through Molecular Dynamics Simulation

Abstract
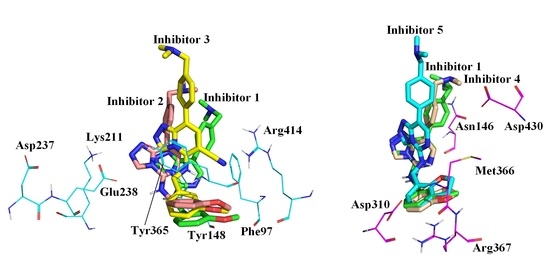
:
1. Introduction
2. Results
2.1. Molecular Docking Results
2.2. Stability Analysis of the Simulation System
2.3. Free Energy Landscape and Sampling
2.4. Sample Conformation Analysis
2.5. Hydrogen Bond Analysis
2.6. Binding Free Energy Analysis
3. Discussion
3.1. Comparison of Inhibitors and Natural Substrate Binding Modes
3.2. Differences in the Inhibitory Capacity of the Six Inhibitors
4. Materials and Methods
4.1. Acquisition of the EED Protein and Inhibitor Structures
4.2. Molecular Docking
4.3. Molecular Dynamics Simulation
4.4. Binding Free Energy Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glancy, E.; Ciferri, C.; Bracken, A.P. Structural basis for PRC2 engagement with chromatin. Curr. Opin. Struct. Biol. 2021, 67, 135–144. [Google Scholar] [CrossRef]
- Conway, E.; Healy, E.; Bracken, A.P. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr. Opin. Cell Biol. 2015, 37, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Hojfeldt, J.W.; Laugesen, A.; Willumsen, B.M.; Damhofer, H.; Hedehus, L.; Tvardovskiy, A.; Mohammad, F.; Jensen, O.N.; Helin, K. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 2018, 25, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Lavarone, E.; Barbieri, C.M.; Pasini, D. Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat. Commun. 2019, 10, 1679. [Google Scholar] [CrossRef]
- Zhao, Y.; Guan, Y.; Zhao, F.; Yu, T.; Zhang, S.; Zhang, Y.; Duan, Y.; Zhou, X. Recent strategies targeting Embryonic Ectoderm Development (EED) for cancer therapy: Allosteric inhibitors, PPI inhibitors, and PROTACs. Eur. J. Med. Chem. 2022, 231, 114–144. [Google Scholar] [CrossRef] [PubMed]
- Jürg, M.; Verrijzer, P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr. Opin. Genet. Dev. 2009, 19, 150–158. [Google Scholar]
- Ku, S.; Spencer, R.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Sato, T.; Kaneda, A.; Tsuji, S.; Isagawa, T.; Yamamoto, S.; Fujita, T.; Yamanaka, R.; Tanaka, Y.; Nukiwa, T.; Marquez, V.E.; et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci. Rep. 2013, 3, 1911. [Google Scholar] [CrossRef]
- Zhang, H.; Qi, J.; Reyes, J.M.; Li, L.; Rao, P.K.; Li, F.; Lin, C.Y.; Perry, J.A.; Lawlor, M.A.; Federation, A.; et al. Oncogenic Deregulation of EZH2 as an Opportunity for Targeted Therapy in Lung Cancer. Cancer Discov. 2016, 6, 1006–1021. [Google Scholar] [CrossRef]
- Puppe, J.; Opdam, M.; Schouten, P.C.; Jóźwiak, K.; Lips, E.; Severson, T.; Van de Ven, M.; Brambillasca, C.; Bouwman, P.; Van Tellingen, O.; et al. EZH2 Is Overexpressed in BRCA1-like Breast Tumors and Predictive for Sensitivity to High-Dose Platinum-Based Chemotherapy. Clin. Cancer Res. 2019, 25, 4351–4362. [Google Scholar] [CrossRef]
- Gao, S.B.; Zheng, Q.F.; Xu, B.; Pan, C.B.; Li, K.L.; Zhao, Y.; Zheng, Q.L.; Lin, X.; Xue, L.X.; Jin, G.H. EZH2 represses target genes through H3K27-dependent and H3K27-independent mechanisms in hepatocellular carcinoma. Mol. Cancer Res. 2014, 12, 1388–1397. [Google Scholar] [CrossRef]
- Gan, L.; Xu, M.; Hua, R.; Tan, C.; Zhang, J.; Gong, Y.; Wu, Z.; Weng, W.; Sheng, W.; Guo, W. The polycomb group protein EZH2 induces epithelial–mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J. Hematol. Oncol. 2018, 11, 9. [Google Scholar] [CrossRef]
- Jones, B.A.; Varambally, S.; Arend, R.C. Histone Methyltransferase EZH2: A Therapeutic Target for Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Wassef, M.; Margueron, R. The Multiple Facets of PRC2 Alterations in Cancers. J. Mol. Biol. 2017, 429, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- Højfeldt, L.A.; Westergaard, J.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar]
- Dockerill, M.; Gregson, C.; O’Donovan, D.H. Targeting PRC2 for the treatment of cancer: An updated patent review. Expert Opin. Ther. Pat. 2020, 31, 119–135. [Google Scholar] [CrossRef]
