
Inhibition of Klf10 Attenuates Oxidative Stress-Induced Senescence of Chondrocytes via Modulating Mitophagy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Klf10 Was Overexpressed in Senescent Chondrocytes and OA Cartilage
2.2. Knocking down Klf10 Attenuated TBHP-Induced Senescence
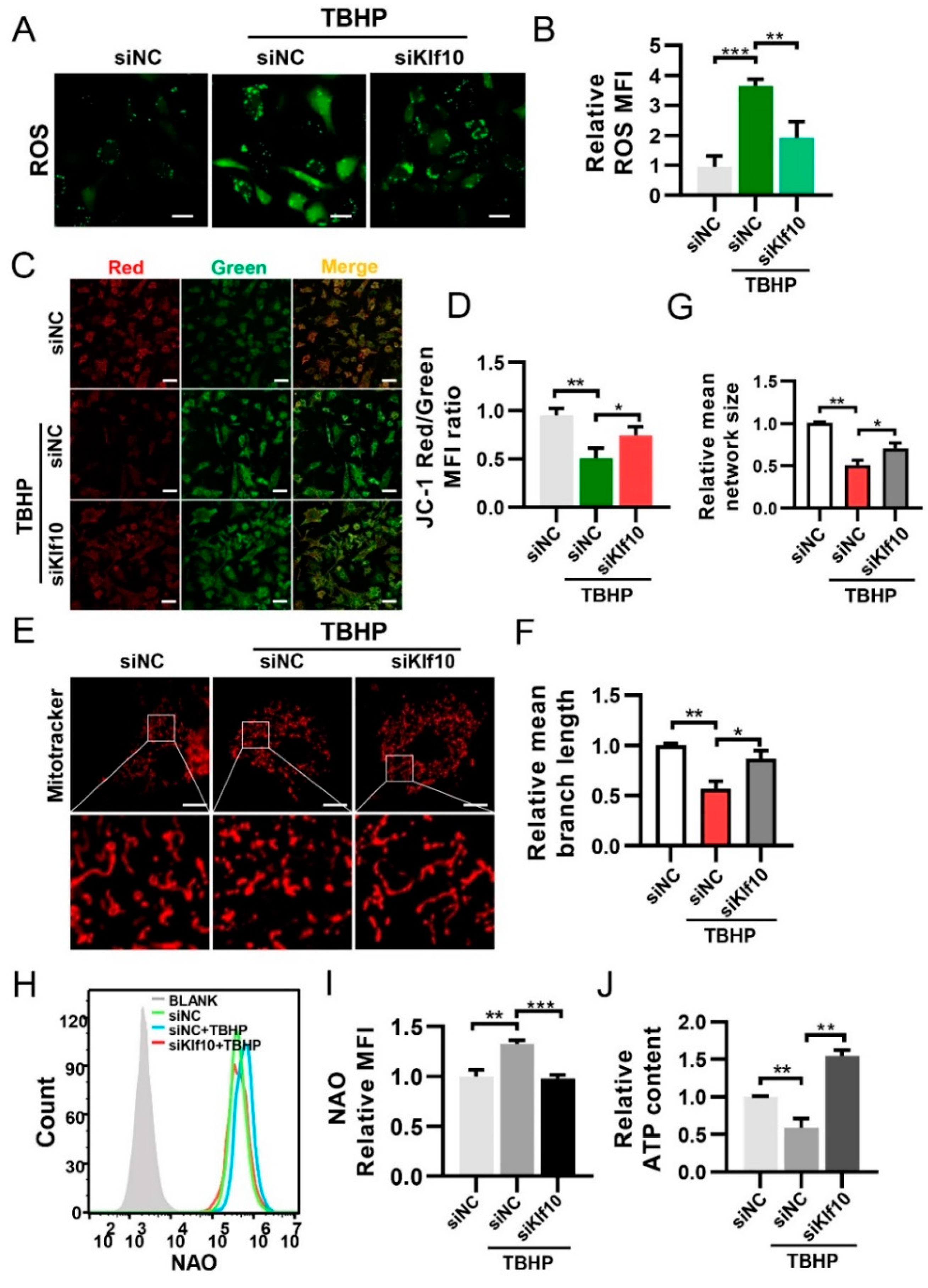
2.3. Downregulating Klf10 Attenuated Senescence via Decreasing ROS Production and Sustaining Mitochondrial Homeostasis
2.4. Downregulating Klf10 Restored Autophagy Flux Suppressed in TBHP-Induced Senescent Chondrocytes
2.5. Downregulation of Klf10 Promoted Mitophagy by Upregulating Bnip3
3. Materials and Methods
3.1. Isolation and Culture of Murine Chondrocytes
3.2. Chondrocyte Treatment and siRNA Transfection
3.3. Western Blot Analysis
3.4. RNA Extraction and Quantitative Real-Time PCR (RT-qPCR)
3.5. SA-β-Gal Staining
3.6. EdU (5-Ethynyl-2′-deoxyuridine) Assay
3.7. Determination of Intracellular ATP
3.8. Transmission Electron Microscopy (TEM)
3.9. Immunofluorescence
3.10. Assessment of Mitochondria and Lysosome Co-Localization
3.11. Measurement of Cellular ROS
3.12. Mitochondrial Mass Analysis
3.13. Mitochondrial Membrane Potential (Δψm) Detection
3.14. Human Samples Collection and Construction of Mouse DMM Model of OA
3.15. Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Cui, A.; Li, H.; Wang, D.; Zhong, J.; Chen, Y.; Lu, H. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine 2020, 29–30, 100587. [Google Scholar] [CrossRef] [PubMed]
- Vina, E.R.; Kwoh, C.K. Epidemiology of osteoarthritis: Literature update. Curr. Opin. Rheumatol. 2018, 30, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Development 2019, 146, dev151837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Faust, H.J.; Zhang, H.; Han, J.; Wolf, M.T.; Jeon, O.H.; Sadtler, K.; Peña, A.N.; Chung, L.; Maestas, D.R., Jr.; Tam, A.J.; et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J. Clin. Investig. 2020, 130, 5493–5507. [Google Scholar] [CrossRef]
- Loeser, R.F.; Kelley, K.L.; Armstrong, A.; Collins, J.A.; Diekman, B.O.; Carlson, C.S. Deletion of JNK Enhances Senescence in Joint Tissues and Increases the Severity of Age-Related Osteoarthritis in Mice. Arthritis Rheumatol. 2020, 72, 1679–1688. [Google Scholar] [CrossRef]
- Martin, J.A.; Buckwalter, J.A. The role of chondrocyte senescence in the pathogenesis of osteoarthritis and in limiting cartilage repair. J. Bone Jt. Surg. Am. Vol. 2003, 85-A (Suppl. 2), 106–110. [Google Scholar] [CrossRef]
