
Tuning Photochemical and Photophysical Properties of P(V) Phthalocyanines

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
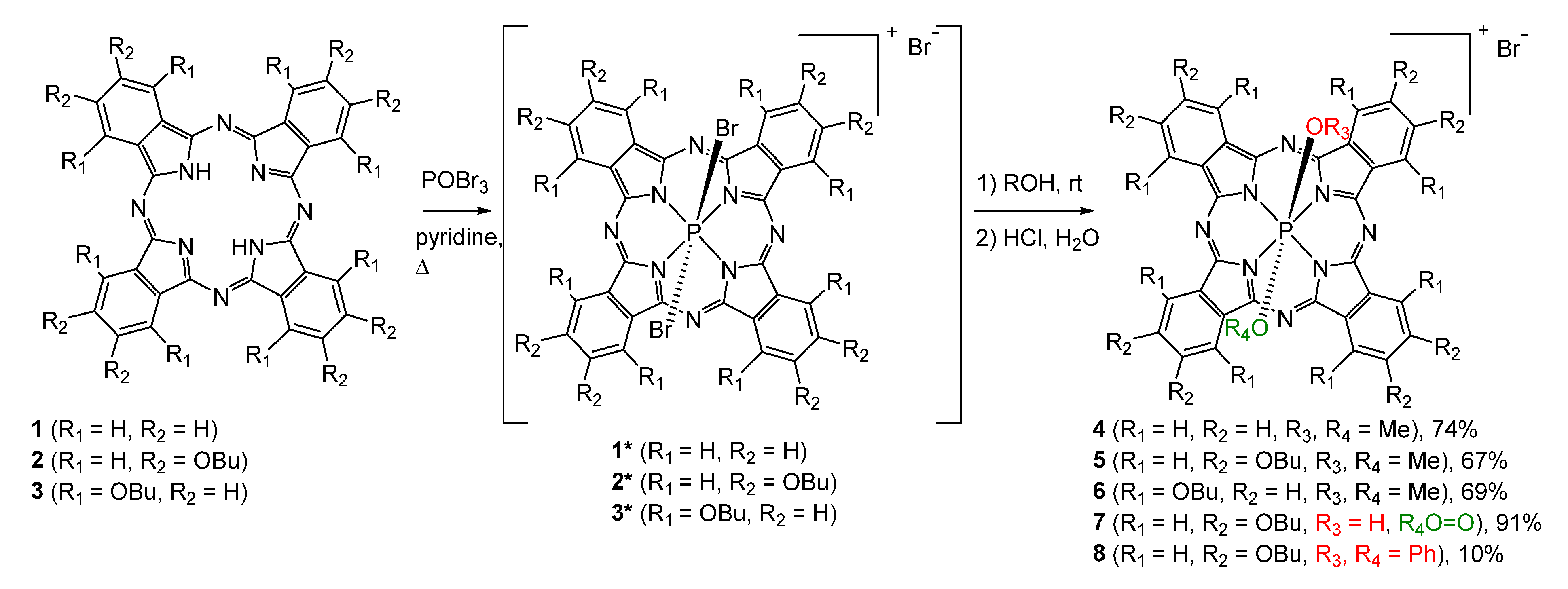
2.1. Synthesis
2.2. UV-Vis Spectroscopy
2.3. NMR Spectroscopy
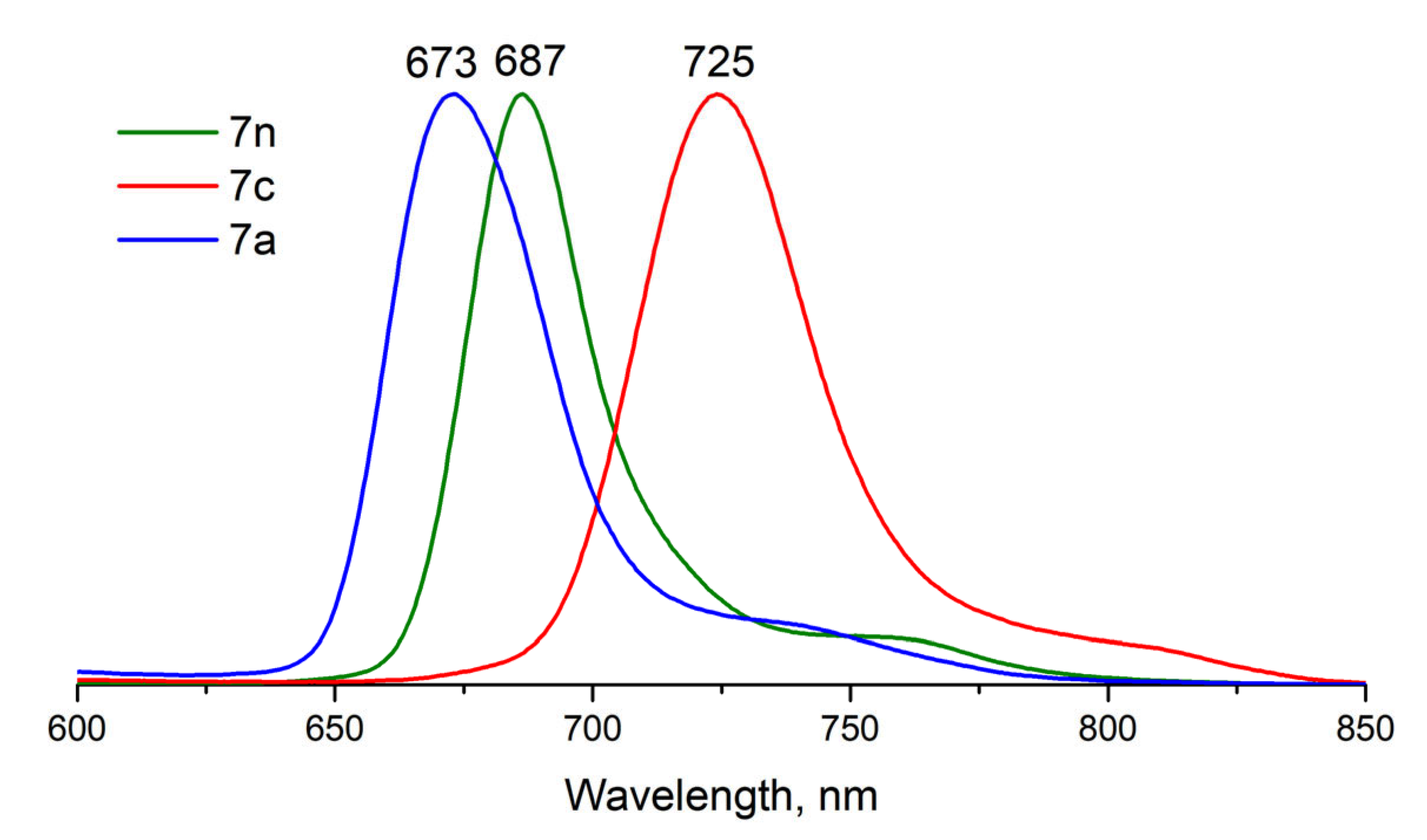
2.4. Fluorescence
2.5. Singlet Oxygen Generation
2.6. Photostability
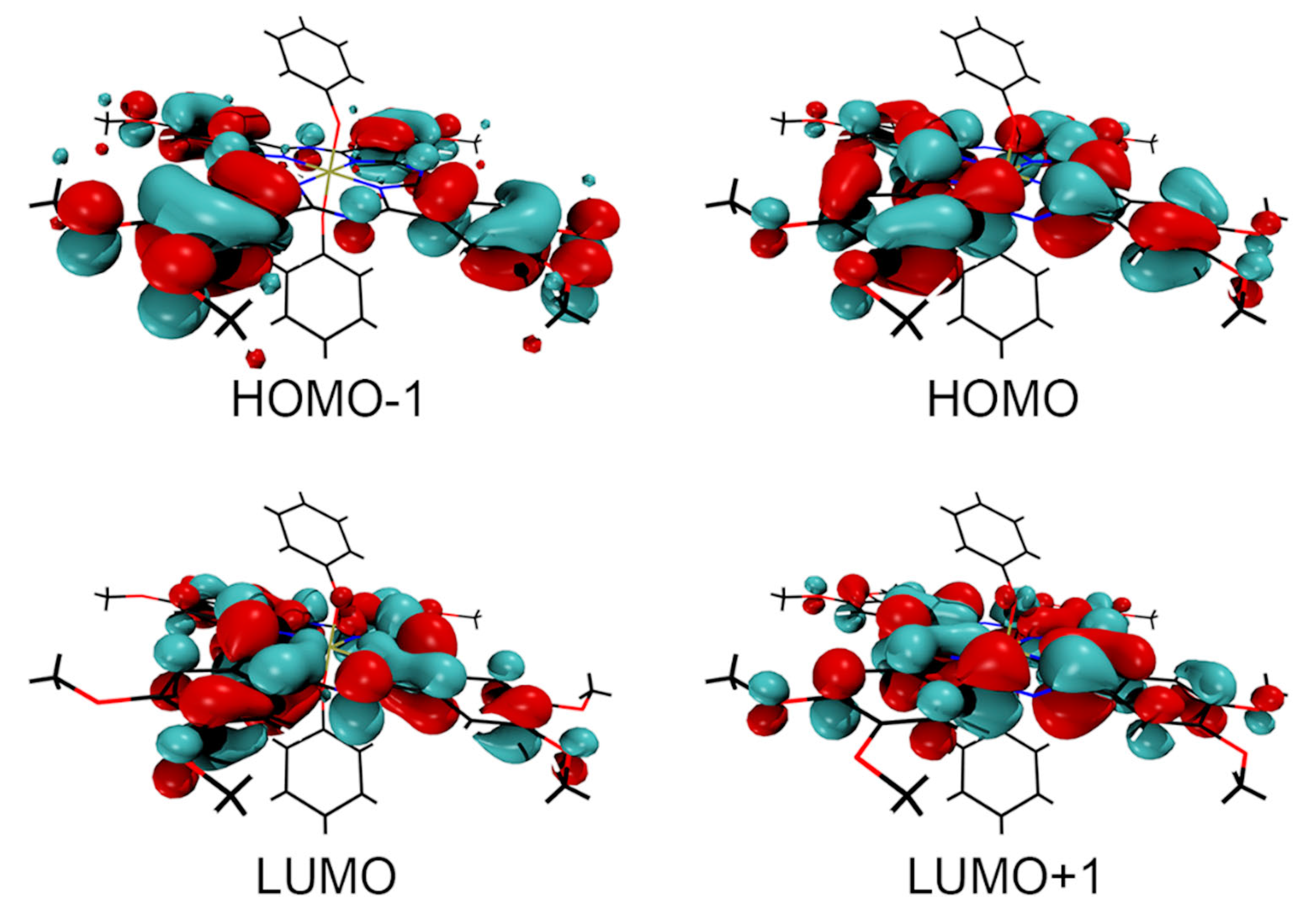
2.7. Theoretical Calculations
3. Materials and Methods
3.1. Chemicals and Instrumentation
3.2. Singlet Oxygen Generation Measurements
3.3. Fluorescence Quantum Yields
3.4. Fluorescence Lifetime Measurements
3.5. DFT Computations
3.6. Experimental Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fukuda, T.; Kobayashi, N. UV-Visible Absorption Spectroscopic Properties of Phthalocyanines and Related Macrocycles. In Handbook of Porphyrin Science; World Scientific Publishing: Singapore, 2010; pp. 1–644. [Google Scholar]
- Nyokong, T. Effects of Substituents on the Photochemical and Photophysical Properties of Main Group Metal Phthalocyanines. Coord. Chem. Rev. 2007, 251, 1707–1722. [Google Scholar] [CrossRef]
- Claessens, C.G.; Hahn, U.; Torres, T. Phthalocyanines: From Outstanding Electronic Properties to Emerging Applications. Chem. Rec. 2008, 8, 75–97. [Google Scholar] [CrossRef]
- Nyokong, T.; Antunes, E.M. Photochemical and Photophysical Properties of Metallophthalocyanines. In Handbook of Porphyrin Science; World Scientific Publishing: Singapore, 2010; pp. 247–357. ISBN 9789814307246 9814307246. [Google Scholar]
- Machacek, M.; Cidlina, A.; Novakova, V.; Svec, J.; Rudolf, E.; Miletin, M.; Kučera, R.; Simunek, T.; Zimcik, P. Far-Red-Absorbing Cationic Phthalocyanine Photosensitizers: Synthesis and Evaluation of the Photodynamic Anticancer Activity and the Mode of Cell Death Induction. J. Med. Chem. 2015, 58, 1736–1749. [Google Scholar] [CrossRef]