- Kasinath, V.; Faini, M.; Poepsel, S.; Reif, D.; Feng, X.A.; Stjepanovic, G.; Aebersold, R.; Nogales, E. Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 2018, 359, 940–944. [Google Scholar] [CrossRef]
- Van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumors: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Gibaja, V.; Shen, F.; Harari, J.; Korn, J.; Ruddy, D.; Saenz-Vash, V.; Zhai, H.; Rejtar, T.; Paris, C.G.; Yu, Z.; et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 2016, 35, 558–566. [Google Scholar] [CrossRef]
- Eich, M.L.; Athar, M.; Ferguson, J.E., 3rd; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef]
- Baker, T.; Nerle, S.; Pritchard, J.; Zhao, B.; Rivera, V.M.; Garner, A.; Gonzalvez, F. Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget 2015, 6, 32646–32655. [Google Scholar] [CrossRef]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Drury, W.J., 3rd; Voigt, P.; Martin, S.R.; Taylor, W.R.; De Marco, V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef]
- Jain, B.P.; Pandey, S. WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J. 2018, 37, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Sanulli, S.; Justin, N.; Teissandier, A.; Ancelin, K.; Portoso, M.; Caron, M.; Michaud, A.; Lombard, B.; Da Rocha, S.T.; Offer, J.; et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol. Cell 2015, 57, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Xing, X.; Hu, M.; Zhang, Y.; Liu, P.; Chai, J. Structural basis of EZH2 recognition by EED. Structure 2007, 15, 1306–1315. [Google Scholar] [CrossRef]
- Huang, Y.; Sendzik, M.; Zhang, J.; Gao, Z.; Sun, Y.; Wang, L.; Gu, J.; Zhao, K.; Yu, Z.; Zhang, L.; et al. Discovery of the Clinical Candidate MAK683: An EED-Directed, Allosteric, and Selective PRC2 Inhibitor for the Treatment of Advanced Malignancies. J. Med. Chem. 2022, 65, 5317–5333. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Sendzik, M.; Huang, Y.; Shultz, M.D.; Cantwell, J.; Dillon, M.P.; Fu, X.; Fuller, J.; Gabriel, T.; Gu, J.; et al. Structure-Guided Design of EED Binders Allosterically Inhibiting the Epigenetic Polycomb Repressive Complex 2 (PRC2) Methyltransferase. J. Med. Chem. 2017, 60, 415–427. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, J.; Yu, Z.; Zhang, H.; Wang, Y.; Lingel, A.; Qi, W.; Gu, J.; Zhao, K.; Shultz, M.D.; et al. Discovery of First-in-Class, Potent, and Orally Bioavailable Embryonic Ectoderm Development (EED) Inhibitor with Robust Anticancer Efficacy. J. Med. Chem. 2017, 60, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tang, Z.; Fang, H.; Shi, W. Inhibition of hepatocellular carcinoma cell proliferation by embryonic ectodermal development protein inhibitor and its molecular mechanism. Chin. J. Exp. Surg. 2021, 38, 648–650. [Google Scholar]
- Rej, R.K.; Wang, C.; Lu, J.; Wang, M.; Petrunak, E.; Zawacki, K.P.; McEachern, D.; Fernandez-Salas, E.; Yang, C.Y.; Wang, L.; et al. EEDi-5285: An Exceptionally Potent, Efficacious, and Orally Active Small-Molecule Inhibitor of Embryonic Ectoderm Development. J. Med. Chem. 2020, 63, 7252–7267. [Google Scholar] [CrossRef]
- Rej, R.K.; Wang, C.; Lu, J.; Wang, M.; Petrunak, E.; Zawacki, K.P.; McEachern, D.; Yang, C.Y.; Wang, L.; Li, R.; et al. Discovery of EEDi-5273 as an Exceptionally Potent and Orally Efficacious EED Inhibitor Capable of Achieving Complete and Persistent Tumor Regression. J. Med. Chem. 2021, 64, 14540–14556. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inf. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Tanchuk, V.Y.; Tanin, V.O.; Vovk, A.I.; Poda, G. A New, Improved Hybrid Scoring Function for Molecular Docking and Scoring Based on AutoDock and AutoDock Vina. Chem. Biol. Drug Des. 2016, 87, 618–625. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; De Groot, B.L.; Grubmüller, H. Best bang for your buck: GPU nodes for GROMACS biomolecular simulations. J. Comput. Chem. 2015, 36, 1990–2008. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Dickson, C.J.; Rosso, L.; Betz, R.M.; Walker, R.C.; Gould, I.R. GAFFlipid: A General Amber Force Field for the accurate molecular dynamics simulation of phospholipid. Soft Matter 2012, 8, 9617–9627. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L.J. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Valdes-Tresanco, M.S.; Valdes-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Bonding Residue | Occurrence (%) |
