
Synthesis and Biological Evaluation of Novel Dispiro-Indolinones with Anticancer Activity

 ,
, 
Abstract
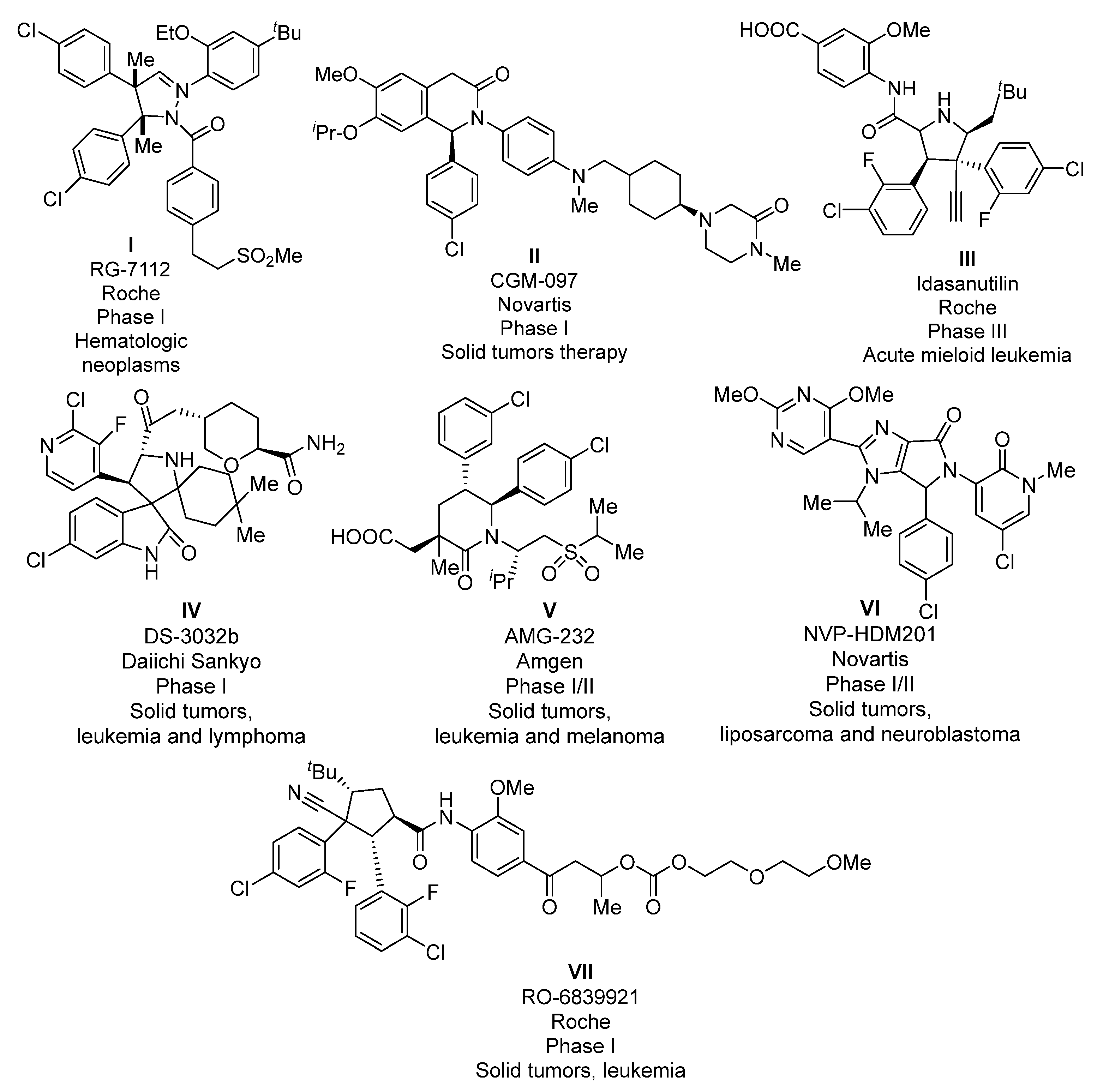
:1. Introduction
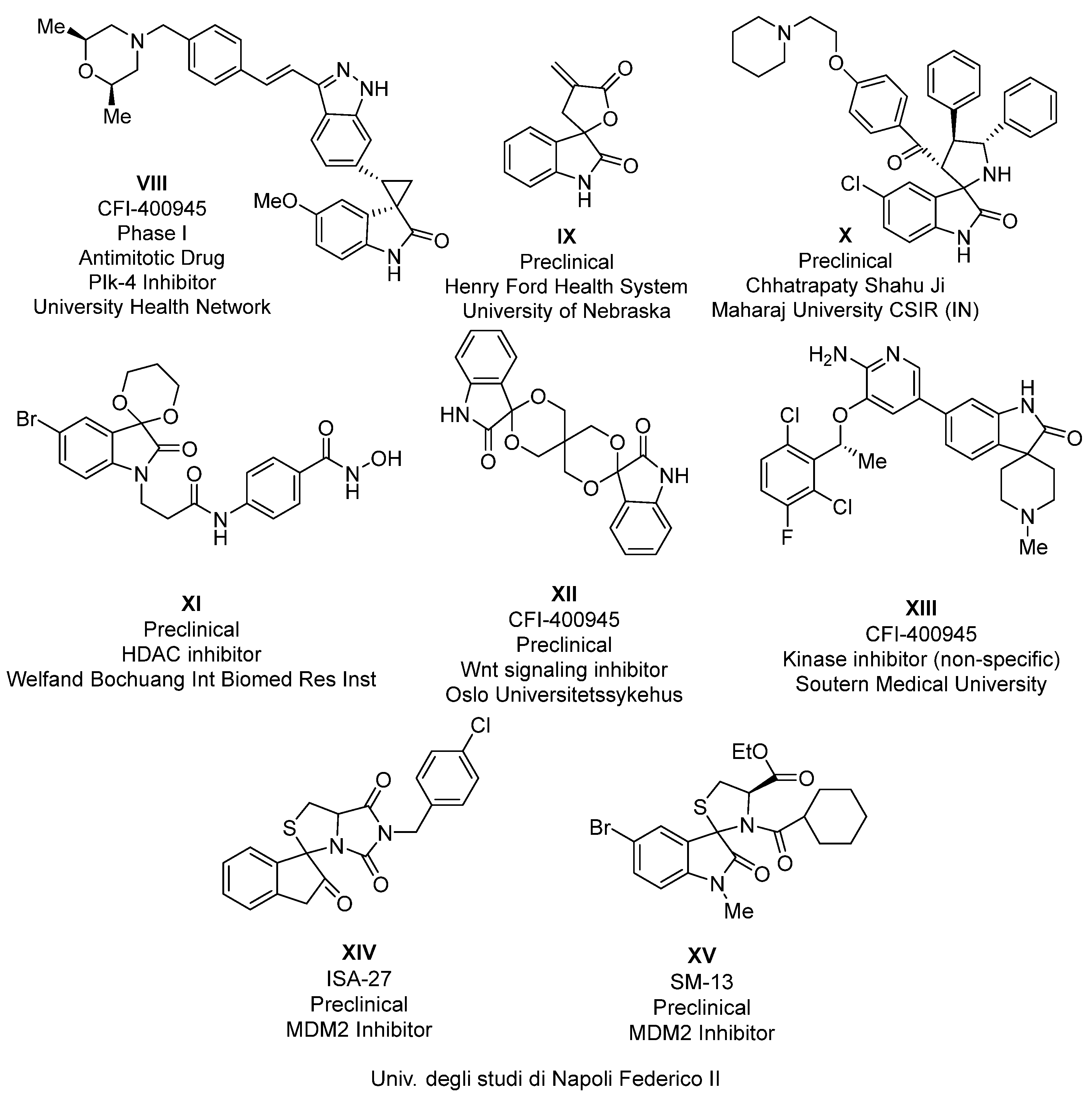
2. Results and Discussion
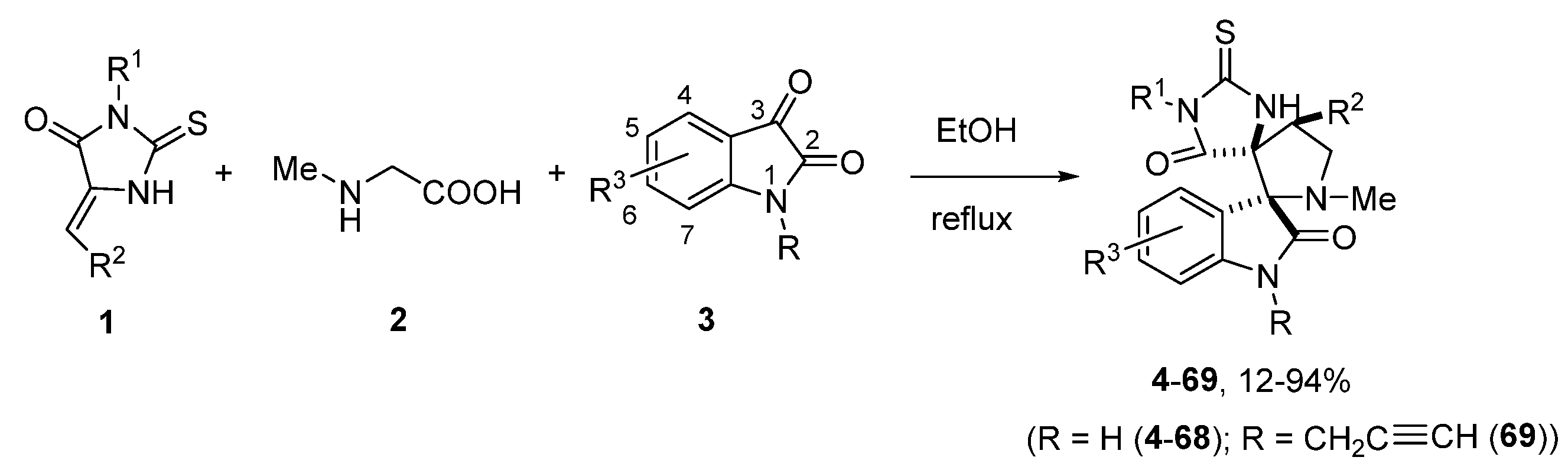
2.1. Synthesis
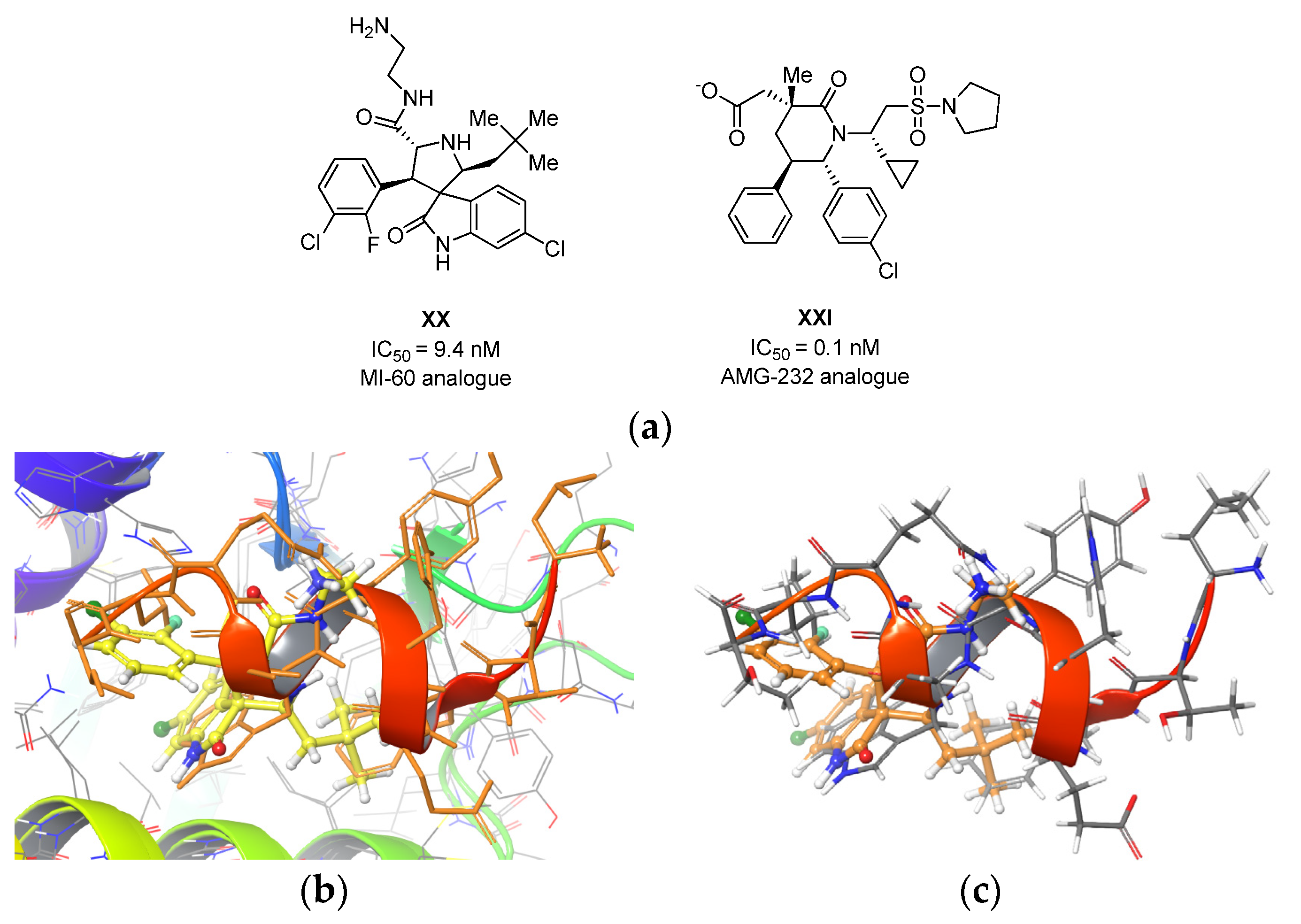
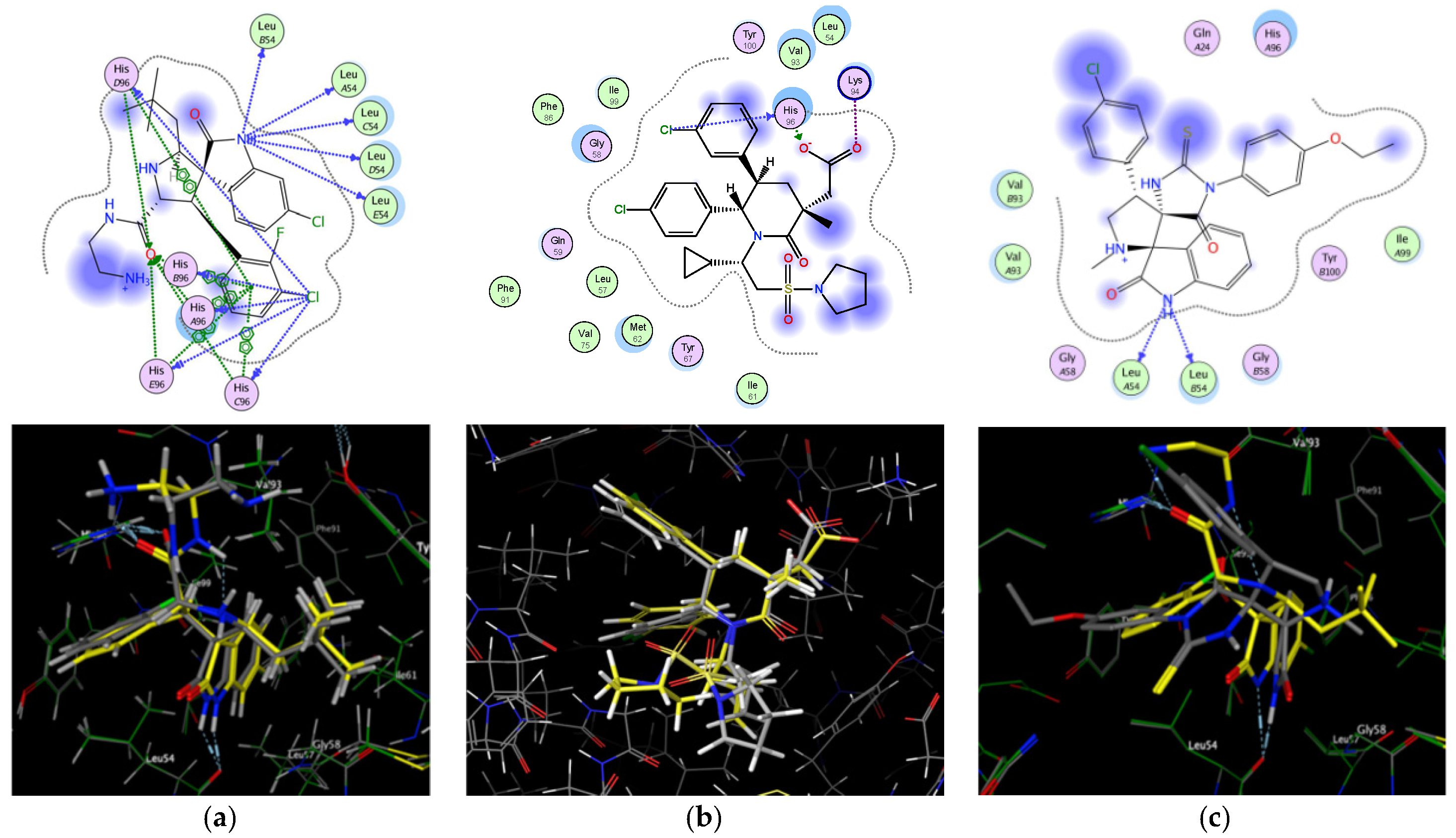
2.2. Molecular Docking Study
2.3. Biological Evaluation In Vitro
2.3.1. Cytotoxicity
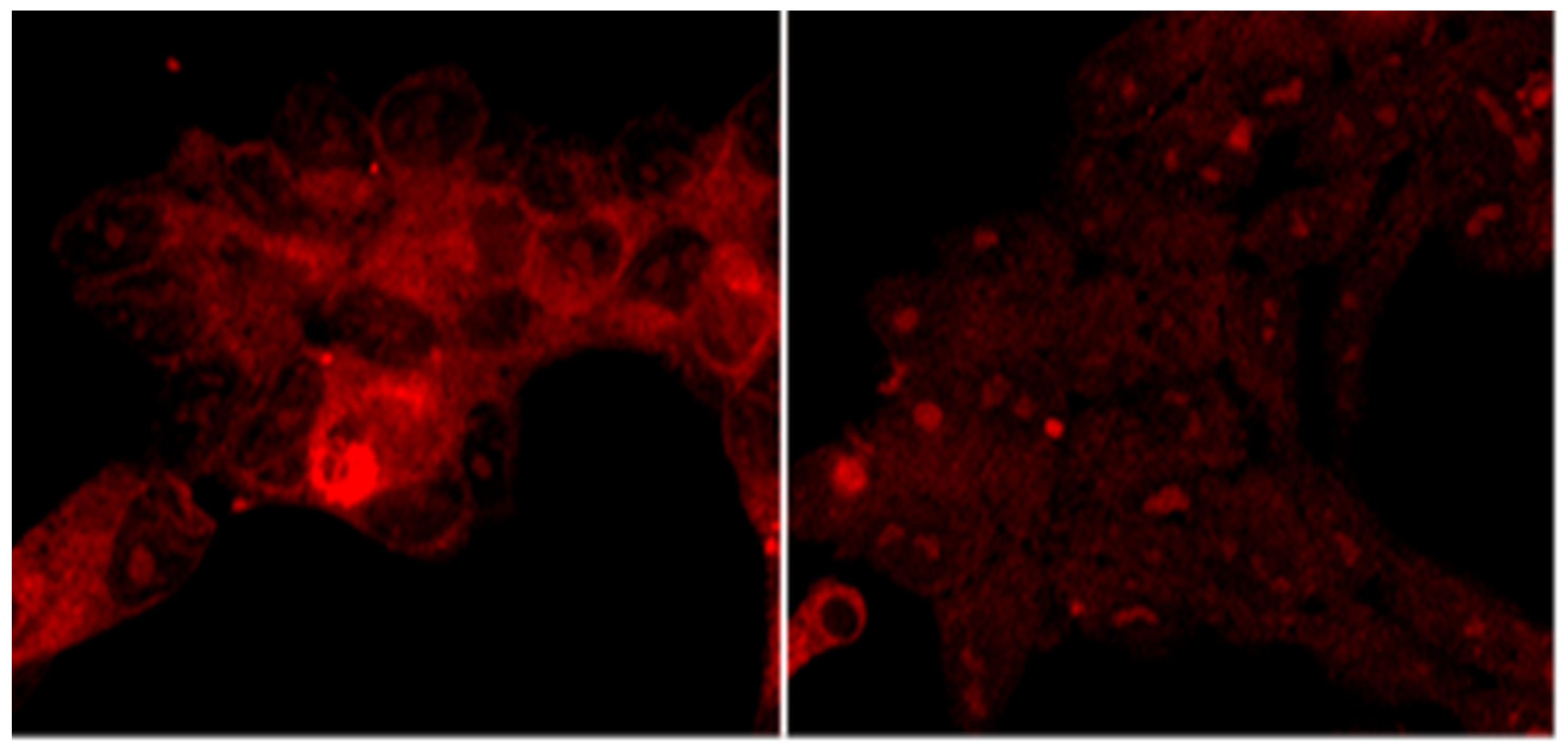
2.3.2. Intracellular Accumulation
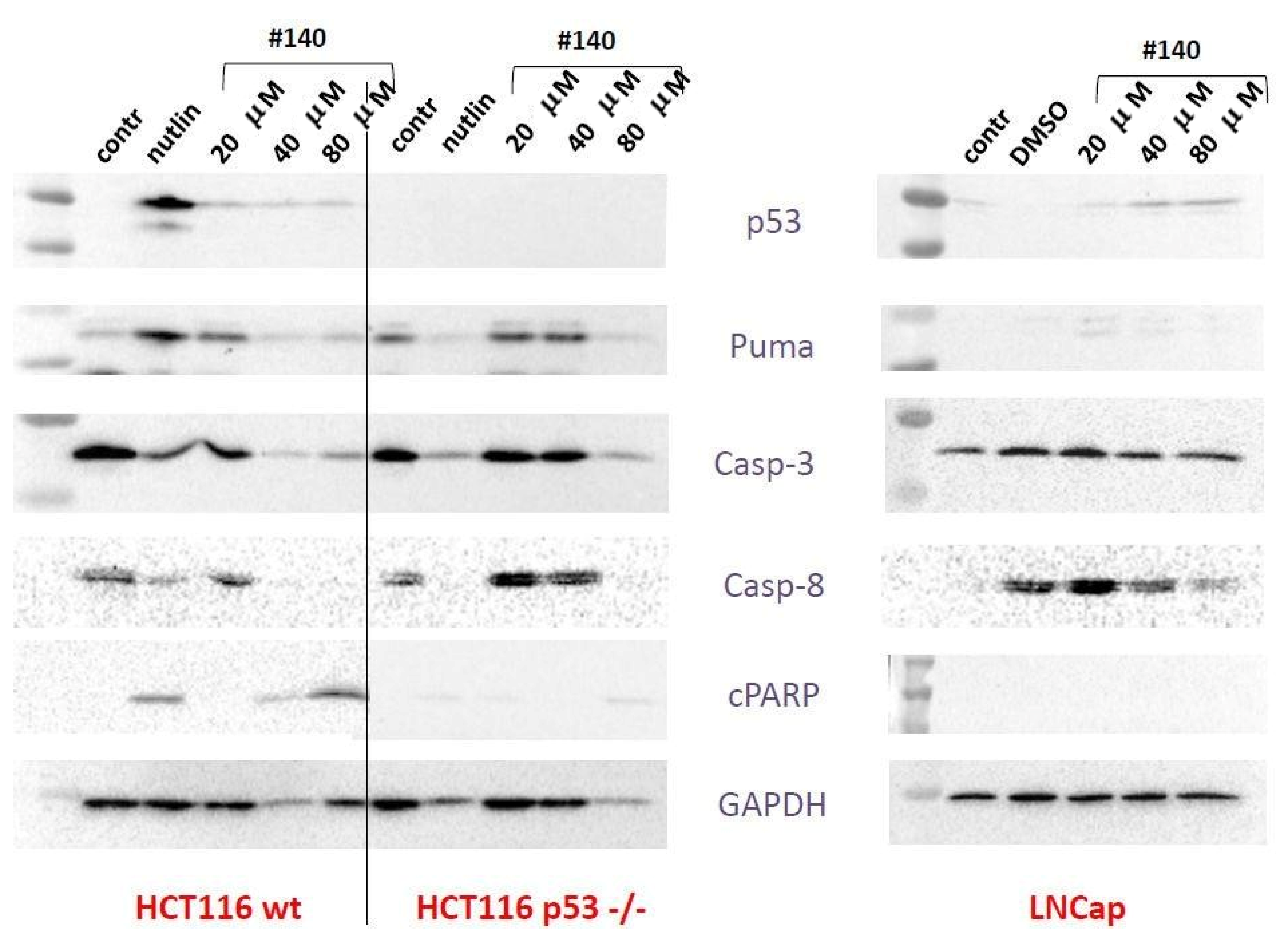
2.3.3. Western Blot, Mechanism of Cell Death, and 2D Electrophoresis
2.4. Biological Evaluation In Vivo
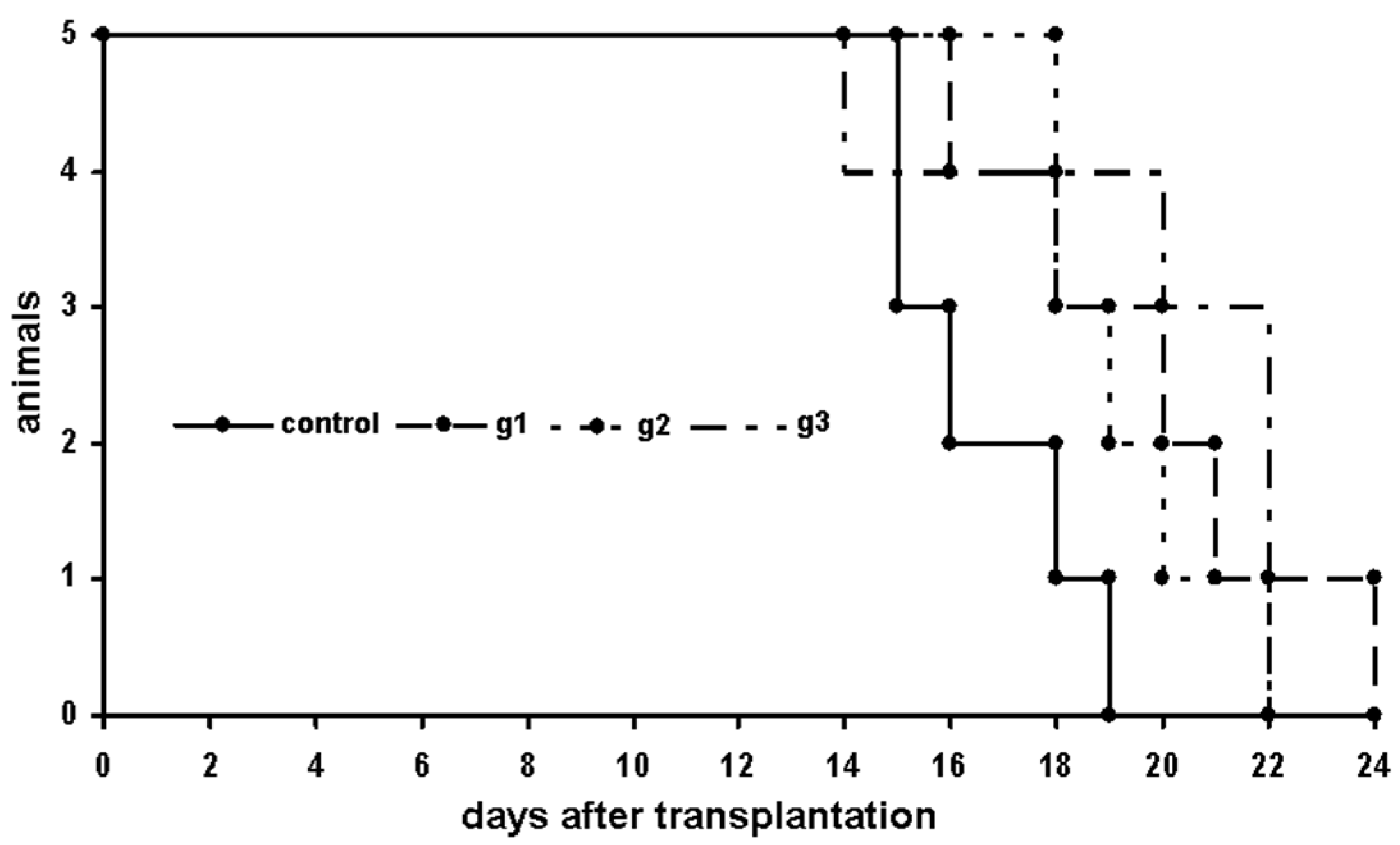
2.4.1. P388 Model
First Trial
Second Trial
Third Trial
Fourth Trial
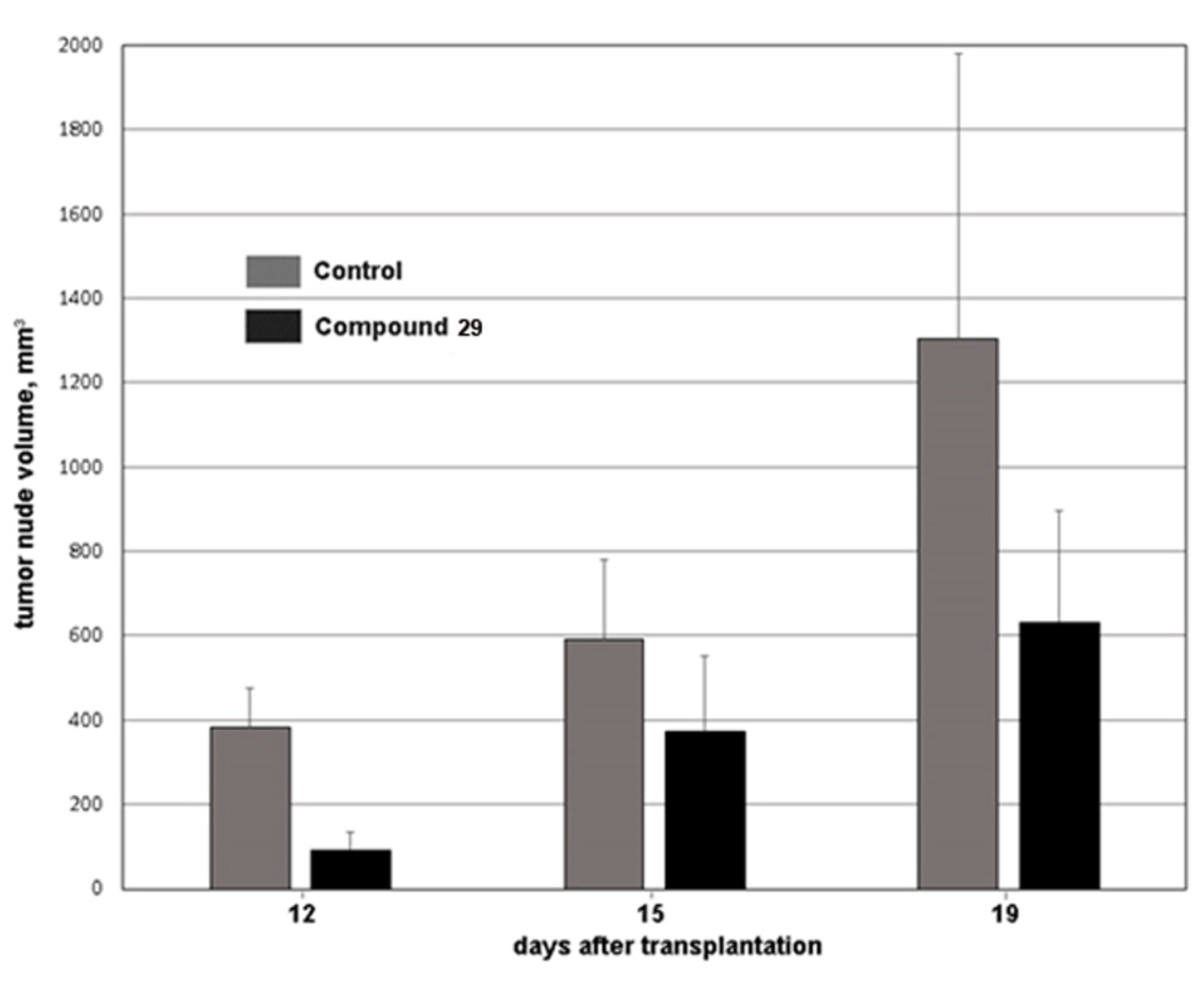
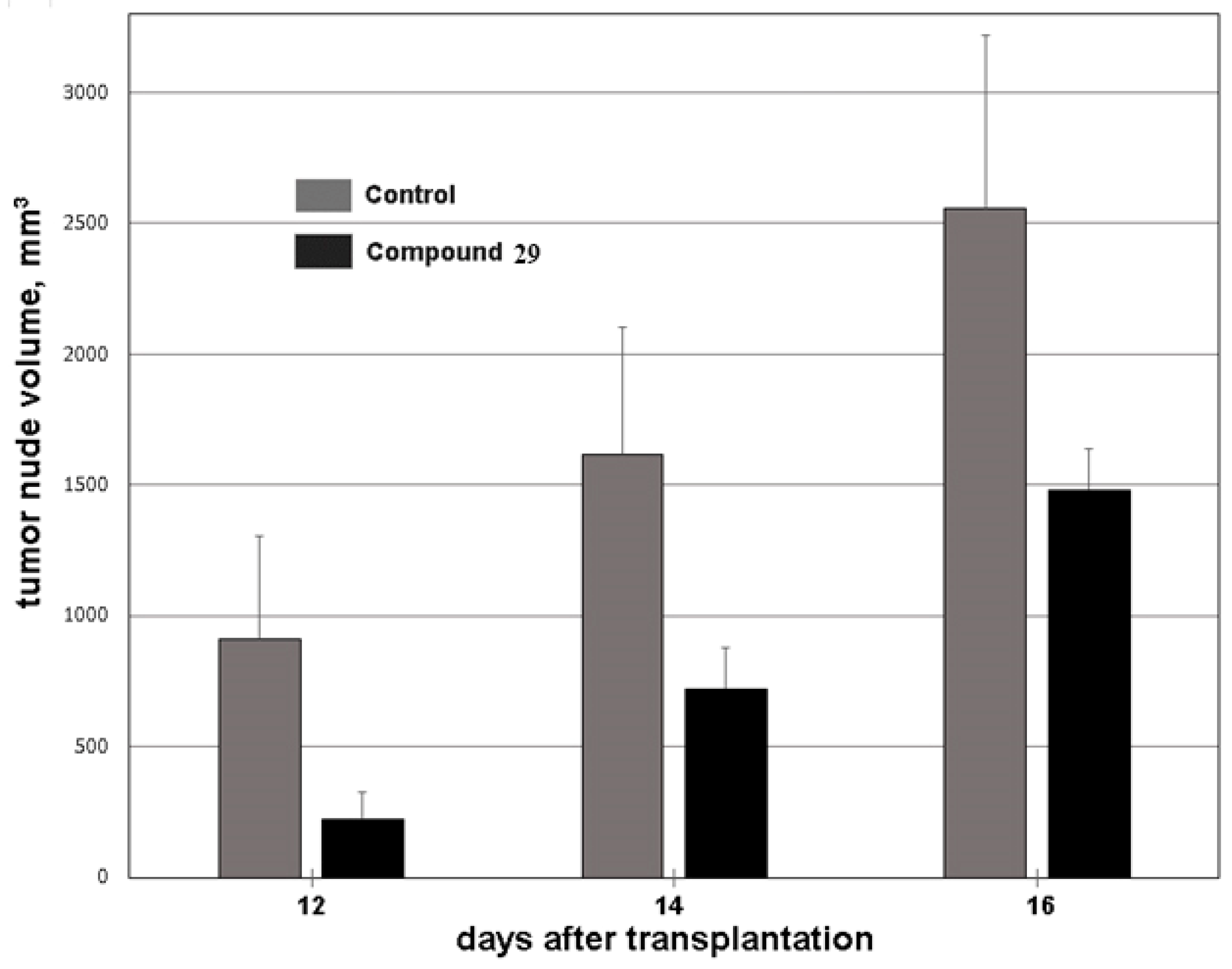
2.4.2. HCT116 Model
2.4.3. Acute Toxicity
3. Materials and Methods
3.1. General Procedure for the Synthesis of Compounds 4–69
3.2. Molecular Docking
3.3. Biological Evaluation
3.3.1. In Vitro Assay
3.3.2. MTT test
3.3.3. Intracellular Accumulation
3.3.4. Two-Dimensional (2D) Protein Gel Electrophoresis
3.3.5. In Vivo Cytotoxicity Assay
Animals
Compound Formulation
3.3.6. Acute Toxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Stiewe, T. The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer 2007, 7, 165–168. [Google Scholar] [CrossRef]
- Kemp, C.J.; Donehower, L.A.; Bradley, A.; Balmain, A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 1993, 74, 813–822. [Google Scholar] [CrossRef]
- Feki, A.; Irminger-Finger, I. Mutational spectrum of p53 mutations in primary breast and ovarian tumors. Crit. Rev. Oncol. Hematol. 2004, 52, 103–116. [Google Scholar] [CrossRef]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef]
- Montes de Oca, L.R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef] [Green Version]
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004, 119, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, C.; Bressac-de Paillerets, B.; Lefrere, I.; Ronsin, M.; Feunteun, J.; Tursz, T.; Wiels, J. Overexpression of MDM2, due to enhanced translation, results in inactivation of wild-type p53 in Burkitt’s lymphoma cells. Oncogene 1998, 16, 1603–1610. [Google Scholar] [CrossRef] [Green Version]
- Momand, J.; Wu, H.H.; Dasgupta, G. MDM2—master regulator of the p53 tumor suppressor protein. Gene 2000, 242, 15–29. [Google Scholar]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef] [Green Version]