- Openda, Y.I.; Nyokong, T. Detonation Nanodiamonds-Phthalocyanine Photosensitizers with Enhanced Photophysicochemical Properties and Effective Photoantibacterial Activity. Photodiagn. Photodyn. Ther. 2020, 32, 102072. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zheng, B.; Peng, X.; Li, S.; Ying, J.; Zhao, Y.; Huang, J.-D.; Yoon, J. Phthalocyanines as Medicinal Photosensitizers: Developments in the Last Five Years. Coord. Chem. Rev. 2019, 379, 147–160. [Google Scholar] [CrossRef]
- Li, X.; Lee, S.; Yoon, J. Supramolecular Photosensitizers Rejuvenate Photodynamic Therapy. Chem. Soc. Rev. 2018, 47, 1174–1188. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.C.; Rodríguez-Morgade, M.S.; Pandey, R.K.; Ng, D.K.P.; Torres, T.; Dumoulin, F. The Unique Features and Promises of Phthalocyanines as Advanced Photosensitisers for Photodynamic Therapy of Cancer. Chem. Soc. Rev. 2020, 49, 1041–1056. [Google Scholar] [CrossRef]
- Santos, K.L.M.; Barros, R.M.; da Silva Lima, D.P.; Nunes, A.M.A.; Sato, M.R.; Faccio, R.; de Lima Damasceno, B.P.G.; Oshiro-Junior, J.A. Prospective Application of Phthalocyanines in the Photodynamic Therapy against Microorganisms and Tumor Cells: A Mini-Review. Photodiagn. Photodyn. Ther. 2020, 32, 102032. [Google Scholar] [CrossRef]
- Teixeira, R.; Serra, V.V.; Botequim, D.; Paulo, P.M.R.; Andrade, S.M.; Costa, S.M.B. Fluorescence Spectroscopy of Porphyrins and Phthalocyanines: Some Insights into Supramolecular Self-Assembly, Microencapsulation, and Imaging Microscopy. Molecules 2021, 26, 4264. [Google Scholar] [CrossRef] [PubMed]
- Pansare, V.J.; Hejazi, S.; Faenza, W.J.; Prud’homme, R.K. Review of Long-Wavelength Optical and NIR Imaging Materials: Contrast Agents, Fluorophores, and Multifunctional Nano Carriers. Chem. Mater. 2012, 24, 812–827. [Google Scholar] [CrossRef]