- Gao, S.G.; Zeng, C.; Li, L.J.; Luo, W.; Zhang, F.J.; Tian, J.; Cheng, C.; Tu, M.; Xiong, Y.L.; Jiang, W.; et al. Correlation between senescence-associated beta-galactosidase expression in articular cartilage and disease severity of patients with knee osteoarthritis. Int. J. Rheum. Dis. 2016, 19, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Wang, G.; Wu, B.; Chou, W.C.; Cheng, L.; Zhou, C.; Lou, J.; Wu, D.; Su, L.; Zheng, J.; et al. DCAF1 regulates Treg senescence via the ROS axis during immunological aging. J. Clin. Investig. 2020, 130, 5893–5908. [Google Scholar] [CrossRef]
- Niu, F.; Cui, X.; Zhao, P.; Sun, M.; Yang, B.; Deyholos, M.K.; Li, Y.; Zhao, X.; Jiang, Y.Q. WRKY42 transcription factor positively regulates leaf senescence through modulating SA and ROS synthesis in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2020, 104, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Zhang, H.; Simon, A.K. Polyamines reverse immune senescence via the translational control of autophagy. Autophagy 2020, 16, 181–182. [Google Scholar] [CrossRef]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Muciño-Hernández, G.; Gerónimo-Olvera, C.; Maciel-Barón, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregón, S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef]
- Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis in organismal senescence and neurodegeneration. Mech. Ageing Dev. 2020, 191, 111345. [Google Scholar] [CrossRef]
- Janikiewicz, J.; Szymański, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszyński, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callender, L.A.; Carroll, E.C.; Bober, E.A.; Akbar, A.N.; Solito, E.; Henson, S.M. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell 2020, 19, e13067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschênes-Simard, X.; Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araya, J.; Tsubouchi, K.; Sato, N.; Ito, S.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 2019, 15, 510–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Dai, H.; Yuan, J.; Chen, J.; Lin, L.; Zhang, W.; Wang, L.; Zhang, J.; Li, K.; He, Y. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, M.; Harris, S.A.; Oursler, M.J.; Rasmussen, K.; Riggs, B.L.; Spelsberg, T.C. Identification of a novel TGF-beta-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 1995, 23, 4907–4912. [Google Scholar] [CrossRef] [Green Version]