- Ganguli, G.; Abecassis, J.; Wasylyk, B. MDM2 induces hyperplasia and premalignant lesions when expressed in the basal layer of the epidermis. EMBO J. 2000, 19, 5135–5147. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P.; Hall, P.A. MDM2-arbiter of p53's destruction. Trends Biochem. Sci. 1997, 22, 372–374. [Google Scholar] [CrossRef]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Cai, L.; Chen, C.; Xie, X.; Zhao, Q.; Zhao, X.; Zhou, H.; Han, B.; Peng, C. Computational analysis of spiro-oxindole inhibitors of the MDM2-p53 interaction: Insights and selection of novel inhibitors. J. Biomol. Struct. Dyn. 2016, 34, 341–351. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef]
- Riedinger, C.; McDonnell, J.M. Inhibitors of MDM2 and MDMX: A structural perspective. Future Med. Chem. 2009, 1, 1075–1094. [Google Scholar] [CrossRef]
- Millard, M.; Pathania, D.; Grande, F.; Xu, S.; Neamati, N. Small-molecule inhibitors of p53-MDM2 interaction: The 2006–2010 update. Curr. Pharm. Des 2011, 17, 536–559. [Google Scholar] [CrossRef]
- Vassilev, L.T. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007, 13, 23–31. [Google Scholar] [CrossRef]
- Khoury, K.; Popowicz, G.M.; Holak, T.A.; Domling, A. The p53-MDM2/MDMX axis—a chemotype perspective. Med. Chem. Comm. 2011, 2, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Chen, J.; Elenbaas, B.; Levine, A.J. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 1994, 8, 1235–1246. [Google Scholar] [CrossRef] [Green Version]
- Lemos, A.; Leão, M.; Soares, J.; Palmeira, A.; Pinto, M.; Saraiva, L.; Sousa, M.E. Medicinal Chemistry Strategies to Disrupt the p53-MDM2/MDMX Interaction. Med. Res. Rev. 2016, 36, 789–844. [Google Scholar] [CrossRef]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Ortiz, N.; Neochoritis, C.G.; Dömling, A. How To Design a Successful p53-MDM2/X Interaction Inhibitor: A Thorough Overview Based on Crystal Structures. ChemMedChem. 2016, 11, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Beloglazkina, A.; Zyk, N.; Majouga, A.; Beloglazkina, E. Recent Small-Molecule Inhibitors of the p53–MDM2 Protein–Protein Interaction. Molecules 2020, 25, 1211. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y.; et al. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef]