- Lobo, A.C.S.; Silva, A.D.; Tomé, V.A.; Pinto, S.M.A.; Silva, E.F.F.; Calvete, M.J.F.; Gomes, C.M.F.; Pereira, M.M.; Arnaut, L.G. Phthalocyanine Labels for Near-Infrared Fluorescence Imaging of Solid Tumors. J. Med. Chem. 2016, 59, 4688–4696. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Furuyama, T.; Satoh, K. Rationally Designed Phthalocyanines Having Their Main Absorption Band beyond 1000 Nm. J. Am. Chem. Soc. 2011, 133, 19642–19645. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Satoh, K.; Kushiya, T.; Kobayashi, N. Design, Synthesis, and Properties of Phthalocyanine Complexes with Main-Group Elements Showing Main Absorption and Fluorescence beyond 1000 Nm. J. Am. Chem. Soc. 2014, 136, 765–776. [Google Scholar] [CrossRef]
- Furuyama, T.; Kobayashi, N. Azaporphyrin Phosphorus(v) Complexes: Synthesis, Structure, and Modification of Optical Properties. Phys. Chem. Chem. Phys. 2017, 19, 15596–15612. [Google Scholar] [CrossRef] [PubMed]
- Meshkov, I.N.; Bulach, V.; Gorbunova, Y.G.; Gostev, F.E.; Nadtochenko, V.A.; Tsivadze, A.Y.; Hosseini, M.W. Tuning Photochemical Properties of Phosphorus(V) Porphyrin Photosensitizers. Chem. Commun. 2017, 53, 9918–9921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnikov, I.E.; Kurochkin, M.A.; Meshkov, I.N.; Akasov, R.A.; Kalinichev, A.A.; Kolesnikov, E.Y.; Gorbunova, Y.G.; Lähderanta, E. Water-Soluble Multimode Fluorescent Thermometers Based on Porphyrins Photosensitizers. Mater. Des. 2021, 203, 109613. [Google Scholar] [CrossRef]
- Kolesnikov, I.E.; Kalinichev, A.A.; Solomatina, A.I.; Kurochkin, M.A.; Meshkov, I.N.; Kolesnikov, E.Y.; Gorbunova, Y.G. Thermosensitive Phosphorus(V) Porphyrin: Toward Subcellular Ratiometric Optical Temperature Sensing. Sens. Actuators A Phys. 2022, 347, 113917. [Google Scholar] [CrossRef]