- Parakati, R.; DiMario, J.X. Repression of myoblast proliferation and fibroblast growth factor receptor 1 promoter activity by KLF10 protein. J. Biol. Chem. 2013, 288, 13876–13884. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.F.; Sui, C.L.; Wu, W.C.; Wang, J.J.; Yang, D.H.; Chen, Y.C.; Yu, W.C.; Chang, H.S. Klf10 induces cell apoptosis through modulation of BI-1 expression and Ca2+ homeostasis in estrogen-responding adenocarcinoma cells. Int. J. Biochem. Cell Biol. 2011, 43, 666–673. [Google Scholar] [CrossRef]
- Chen, Z.; Li, W.; Wang, H.; Wan, C.; Luo, D.; Deng, S.; Chen, H.; Chen, S. Klf10 regulates odontoblast differentiation and mineralization via promoting expression of dentin matrix protein 1 and dentin sialophosphoprotein genes. Cell Tissue Res. 2016, 363, 385–398. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhang, L.; Zheng, B.; Zhang, L.; Qin, Y.; Zhang, X.; Yang, Z.; Nie, Z.; Yang, G.; Yu, J.; et al. Salvia miltiorrhiza bunge exerts anti-oxidative effects through inhibiting KLF10 expression in vascular smooth muscle cells exposed to high glucose. J. Ethnopharmacol. 2020, 262, 113208. [Google Scholar] [CrossRef]
- Chang, V.H.; Tsai, Y.C.; Tsai, Y.L.; Peng, S.L.; Chen, S.L.; Chang, T.M.; Yu, W.C.; Ch’ang, H.J. Krüpple-like factor 10 regulates radio-sensitivity of pancreatic cancer via UV radiation resistance-associated gene. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2017, 122, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosset, M.; Berenbaum, F.; Thirion, S.; Jacques, C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008, 3, 1253–1260. [Google Scholar] [CrossRef]
- Wedel, S.; Martic, I.; Hrapovic, N.; Fabre, S.; Madreiter-Sokolowski, C.T.; Haller, T.; Pierer, G.; Ploner, C.; Jansen-Dürr, P.; Cavinato, M. tBHP treatment as a model for cellular senescence and pollution-induced skin aging. Mech. Ageing Dev. 2020, 190, 111318. [Google Scholar] [CrossRef] [PubMed]
- Doherty, E.; Perl, A. Measurement of Mitochondrial Mass by Flow Cytometry during Oxidative Stress. React. Oxyg. Species 2017, 4, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Bradley, E.W.; Weivoda, M.M.; Hwang, S.M.; Pirtskhalava, T.; Decklever, T.; Curran, G.L.; Ogrodnik, M.; Jurk, D.; Johnson, K.O.; et al. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiology. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Yang, W.Y.; Chan, K.; Lavallee, M.; Xu, K.; Gao, T.; Wang, H.; Yang, X. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can. J. Physiol. Pharmacol. 