- Ivanenkov, Y.A.; Vasilevski, S.V.; Beloglazkina, E.K.; Kukushkin, M.E.; Machulkin, A.E.; Veselov, M.S.; Chufarova, N.V.; Chernyagina, E.S.; Vanzcool, A.S.; Zyk, N.V.; et al. Design, synthesis and biological evaluation of novel potent MDM2/p53 small-molecule inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 404–409. [Google Scholar] [CrossRef]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef]
- Watson, A.F.; Liu, J.; Bennaceur, K.; Drummond, C.J.; Endicott, J.A.; Golding, B.T.; Griffin, R.J.; Haggerty, K.; Lu, X.; McDonnell, J.M.; et al. MDM2-p53proteine-protein interaction inhibitors: A-ring substituted isoindolinones. Bioorg. Med. Chem. Lett. 2011, 21, 5916–5919. [Google Scholar] [CrossRef]
- Stoll, R.; Renner, C.; Hansen, S.; Palme, S.; Klein, C.; Belling, A.; Zeslawski, W.; Kamionka, M.; Rehm, T.; Mühlhahn, P.; et al. Chalcone derivatives antagonize interactionsbetween the human oncoprotein MDM2 and p53. Biochemistry 2001, 40, 336–344. [Google Scholar] [CrossRef]
- Rew, Y.; Sun, D.; Gonzalez-Lopez De Turiso, F.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B.M.; et al. Structure-based design of novel inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2012, 55, 4936–4954. [Google Scholar] [CrossRef]
- Zhuang, C.; Miao, Z.; Zhu, L.; Dong, G.; Guo, Z.; Wang, S.; Zhang, Y.; Wu, Y.; Yao, J.; Sheng, C.; et al. Discovery, synthesis, and biological evaluation of orally active pyrrolidone derivatives as novel inhibitors of p53-MDM2 proteinprotein interaction. J. Med. Chem. 2012, 55, 9630–9642. [Google Scholar] [CrossRef]
- Lee, J.H.; Zhang, Q.; Jo, S.; Chai, S.C.; Oh, M.; Im, W.; Lu, H.; Lim, H.S. Novel pyrrolopyrimidine-based a-Helix mimetics: Cell-permeable inhibitors of protein-protein interactions. J. Am. Chem. Soc. 2011, 133, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Beck, H.P.; De Graffenreid, M.; Fox, B.; Allen, J.G.; Rew, Y.; Schneider, S.; Saiki, A.Y.; Yu, D.; Oliner, J.D.; Salyers, K.; et al. Improvement of the synthesis and pharmacokinetic properties of chromenotriazolopyrimidine MDM2-p53 protein-protein inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2752–2755. [Google Scholar] [CrossRef]
- Leão, M.; Pereira, C.; Bisio, A.; Ciribilli, Y.; Paiva, A.M.; Machado, N.; Palmeira, A.; Fernandes, M.X.; Sousa, E.; Pinto, M.; et al. Discovery of a new small-molecule inhibitor of p53eMDM2 interaction using a yeast-based approach. Biochem. Pharmacol. 2013, 85, 1234–1245. [Google Scholar] [CrossRef]