- Isago, H.; Fujita, H.; Hirota, M.; Sugimori, T.; Kagaya, Y. Synthesis, Spectral and Electrochemical Properties of a Novel Phosphorous(V)-Phthalocyanine. J. Porphyr. Phthalocyanines 2013, 17, 763–771. [Google Scholar] [CrossRef]
- Isago, H.; Fujita, H.; Sugimori, T. Amphoteric Phosphorous(V)-Phthalocyanines as Proton-Driven Switchable Fluorescers toward Deep-Tissue Bio-Imaging. J. Inorg. Biochem. 2018, 180, 222–229. [Google Scholar] [CrossRef]
- Furuyama, T.; Harako, R.; Kobayashi, N. Structural Changes in Non-Planar Octaaryl Substituted Phthalocyanine Phosphorus Complexes. J. Porphyr. Phthalocyanines 2015, 19, 500–509. [Google Scholar] [CrossRef]
- Kolomeychuk, F.M.; Safonova, E.A.; Polovkova, M.A.; Sinelshchikova, A.A.; Martynov, A.G.; Shokurov, A.V.; Kirakosyan, G.A.; Efimov, N.N.; Tsivadze, A.Y.; Gorbunova, Y.G. Switchable Aromaticity of Phthalocyanine via Reversible Nucleophilic Aromatic Addition to an Electron-Deficient Phosphorus(V) Complex. J. Am. Chem. Soc. 2021, 143, 14053–14058. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.P.; Goldberg, D.P. Octalkoxy-Substituted Phosphorus(V) Triazatetrabenzcorroles via Ring Contraction of Phthalocyanine Precursors. Inorg. Chem. 2003, 42, 8181–8191. [Google Scholar] [CrossRef]
- Meshkov, I.N.; Bulach, V.; Gorbunova, Y.G.; Kyritsakas, N.; Grigoriev, M.S.; Tsivadze, A.Y.; Hosseini, M.W. Phosphorus(V) Porphyrin-Based Molecular Turnstiles. Inorg. Chem. 2016, 55, 10774–10782. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Subramanian, L.R.; Hanack, M. Studies on Phosphorus Phthalocyanines and Triazatetrabenzcorroles. European J. Org. Chem. 1998, 2759–2767. [Google Scholar] [CrossRef]