2017, 95, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Di, G.; Li, J.; Chen, Y.; Li, W.; Wu, J.; Cheng, T.; Yao, M.; Shao, Z. TIEG1 induces apoptosis through mitochondrial apoptotic pathway and promotes apoptosis induced by homoharringtonine and velcade. FEBS Lett. 2007, 581, 3826–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, A.; Bronk, S.F.; Roberts, P.J.; Urrutia, R.; Gores, G.J. The transforming growth factor beta(1)-inducible transcription factor TIEG1, mediates apoptosis through oxidative stress. Hepatology 1999, 30, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Kammoun, M.; Piquereau, J.; Nadal-Desbarats, L.; Même, S.; Beuvin, M.; Bonne, G.; Veksler, V.; Le Fur, Y.; Pouletaut, P.; Même, W.; et al. Novel role of Tieg1 in muscle metabolism and mitochondrial oxidative capacities. Acta Physiol. 2020, 228, e13394. [Google Scholar] [CrossRef]
- Wara, A.K.; Wang, S.; Wu, C.; Fang, F.; Haemmig, S.; Weber, B.N.; Aydogan, C.O.; Tesmenitsky, Y.; Aliakbarian, H.; Hawse, J.R.; et al. KLF10 Deficiency in CD4(+) T Cells Triggers Obesity, Insulin Resistance, and Fatty Liver. Cell Rep. 2020, 33, 108550. [Google Scholar] [CrossRef]
- Dombi, E.; Mortiboys, H.; Poulton, J. Modulating Mitophagy in Mitochondrial Disease. Curr. Med. Chem. 2018, 25, 5597–5612. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Hu, S.; Zhang, C.; Ni, L.; Huang, C.; Chen, D.; Shi, K.; Jin, H.; Zhang, K.; Li, Y.; Xie, L.; et al. Stabilization of HIF-1α alleviates osteoarthritis via enhancing mitophagy. Cell Death Dis. 2020, 11, 481. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.Y.; Khan, N.M.; Ahmad, I.; Haqqi, T.M. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthr. Cartil. 2018, 26, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, J.; Lin, N.; Peng, R.; Jiang, N.; Wu, B.; Xing, B.; Lin, S.; Xu, X.; Lu, H. Inhibition of Klf10 Attenuates Oxidative Stress-Induced Senescence of Chondrocytes via Modulating Mitophagy. Molecules 2023, 28, 924. https://doi.org/10.3390/molecules28030924
Shang J, Lin N, Peng R, Jiang N, Wu B, Xing B, Lin S, Xu X, Lu H. Inhibition of Klf10 Attenuates Oxidative Stress-Induced Senescence of Chondrocytes via Modulating Mitophagy. Molecules. 2023; 28(3):924. https://doi.org/10.3390/molecules28030924
Chicago/Turabian StyleShang, Jie, Nan Lin, Rong Peng, Ning Jiang, Biao Wu, Baizhou Xing, Shiyuan Lin, Xianghe Xu, and Huading Lu. 2023. "Inhibition of Klf10 Attenuates Oxidative Stress-Induced Senescence of Chondrocytes via Modulating Mitophagy" Molecules 28, no. 3: 924. https://doi.org/10.3390/molecules28030924
APA StyleShang, J., Lin, N., Peng, R., Jiang, N., Wu, B., Xing, B., Lin, S., Xu, X., & Lu, H. (2023). Inhibition of Klf10 Attenuates Oxidative Stress-Induced Senescence of Chondrocytes via Modulating Mitophagy. Molecules, 28(3), 924. https://doi.org/10.3390/molecules28030924