- Stachyra-Valat, T.; Baysang, F.; D’Alessandro, A.C.; Dirk, E.; Furet, P.; Guagnano, V.; Kallen, J.; Leder, L.; Mah, R.; Masuya, K.; et al. NVP-HDM201: Biochemical and biophysical profile of a novel highly potent and selective PPI inhibitor of p53-Mdm2. Cancer Res. 2016, 76, 1239. [Google Scholar]
- Sarek, G.; Kurki, S.; Enbäck, J.; Iotzova, G.; Haas, J.; Laakkonen, P.; Laiho, M.; Ojala, P.M. Reactivation of the p53 pathway as a treatment modality for KSHVinduced lymphomas. J. Clin. Invest. 2007, 117, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef]
- Wang, W.S.; Zhu, X.L.; Hong, X.Q.; Zheng, L.; Zhu, H.; Hu, Y.Z. Identification of novel inhibitors of p53-MDM2 interaction facilitated by pharmacophore-based virtual screening combining molecular docking strategy. Med. Chem. Commun. 2013, 4, 411–416. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Hu, Y. Pharmacophore Model Construction of p53-MDM2 Binding Inhibitors and its Application in the Discovery of a Novel Lead Compound. In Proceedings of the 22nd International Symposium on Medicinal Chemistry, Berlin, Germany, 2–6 September 2012; p. 206. [Google Scholar]
- Available online: http://clinicaltrials.gov/ct2/show/NCT01462175?term¼RO5503781&rank¼1 (accessed on 10 October 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT00559533 (accessed on 10 October 2022).
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the phase i trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Xia, B.H.M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; To, K.-H.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef] [Green Version]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT02545283 (accessed on 10 October 2022).
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Yu, D.Q.; Liu, H.M. Spirooxindoles: Promising scaffolds for anticancer agents. Eur. J. Med. Chem. 2015, 97, 673–698. [Google Scholar] [CrossRef]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, L.; Sun, W.; Lu, J.; McEachern, D.; Li, X.; Yu, S.; Bernard, D.; Ochsenbein, P.; Ferey, V.; et al. Diastereomericspirooxindoles as highly potent and efficacious MDM2 inhibitors. J. Am. Chem. Soc. 2013, 135, 7223–7234. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Del Giudice, C.; Bertamino, A.; Ciccarelli, M.; Gomez-Monterrey, I.; Campiglia, P.; Novellino, E.; Illario, M.; Trimarco, B.; De Luca, N.; et al. New small molecules, ISA27 and SM13, inhibit tumour growth inducing mitochondrial effects of p53. Br. J. Cancer. 2015, 112, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Patent WO 2012065022. Available online: https://pubchem.ncbi.nlm.nih.gov/patent/WO-2012065022-A3 (accessed on 11 November 2011).
