
Design and Synthesis of 3-(β-d-Glucopyranosyl)-4-amino/4-guanidino Pyrazole Derivatives and Analysis of Their Glycogen Phosphorylase Inhibitory Potential



Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Glycogen Phosphorylase Binding Assays
2.3. In-Silico Studies
3. Conclusions
4. Experimental Section
4.1. Synthetic Methods








4.2. Computational Details
4.2.1. Protein Preparation
4.2.2. Ligand Preparation
4.2.3. Docking
4.2.4. MM-GBSA Calculations
4.2.5. Determination of Inhibitory Constants (Ki) for Glycogen Phosphorylase
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Oikonomakos, N.G. Glycogen phosphorylase as a molecular target for type 2 diabetes therapy. Curr. Protein. Pept. Sci. 2002, 3, 561–586. [Google Scholar] [CrossRef]
- IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021.
- Martin, W.H.; Hoover, D.J.; Armento, S.J.; Stock, I.A.; McPherson, R.K.; Danley, D.E.; Stevenson, R.W.; Barrett, E.J.; Treadway, J.L. Discovery of a human liver glycogen phosphorylase inhibitor that lowers blood glucose in vivo. Prac. Natl. Acad. Sci. USA 1998, 95, 1776–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, L.; Marton, J.; Vida, A.; Kis, G.; Bokor, É.; Kun, S.; Gönczi, M.; Docsa, T.; Tóth, A.; Antal, M.; et al. Glycogen phosphorylase inhibition improves beta cell function. Brit. J. Pharmacol. 2018, 175, 301–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Sun, H. Pharmacological manipulation of brain glycogenolysis as a therapeutic approach to cerebral ischemia. Mini Rev. Med. Chem. 2010, 10, 1188–1193. [Google Scholar] [CrossRef]
- Guan, T.; Qian, Y.S.; Tang, X.Z.; Huang, M.H.; Huang, L.F.; Li, Y.M.; Sun, H.B. Maslinic Acid, a Natural Inhibitor of Glycogen Phosphorylase, Reduces Cerebral Ischemic Injury in Hyperglycemic Rats by GLT-1 Up-Regulation. J. Neurosci. Res. 2011, 89, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.L.; Helou, Y.; Batlle, R.; Liu, X.J.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.P.; Snell, C.; Steers, G.; Turley, H.; Li, J.L.; Gunther, U.L.; et al. Glucose Utilization via Glycogen Phosphorylase Sustains Proliferation and Prevents Premature Senescence in Cancer Cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Zois, C.E.; Hendriks, A.M.; Haider, S.; Pires, E.; Bridges, E.; Kalamida, D.; Voukantsis, D.; Lagerholm, B.C.; Fehrmann, R.S.N.; den Dunnen, W.F.A.; et al. Liver glycogen phosphorylase is upregulated in glioblastoma and provides a metabolic vulnerability to high dose radiation. Cell Death Dis. 2022, 13, 573. [Google Scholar] [CrossRef]
- Chang, K.; Zhang, B.Q.; Guo, X.T.; Zong, M.; Rahman, R.; Sanchez, D.; Winder, N.; Reardon, D.A.; Zhao, B.S.; Wen, P.Y.; et al. Multimodal imaging patterns predict survival in recurrent glioblastoma patients treated with bevacizumab. Neuro-Oncology 2016, 18, 1680–1687. [Google Scholar] [CrossRef] [Green Version]
- Somsák, L.; Czifrák, K.; Tóth, M.; Bokor, É.; Chrysina, E.D.; Alexacou, K.M.; Hayes, J.M.; Tiraidis, C.; Lazoura, E.; Leonidas, D.D.; et al. New inhibitors of glycogen phosphorylase as potential antidiabetic agents. Curr. Med. Chem. 2008, 15, 2933–2983. [Google Scholar] [CrossRef] [Green Version]