|---|---|---|
| Complex 1 | Asn194 | 85.34 |
| ARG367 | 8.47 | |
| ASN146 | 1.33 | |
| Complex 2 | ASN194 | 98.24 |
| ARG367 | 8.34 | |
| LYS211 | 6.06 | |
| Complex 3 | ASN194 | 37.28 |
| ARG414 | 19.18 | |
| ARG367 | 4.14 | |
| Complex 4 | ASN194 | 108.51 |
| ARG367 | 6.65 | |
| LYS211 | 3.70 | |
| Complex 5 | ASN194 | 63.60 |
| ARG414 | 4.57 | |
| LYS211 | 4.10 | |
| Complex 6 | ASN194 | 104.13 |
| ASN146 | 17.61 | |
| ARG367 | 6.12 |
| Energy Contribution | Complex 1 | Complex 2 | Complex 3 | Complex 4 | Complex 5 | Complex 6 |
|---|---|---|---|---|---|---|
| ΔEvdW | −209.73 | −192.71 | −150.48 | −227.44 | −176.55 | −219.95 |
| ΔEelec | −76.93 | −79.11 | −45.68 | −83.26 | −37.68 | −93.09 |
| ΔGPB | 151.89 | 145.65 | 100.42 | 154.29 | 111.65 | 159.39 |
| ΔGSA | −17.66 | −16.22 | −12.07 | −16.87 | −13.74 | −17.34 |
| ΔGpolar a | 74.96 | 66.54 | 54.74 | 71.03 | 73.97 | 66.30 |
| ΔGnonpolar b | −227.39 | −208.93 | −162.55 | −244.31 | −190.29 | −237.29 |
| ΔGbinding c | −152.43 | −142.39 | −107.81 | −173.28 | −116.32 | −170.99 |
| Residue | Complex 1 | Complex 2 | Complex 3 | Complex 4 | Complex 5 | Complex 6 |
|---|---|---|---|---|---|---|
| Tyr365 | −22.04 | −25.58 | −21.86 | −27.71 | −22.18 | −27.76 |
| Tyr148 | −16.80 | −14.11 | −13.19 | −17.36 | −12.77 | −18.74 |
| Phe97 | −16.03 | −12.24 | −8.53 | −14.50 | −10.94 | −17.06 |
| Trp364 | −1.38 | −2.21 | −1.26 | −3.67 | −2.02 | −1.74 |
| Arg367 | −8.80 | −4.83 | −7.61 | −9.99 | −0.99 | −7.65 |
| Asn194 | −20.71 | −20.27 | −6.99 | −22.94 | −11.15 | −23.50 |
| Lys211 | −5.56 | −21.37 | −5.24 | −11.64 | −12.93 | −5.90 |
| Glu238 | −3.29 | 4.28 | −0.84 | −0.92 | 5.41 | −5.84 |
| Asp237 | 0.53 | 2.65 | 0.10 | 1.02 | 1.37 | −0.20 |
| Arg414 | −3.53 | −1.91 | −6.86 | −2.80 | −5.63 | −5.02 |
| Asp310 | −3.54 | −4.21 | −3.34 | −3.34 | −10.85 | −4.37 |
| Met366 | −3.61 | −4.02 | −2.35 | −5.83 | −1.05 | −5.59 |
| Asn146 | −4.68 | −2.39 | −2.84 | −1.71 | −1.55 | −4.15 |
| Asp430 | −1.25 | −1.68 | 0.20 | −3.95 | −0.89 | 1.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, J.; Zhang, H.; Guan, S.; Liu, C.; Du, J.; Shen, X.; Wang, S. Insight into the Inhibitory Mechanism of Embryonic Ectoderm Development Subunit by Triazolopyrimidine Derivatives as Inhibitors through Molecular Dynamics Simulation. Molecules 2023, 28, 7997. https://doi.org/10.3390/molecules28247997
Ju J, Zhang H, Guan S, Liu C, Du J, Shen X, Wang S. Insight into the Inhibitory Mechanism of Embryonic Ectoderm Development Subunit by Triazolopyrimidine Derivatives as Inhibitors through Molecular Dynamics Simulation. Molecules. 2023; 28(24):7997. https://doi.org/10.3390/molecules28247997
Chicago/Turabian StyleJu, Jianan, Hao Zhang, Shanshan Guan, Chang Liu, Juan Du, Xiaoli Shen, and Song Wang. 2023. "Insight into the Inhibitory Mechanism of Embryonic Ectoderm Development Subunit by Triazolopyrimidine Derivatives as Inhibitors through Molecular Dynamics Simulation" Molecules 28, no. 24: 7997. https://doi.org/10.3390/molecules28247997
APA StyleJu, J., Zhang, H., Guan, S., Liu, C., Du, J., Shen, X., & Wang, S. (2023). Insight into the Inhibitory Mechanism of Embryonic Ectoderm Development Subunit by Triazolopyrimidine Derivatives as Inhibitors through Molecular Dynamics Simulation. Molecules, 28(24), 7997. https://doi.org/10.3390/molecules28247997