- Voruganti, S.; Qin, J.J.; Sarkar, S.; Nag, S.; Walbi, I.A.; Wang, S.; Zhao, Y.; Wang, W.; Zhang, R. Oral nano-delivery of anticancer ginsenoside 25-OCH3-PPD, a natural inhibitor of the MDM2 oncogene: Nanoparticle preparation, characterization, in vitro and in vivo anti-prostate cancer activity, and mechanisms of action. Oncotarget 2015, 6, 21379–21394. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Li, Z.; Rew, Y.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; Deignan, J.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable inhibitor of the MDM2-p53 interaction. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef]
- Allen, J.G.; Bourbeau, M.P.; Wohlhieter, G.E.; Bartberger, M.D.; Michelsen, K.; Hungate, R.; Gadwood, R.C.; Gaston, R.D.; Evans, B.; Mann, L.W.; et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2-tumor protein 53 protein−protein interaction. J. Med. Chem. 2009, 52, 7044–7053. [Google Scholar] [CrossRef]
- Lucas, B.S.; Fisher, B.; McGee, L.R.; Olson, S.H.; Medina, J.C.; Cheung, E. An expeditious synthesis of the MDM2-p53 inhibitor AM-8553. J. Am. Chem. Soc. 2012, 134, 12855–12860. [Google Scholar] [CrossRef]
- Gonzalez-Lopez de Turiso, F.; Sun, D.; Rew, Y.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Correll, T.L.; Huang, X.; et al. Rational design and binding mode duality of MDM2-p53 inhibitors. J. Med. Chem. 2013, 56, 4053–4070. [Google Scholar] [CrossRef]
- Bernard, D.; Zhao, Y.; Wang, S. AM-8553: A novel MDM2 inhibitor with a promising outlook for potential clinical development. J. Med. Chem. 2012, 55, 4934–4935. [Google Scholar] [CrossRef]
- Gonzalez, A.Z.; Eksterowicz, J.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Fox, B.M.; Fu, J.; et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J. Med. Chem. 2014, 57, 2472–2488. [Google Scholar] [CrossRef]
- Gonzalez, A.Z.; Li, Z.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Eksterowicz, J.; Fox, B.M.; Fu, J.; et al. Novel Inhibitors of the MDM2-p53 Interaction Featuring Hydrogen Bond Acceptors as Carboxylic Acid Isosteres. J. Med. Chem. 2014, 57, 2963–2988. [Google Scholar] [CrossRef]
- Rew, Y.; Sun, D.; Yan, X.; Beck, H.P.; Canon, J.; Chen, A.; Duquette, J.; Eksterowicz, J.; Fox, B.M.; Fu, J.; et al. Discovery of AM-7209, a potent and selective 4-amidobenzoic acid inhibitor of the MDM2-p53 interaction. J. Med. Chem. 2014, 57, 10499–10511. [Google Scholar] [CrossRef]