- Kantsadi, A.L.; Apostolou, A.; Theofanous, S.; Stravodimos, G.A.; Kyriakis, E.; Gorgogietas, V.A.; Chatzileontiadou, D.S.; Pegiou, K.; Skamnaki, V.T.; Stagos, D.; et al. Biochemical and biological assessment of the inhibitory potency of extracts from vinification byproducts of Vitis vinifera extracts against glycogen phosphorylase. Food Chem. Toxicol. 2014, 67, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Chrysina, E.D.; Kosmopoulou, M.N.; Tiraidis, C.; Kardakaris, R.; Bischler, N.; Leonidas, D.D.; Hadady, Z.; Somsák, L.; Docsa, T.; Gergely, P.; et al. Kinetic and crystallographic studies on 2-(β-d-glucopyranosyl)-5-methyl-1, 3, 4-oxadiazole, -benzothiazole, and -benzimidazole, inhibitors of muscle glycogen phosphorylase b. Evidence for a new binding site. Protein Sci. 2005, 14, 873–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somsák, L. Glucose derived inhibitors of glycogen phosphorylase. C. R. Chim. 2011, 14, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.M.; Kantsadi, A.L.; Leonidas, D.D. Natural products and their derivatives as inhibitors of glycogen phosphorylase: Potential treatment for type 2 diabetes. Phytochem. Rev. 2014, 13, 471–498. [Google Scholar] [CrossRef]
- Martin, J.L.; Veluraja, K.; Ross, K.; Johnson, L.N.; Fleet, G.W.J.; Ramsden, N.G.; Bruce, I.; Orchard, M.G.; Oikonomakos, N.G.; Papageorgiou, A.C.; et al. Glucose Analog Inhibitors of Glycogen-Phosphorylase—The Design of Potential-Drugs for Diabetes. Biochemistry 1991, 30, 10101–10116. [Google Scholar] [CrossRef]
- Somsák, L.; Bokor, É.; Juhász, L.; Kun, S.; Lázár, L.; Juhász-Tóth, E.; Tóth, M. New syntheses towards C-glycosyl type glycomimetics. Pure Appl. Chem. 2019, 91, 1159–1175. [Google Scholar] [CrossRef]
- Barr, D.; Szennyes, E.; Bokor, É.; Al-Oanzi, Z.H.; Moffatt, C.; Kun, S.; Docsa, T.; Sipos, A.; Davies, M.P.; Mathomes, R.T.; et al. Identification of C-β-d-Glucopyranosyl Azole-Type Inhibitors of Glycogen Phosphorylase That Reduce Glycogenolysis in Hepatocytes: In Silico Design, Synthesis, in Vitro Kinetics, and ex Vivo Studies. ACS Chem. Biol. 2019, 14, 1460–1470. [Google Scholar] [CrossRef]
- Bokor, É. N- and C-Glycopyranosyl heterocycles as glycogen phosphorylase inhibitors. In Recent Trends in Carbohydrate Chemistry—Synthesis, Structure and Function of Carbohydrates; Rauter, A.P., Somsák, L., Christensen, B.E., Kosma, P., Adamo, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 1, pp. 253–300. [Google Scholar]
- Bokor, É.; Kun, S.; Docsa, T.; Gergely, P.; Somsák, L. 4(5)-Aryl-2-C-glucopyranosyl-imidazoles as New Nanomolar Glucose Analogue Inhibitors of Glycogen Phosphorylase. ACS Med. Chem. Lett. 2015, 6, 1215–1219. [Google Scholar] [CrossRef] [Green Version]
- Sipos, Á.; Szennyes, E.; Hajnal, N.E.; Kun, S.; Szabó, K.E.; Uray, K.; Somsák, L.; Docsa, T.; Bokor, É. Dual-Target Compounds against Type 2 Diabetes Mellitus: Proof of Concept for Sodium Dependent Glucose Transporter (SGLT) and Glycogen Phosphorylase (GP) Inhibitors. Pharmaceuticals 2021, 14, 364. [Google Scholar] [CrossRef]
- Avenoza, A.; Cativiela, C.; Mayoral, J.A.; Peregrina, J.M.; Sinou, D. Reaction of Cyclopentadiene with (E)-2-Cyanocinnamate of (S)-Ethyl Lactate. Tetrahedron-Asymmetr 1990, 1, 765–768. [Google Scholar] [CrossRef]
- Rao, H.S.P.; Muthanna, N. Variations in the Blaise Reaction: Conceptually New Synthesis of 3-Amino Enones and 1,3-Diketones. Eur. J. Org. Chem. 2015, 2015, 1525–1532. [Google Scholar] [CrossRef]