- Kasuga, K.; Lin, L.; Handa, M.; Sugimori, T.; Isa, K.; Matsuura, K.; Takinami, Y. Preparation and Some Properties of a Phosphorus(V) Complex of Tetra- Tert -Butylphthalocyanine. Inorg. Chem. 1999, 38, 4174–4176. [Google Scholar] [CrossRef]
- Antunes, E.M.; Nyokong, T. Synthesis and Photophysical Behavior of Axially Substituted Phthalocyanine, Tetrabenzotriazaporphyrin, and Triazatetrabenzcorrole Phosphorous Complexes. J. Porphyr. Phthalocyanines 2009, 13, 153–160. [Google Scholar] [CrossRef]
- Durmuş, M.; Nyokong, T. Synthesis, Photophysical and Photochemical Properties of Aryloxy Tetra-Substituted Gallium and Indium Phthalocyanine Derivatives. Tetrahedron 2007, 63, 1385–1394. [Google Scholar] [CrossRef]
- Jacques, P.; Braun, A.M. Laser Flash Photolysis of Phthalocyanines in Solution and Microemulsion. Helv. Chim. Acta 1981, 64, 1800–1806. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8. [Google Scholar] [CrossRef]
- Linstead, R.P.; Lowe, A.R. 214. Phthalocyanines. Part III. Preliminary Experiments on the Preparation of Phthalocyanines from Phthalonitrile. J. Chem. Soc. 1934, 1022–1027. [Google Scholar] [CrossRef]
- Oluwole, D.O.; Yagodin, A.V.; Mkhize, N.C.; Sekhosana, K.E.; Martynov, A.G.; Gorbunova, Y.G.; Tsivadze, A.Y.; Nyokong, T. First Example of Nonlinear Optical Materials Based on Nanoconjugates of Sandwich Phthalocyanines with Quantum Dots. Chem. A Eur. J. 2017, 23, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Safonova, E.A.; Meshkov, I.N.; Polovkova, M.A.; Volostnykh, M.V.; Tsivadze, A.Y.; Gorbunova, Y.G. Photophysical and Photochemical Properties of Non-Peripheral Butoxy-Substituted Phthalocyanines with Absorption in NIR Range. Mendeleev Commun. 2018, 28, 275–277. [Google Scholar] [CrossRef]