- Somsak, L.; Kovacs, L.; Toth, M.; Osz, E.; Szilagyi, L.; Gyoergydeak, Z.; Dinya, Z.; Docsa, T.; Toth, B.; Gergely, P. Synthesis of and a comparative study on the inhibition of muscle and liver glycogen phosphorylases by epimeric pairs of d-gluco-and d-xylopyranosylidene-spiro-(thio) hydantoins and N-(d-glucopyranosyl) amides. J. Med. Chem. 2001, 44, 2843–2848. [Google Scholar] [CrossRef]
- Jung, M.E.; Yoo, D.; Sawyers, C.L.; Tran, C.; Wongvipat, J. Diarylthiohydantoin compounds. U.S. Patent US8648105B2, 11 February 2014. [Google Scholar]
- Jung, M.E.; Ouk, S.; Yoo, D.; Sawyers, C.L.; Chen, C.; Tran, C.; Wongvipat, J. Structure−activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J. Med. Chem. 2010, 53, 2779–2796. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.E.; Sawyers, C.L.; Ouk, S.; Tran, C.; Wongvipat, J. Androgen receptor modulator for the treatment of prostate cancer and androgen receptor-associated diseases. U.S. Patent 8445507B2, 21 May 2013. [Google Scholar]
- He, J.; Ouyang, G.; Yuan, Z.; Tong, R.; Shi, J.; Ouyang, L. A facile synthesis of functionalized dispirooxindole derivatives via a three-component 1, 3-dipolar cycloaddition reaction. Molecules 2013, 18, 5142–5154. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Sun, Y.; Gong, H.; Xie, Y.J.; Yan, C.G. Facile synthesis of dispirooxindole-fused heterocycles via domino 1, 4-dipolar addition and Diels–Alder reaction of in situ generated Huisgen 1, 4-dipoles. Org. Lett. 2012, 14, 5172–5175. [Google Scholar] [CrossRef]
- Beloglazkina, A.A.; Karpov, N.A.; Mefedova, S.R.; Polyakov, V.S.; Skvortsov, D.A.; Kalinina, M.A.; Tafeenko, V.A.; Majouga, A.G.; Zyk, N.V.; Beloglazkina, E.K. Synthesis of dispirooxindoles containing N-unsubstituted heterocyclic moieties and study of their anticancer activity. Rus. Chem. Bull. 2019, 68, 1006–1013. [Google Scholar] [CrossRef]
- Beloglazkina, A.; Barashkin, A.; Polyakov, V.; Kotovsky, G.; Karpov, N.; Mefedova, S.; Zagribelny, B.; Ivanenkov, Y.; Kalinina, M.; Skvortsov, D.; et al. Synthesis and Biological Evaluation of Novel Dispiro Compounds based on 5-Arylidenehydantoins and Isatins as Inhibitors of p53–MDM2 Protein–Protein Interaction. Chem. Heterocycl. Compd. 2020, 56, 747–755. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Available online: https://www.chemcomp.com (accessed on 10 October 2022).