- Kroitor, A.P.; Dmitrienko, A.A.; Martynov, A.G.; Gorbunova, Y.G.; Sorokin, A.B. Substitution Pattern in Ruthenium Octa-n-Butoxyphthalocyanine Complexes Influence Their Reactivity in N-H Carbene Insertions. Org. Biomol. Chem. 2022, 69–74. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Ring Substituents | Axial Ligands | Solvent | λabs (nm) | λem (nm) | Stocks Shift (nm) | ΦF | ΦΔ | rrel, M/s × 10−10 |
|---|---|---|---|---|---|---|---|---|---|
| 4 | - | OMe | DMSO | 708 | 736 | 28 | 0.10 | 0.27 | 172 |
| 5 | β-BuO | OMe | DMSO | 726 | 754 | 28 | 0.14 | 0.43 | 9 |
| 8 | β-BuO | OPh | DMSO | 728 | 756 | 28 | 0.10 | 0.90 | 14 |
| 7n | β-BuO | (OH)O | DMSO | 679 | 687 | 9 | 0.16 | 0.55 | 0 |
| 7c | β-BuO | (OH)2 | DMSO + TFA | 709 | 725 | 14 | 0.16 | 0.55 | 0 |
| 7a | β-BuO | O2 | DMSO + DBU | 660 | 673 | 13 | 0.02 | 0.05 | 77 |
| 6 | α-BuO | OMe | DMSO | 896 | - | - | - | - | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safonova, E.A.; Kolomeychuk, F.M.; Gvozdev, D.A.; Tsivadze, A.Y.; Gorbunova, Y.G. Tuning Photochemical and Photophysical Properties of P(V) Phthalocyanines. Molecules 2023, 28, 1094. https://doi.org/10.3390/molecules28031094
Safonova EA, Kolomeychuk FM, Gvozdev DA, Tsivadze AY, Gorbunova YG. Tuning Photochemical and Photophysical Properties of P(V) Phthalocyanines. Molecules. 2023; 28(3):1094. https://doi.org/10.3390/molecules28031094
Chicago/Turabian StyleSafonova, Evgeniya A., Filipp M. Kolomeychuk, Daniil A. Gvozdev, Aslan Yu. Tsivadze, and Yulia G. Gorbunova. 2023. "Tuning Photochemical and Photophysical Properties of P(V) Phthalocyanines" Molecules 28, no. 3: 1094. https://doi.org/10.3390/molecules28031094
APA StyleSafonova, E. A., Kolomeychuk, F. M., Gvozdev, D. A., Tsivadze, A. Y., & Gorbunova, Y. G. (2023). Tuning Photochemical and Photophysical Properties of P(V) Phthalocyanines. Molecules, 28(3), 1094. https://doi.org/10.3390/molecules28031094