- Baek, S.; Kutchukian, P.S.; Verdine, G.L.; Huber, R.; Holak, T.A.; Lee, K.W.; Popowicz, G.M. Structure of the stapled p53 peptide bound to Mdm2. J. Am. Chem. Soc. 2012, 134, 103–106. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; Liu, J.J.; Chen, X.; Mihalic, J.; Deignan, J.; Yu, M.; Sun, D.; Kayser, F.; McGee, L.R.; et al. Optimization beyond AMG 232: Discovery and SAR of sulfonamides on a piperidinone scaffold as potent inhibitors of the MDM2-p53 protein-protein interaction. Bioorg. Med. Chem. Lett. 2014, 24, 3782–3785. [Google Scholar] [CrossRef]
- Anil, B.; Riedinger, C.; Endicott, J.A.; Noble, M.E. The structure of an MDM2–Nutlin-3a complex solved by the use of a validated MDM2 surface-entropy reduction mutant. Acta Crystallogr. Sect. D 2013, 69, 1358–1366. [Google Scholar] [CrossRef]
- Ferrari, M.; Fornasiero, M.C.; Isetta, A.M. MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J. Immunol. Methods 1990, 131, 165–172. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Rcsb. Available online: http://www.rcsb.org/ (accessed on 10 October 2022).
- Ronc. Available online: http://www.ronc.ru/ (accessed on 10 October 2022).
- Petrov, R.A.; Mefedova, S.R.; Yamansarov, E.Y.; Maklakova, S.Y.; Grishin, D.A.; Lopatukhina, E.V.; Burenina, O.Y.; Lopukhov, A.V.; Kovalev, S.V.; Timchenko, Y.V.; et al. New Small-Molecule Glycoconjugates of Docetaxel and GalNAc for Targeted Delivery to Hepatocellular Carcinoma. Mol. Pharm. 2021, 18, 461–468. [Google Scholar] [CrossRef]
- Pitomniki Stolbovaya. Available online: http://www.pitomniki-stolbovaya.com/ (accessed on 10 October 2022).
- Beloglazkina, E.K.; Vatsadze, S.Z.; Majouga, A.G.; Frolova, N.A.; Romashkina, R.B.; Zyk, N.V.; Moiseeva, A.A.; Butin, K.P. Synthesis and electrochemical study of complexes of 2-methylthio-5-(pyridylmethylidene)-3,5-dihydro-4H-imidazol-4-ones with transition metals (Co, Ni, and Cu). Molecular structures of CuIIL1Cl2 (L1 is (5Z)-2-methylthio-3-phenyl-5-(α-pyridylmethylidene)-3,5-dihydro-4H-imidazol-4-one) and CoIIL2Cl2 (L2 is (5Z )-3-methyl-2-methylthio-5-(α-pyridylmethylidene)-3,5-dihydro-4H-imidazol-4-one). Russ. Chem. Bull. 2005, 54, 2771–2782. [Google Scholar] [CrossRef]
- Ivanenkov, Y.A.; Majouga, A.G.; Beloglazkina, E.K.; Beloglazkina, A.A.; Veselov, M.S.; Kukushkin, M.E. New dispiro-indolinones, MDM2/p53 interaction inhibitors, method for production and application. RU 2015113026, 9 April 2015. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | R1 | R2 | R3 | Yield, % |
|---|---|---|---|---|
| 4 | Ph- | Ph- | H | 76 |
| 5 | Ph- | Ph- | 5-Br | 72 |
| 6 | Ph- | Ph- | 5-NO2 | 63 |
| 7 | Ph- | Ph- | 7-COOH- | 61 |
| 8 | Ph- | 2-Br-C6H4- | H | 86 |
| 9 | Ph- | 2-Cl-C6H4- | H | 87 |
| 10 | Ph- | 2-Cl-C6H4- | 5-Br | 89 |
| 11 | Ph- | 2-Cl-C6H4- | 5-Cl | 87 |
| 12 | Ph- | 4-Cl-C6H4- | 5-Cl | 69 |
| 13 | Ph- | 4-Cl-C6H4- | 5-Br | 78 |
| 14 | Ph- | 3-Cl-C6H4- | H | 57 |
| 15 | Ph- | 3-Cl-C6H4- | 5-Br | 65 |
| 16 | Ph- | 3,4-Cl-C6H3- | 5-Br | 61 |
| 17 | Ph- | 2-Py- | 5-Br | 65 |
| 18 | Ph- | 2-Py- | 5-Cl | 53 |
| 19 | Ph- | 4-Py- | H- | 87 |
| 20 | Ph- | 4-Py- | 5-Br | 89 |
| 21 | Ph- | 4-Py- | 5-Cl | 89 |
| 22 | Ph- | 3,4-Cl-C6H3- | 5-Br | 61 |
| 23 | Ph- | 3-Cl-C6H4- | 5-Cl | 68 |
| 24 | Ph- | 3-CHO-C6H4- | 5-Cl- | 71 |
| 25 | Ph- | cyclo-C6H11- | H | 36 |
| 26 | Ph- | cyclo-C6H11- | 5-Br | 35 |
| 27 | 4-OC2H5-C6H4- | 3,4-Cl-C6H3- | H- | 79 |
| 28 | 4-OC2H5-C6H4- | 4-Cl-C6H4- | H- | 62 |
| 29 | 4-OC2H5-C6H4- | 4-Cl-C6H4- | 5-Br | 89 |
| 30 | 4-OC2H5-C6H4- | 4-F-C6H4- | H | 72 |
| 31 | 4-OC2H5-C6H4- | 4-F-C6H4- | 5-Br | 68 |
| 32 | 4-OCH3-C6H4- | 3-Cl-C6H4- | H | 59 |
| 33 | 4-OCH3-C6H4- | 3-Cl-C6H4- | 5-Br | 70 |
| 34 | 4-OC2H5-C6H4- | 4-OPropargyl- C6H4- | 5-Br | 59 |
| 35 | 4-OPropargyl-C6H4- | 4-Cl-C6H4- | 5-Br | 94 |
| 36 | 4-Cl-C6H4- | 2-Cl-C6H4- | 5-Cl | 83 |
| 37 | 4-Cl-C6H4- | 4-Cl-C6H4- | 5-Cl | 85 |
| 38 | 4-Cl-C6H4- | 4-F-C6H4- | H | 85 |
| 39 | 4-Cl-C6H4- | 3,4-(OCH3)2-C6H3- | 5-Cl | 81 |
| 40 | 4-F-C6H4- | 4-Cl-C6H4- | 5-Br | 68 |
| 41 | 3-Cl-4-F-C6H3- | 4-OCH3-C6H4- | H | 69 |
| 42 | 3-Cl-4-F-C6H3- | 4-Cl-C6H4- | 5-Cl | 73 |
| 43 | 3-Cl-4-F-C6H3- | 3-Cl-C6H4- | 5-Cl | 79 |
| 44 | 3-Cl-4-F-C6H3- | 3,4-(OCH3)2-C6H3- | H | 68 |
| 45 | 3-Cl-4-F-C6H3- | 3,4-(OCH3)2-C6H3- | 5-Br | 41 |
| 46 | 3-Cl-4-F-C6H3- | 4-C2H5-C6H4- | H | 49 |
| 47 | 3-Cl-4-F-C6H3- | 4-C2H5-C6H4- | Br | 48 |
| 48 | 3-Cl-4-F-C6H3- | 4-OCH3-C6H4- | Br | 68 |
| 49 | 3-Cl-4-F-C6H3- | 4-O(cyclo-C5H9)-C6H4- | Br | 34 |
| 50 | 4-OH-C6H4- | 4-Cl-C6H4- | H | 94 |
| 51 | 4-OH-C6H4- | 4-Cl-C6H4- | 5-Br | 91 |
| 52 | 4-OH-C6H4- | 4-Cl-C6H4- | 5-Cl | 88 |
| 53 | 4-OH-C6H4- | 2-Cl-C6H4- | H | 75 |
| 54 | 4-OH-C6H4- | 2-Cl-C6H4- | 5-Br | 76 |
| 55 | 4-OH-C6H4- | 2-Cl-C6H4- | 5-Cl | 75 |
| 56 | 4-OH-C6H4- | 3,4-OCH3-C6H3- | 5-Br | 69 |
| 57 | 4-OH-C6H4- | 3,4-OCH3-C6H3- | 5-Cl | 53 |
| 58 | 4-OCH3-3-Cl-C6H3- | 3-Cl-C6H4- | H | 54 |
| 59 | 4-OCH3-3-Cl-C6H3- | 3-Cl-C6H4- | 5-Br | 51 |
| 60 | 4-F-C6H4- | 4-OC2H5-C6H4- | H | 38 |
| 61 | 4-F-C6H4- | 4-OC2H5-C6H4- | 5-Br | 36 |
| 62 | CH3CHPh- | 2-Cl-C6H4- | 5-Br | 89 |
| 63 | CH3CHPh- | 3-Cl-C6H4- | 5-Br | 53 |
| 64 | CH3CHPh- | 2-Cl-C6H4- | 5-Cl | 83 |
| 65 | CH3CHPh- | 4-Cl-C6H4- | 5-Br | 65 |
| 66 | CH3CHPh- | 4-Cl-C6H4- | 5-Cl | 62 |
| 67 | Allyl- | 2-Py | H | 12 |
| 68 | Allyl- | 3-Cl-C6H4- | H | 12 |
| 69 | 4-OCH3-3-Cl-C6H3- | 3-Cl-C6H4- | 1-CH2CCH | 36 |
| Compound | Cell Line | |||
|---|---|---|---|---|
| p53 pos | 53 neg | p53 pos | 53 neg | |
| LNCaP | PC3 | HCTwt | HCT−/− | |
| 4 | >100 | >100 | >100 | >100 |
| 5 | 2.2 ± 0.8 | 4.6 ± 2.5 | 10.5 ± 2.3 | 12.5 ± 3.2 |
| 6 | >100 | >100 | >100 | >100 |
| 7 | >100 | >100 | >100 | >100 |
| 8 | >100 | >100 | >100 | >100 |
| 9 | >100 | >100 | 21.7 ± 5.8 | 23.1 ± 6.6 |
| 10 | >100 | >100 | - | - |
| 11 | >100 | >100 | >100 | >100 |
| 12 | >100 | >100 | >100 | >100 |
| 13 | 1.2 ± 0.6 | 11.3 ± 1.2 | 3.5 ± 1.2 | >100 |
| 14 | >100 | >100 | - | - |
| 15 | >100 | >100 | >100 | >100 |
| 16 | >100 | >100 | 7.6 ± 1.6 | 8.8 ± 3.5 |
| 17 | >100 | >100 | 5.6 ± 2.3 | 7.6 ± 3.2 |
| 18 | 2.1 ± 0.3 | 8.2 ± 3.5 | 60.1 ± 8.6 | >100 |
| 19 | >100 | >100 | >100 | >100 |
| 20 | >100 | >100 | >100 | >100 |
| 21 | >100 | >100 | 50.5 ± 10.2 | >100 |
| 22 | >100 | >100 | 20.0 ± 8.9 | 50.0 ± 10.4 |
| 23 | >100 | >100 | 50.7 ± 8.9 | 50.7 ± 12.6 |
| 24 | >100 | >100 | >100 | >100 |
| 25 | >100 | >100 | - | - |
| 26 | >100 | >100 | - | - |
| 27 | >100 | >100 | >100 | >100 |
| 28 | >100 | >100 | >100 | >100 |
| 29 | 3.5 ± 1.9 | 9.8 ± 3.7 | 14.1 ± 4.2 | 70.5 ± 15.2 |
| 30 | >100 | >100 | >100 | >100 |
| 31 | >100 | >100 | >100 | >100 |
| 32 | >100 | >100 | >100 | >100 |
| 33 | >100 | >100 | >100 | >100 |
| 34 | >100 | >100 | >100 | >100 |
| 35 | >100 | >100 | >100 | >100 |
| 36 | >100 | >100 | >100 | >100 |
| 37 | >100 | >100 | >100 | >100 |
| 38 | >100 | >100 | >100 | >100 |
| 39 | >100 | >100 | >100 | >100 |
| 40 | >100 | >100 | >100 | >100 |
| 41 | >100 | >100 | >100 | >100 |
| 42 | >100 | >100 | >100 | >100 |
| 43 | 3.2 ± 1.6 | 5.7 ± 2.6 | 9.3 ± 4.6 | 9.5 ± 4.9 |
| 44 | 8.6 ± 1.8 | 10.2 ± 2.3 | 35.0 ± 16.2 | >100 |
| 45 | >100 | >100 | - | - |
| 46 | >100 | >100 | 6.3 ± 2.5 | 6.5 ± 3.6 |
| 47 | >100 | >100 | >100 | >100 |
| 48 | >100 | >100 | >100 | >100 |
| 49 | >100 | >100 | - | - |
| 50 | 18.0 ± 4.7 | 4.65 ± 2.3 | 6.9 ± 3.1 | >100 |
| 51 | >100 | >100 | >100 | >100 |
| 52 | >100 | >100 | >100 | >100 |
| 53 | >100 | >100 | >100 | >100 |
| 54 | 9.5 ± 2.5 | 20.0 ± 8.4 | >100 | >100 |
| 55 | 7.0 ± 1.0 | 15.1 ± 1.2 | >100 | >100 |
| 56 | 9.0 ± 2.0 | 30.0 ± 10.5 | >100 | >100 |
| 57 | 17.0 ± 1.0 | 20.0 ± 6.9 | >100 | >100 |
| 58 | >100 | >100 | - | - |
| 59 | >50 | >50 | - | - |
| 60 | >100 | >100 | - | - |
| 61 | >100 | >100 | - | - |
| 62 | >100 | >100 | >100 | >100 |
| 63 | 3.4 ± 1.5 | 8.8 ± 3.1 | 8.0 ± 2.5 | >100 |
| 64 | >100 | >100 | 24.0 ± 6.7 | >100 |
| 65 | >100 | >100 | >100 | >100 |
| 66 | >100 | >100 | >100 | >100 |
| 67 | >50 | >50 | - | - |
| 68 | >50 | >50 | - | - |
| 69 | >100 | >100 | >100 | >100 |
| Nutlin-3a | 2.7 ± 0.9 | 28.9 ± 3.7 | 4.5 ± 1.3 | >100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanenkov, Y.A.; Kukushkin, M.E.; Beloglazkina, A.A.; Shafikov, R.R.; Barashkin, A.A.; Ayginin, A.A.; Serebryakova, M.S.; Majouga, A.G.; Skvortsov, D.A.; Tafeenko, V.A.; et al. Synthesis and Biological Evaluation of Novel Dispiro-Indolinones with Anticancer Activity. Molecules 2023, 28, 1325. https://doi.org/10.3390/molecules28031325
Ivanenkov YA, Kukushkin ME, Beloglazkina AA, Shafikov RR, Barashkin AA, Ayginin AA, Serebryakova MS, Majouga AG, Skvortsov DA, Tafeenko VA, et al. Synthesis and Biological Evaluation of Novel Dispiro-Indolinones with Anticancer Activity. Molecules. 2023; 28(3):1325. https://doi.org/10.3390/molecules28031325
Chicago/Turabian StyleIvanenkov, Yan A., Maxim E. Kukushkin, Anastasia A. Beloglazkina, Radik R. Shafikov, Alexander A. Barashkin, Andrey A. Ayginin, Marina S. Serebryakova, Alexander G. Majouga, Dmitry A. Skvortsov, Viktor A. Tafeenko, and et al. 2023. "Synthesis and Biological Evaluation of Novel Dispiro-Indolinones with Anticancer Activity" Molecules 28, no. 3: 1325. https://doi.org/10.3390/molecules28031325
APA StyleIvanenkov, Y. A., Kukushkin, M. E., Beloglazkina, A. A., Shafikov, R. R., Barashkin, A. A., Ayginin, A. A., Serebryakova, M. S., Majouga, A. G., Skvortsov, D. A., Tafeenko, V. A., & Beloglazkina, E. K. (2023). Synthesis and Biological Evaluation of Novel Dispiro-Indolinones with Anticancer Activity. Molecules, 28(3), 1325. https://doi.org/10.3390/molecules28031325