- Mortier, J.; Frederick, R.; Ganeff, C.; Remouchamps, C.; Talaga, P.; Pochet, L.; Wouters, J.; Piette, J.; Dejardin, E.; Masereel, B. Pyrazolo [4,3-c]isoquinolines as potential inhibitors of NF-kappa B activation. Biochem. Pharmacol. 2010, 79, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.; Gowenlock, B.G.; Boyd, A.S.F. Studies in nitrosopyrazoles. 1. Preparative and spectroscopic studies of some 3,5-dialkyl-4-nitrosopyrazoles. J. Chem. Soc. Perk. T. 1996, 2, 2271–2274. [Google Scholar] [CrossRef]
- Bernatowicz, M.S.; Wu, Y.L.; Matsueda, G.R. Urethane Protected Derivatives of 1-Guanylpyrazole for the Mild and Efficient Preparation of Guanidines. Tetrahedron Lett. 1993, 34, 3389–3392. [Google Scholar] [CrossRef]
- Hayes, J.M.; Leonidas, D.D. Computation as a Tool for Glycogen Phosphorylase Inhibitor Design. Mini-Rev. Med. Chem. 2010, 10, 1156–1174. [Google Scholar] [CrossRef]
- Hayes, J.M. Computer-Aided Discovery of Glycogen Phosphorylase Inhibitors Exploiting Natural Products. In Discovery and Development of Antidiabetic Agents from Natural Products: Natural Product Drug Discovery, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 29–62. [Google Scholar]
- Kun, S.; Begum, J.; Kyriakis, E.; Stamati, E.C.V.; Barkas, T.A.; Szennyes, E.; Bokor, É.; Szabó, K.E.; Stravodimos, G.A.; Sipos, Á.; et al. A multidisciplinary study of 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazole derivatives as glycogen phosphorylase inhibitors: Computation, synthesis, crystallography and kinetics reveal new potent inhibitors. Eur. J. Med. Chem. 2018, 147, 266–278. [Google Scholar] [CrossRef]
- Polyák, M.; Varga, G.; Szilágyi, B.; Juhász, L.; Docsa, T.; Gergely, P.; Begum, J.; Hayes, J.M.; Somsák, L. Synthesis, enzyme kinetics and computational evaluation of N-(β-d-glucopyranosyl) oxadiazolecarboxamides as glycogen phosphorylase inhibitors. Bioorgan. Med. Chem. 2013, 21, 5738–5747. [Google Scholar] [CrossRef] [Green Version]
- Martin, Y.C. Let’s not forget tautomers. J. Comput. Aid. Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Goller, A.H. Reliable gas-phase tautomer equilibria of drug-like molecule scaffolds and the issue of continuum solvation. J. Comput. Aid. Mol. Des. 2022, 36, 805–824. [Google Scholar] [CrossRef]
- Milletti, F.; Vulpetti, A. Tautomer Preference in PDB Complexes and its Impact on Structure-Based Drug Discovery. J. Chem. Inf. Model 2010, 50, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.M.; Archontis, G. MM-GB(PB)SA Calculations of Protein-Ligand Binding Free Energies. In Molecular Dynamics: Studies of Synthetic and Biological Macromolecules; Wang, L., Ed.; Intech Open: Rijeka, Croatia, 2012. [Google Scholar]
- Schrödinger Software Suite, Schrödinger, LLC: New York, NY, USA, 2020.
- Blakeley, M.P. Neutron crystallography aids in drug design. Iucrj 2016, 3, 296–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, M.; Kovalevsky, A.Y.; Velazquez, H.; Fisher, S.Z.; Smith, J.C.; McKenna, R. Neutron structure of human carbonic anhydrase II in complex with methazolamide: Mapping the solvent and hydrogen-bonding patterns of an effective clinical drug. Iucrj 2016, 3, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Bax, B.; Chung, C.W.; Edge, C. Getting the chemistry right: Protonation, tautomers and the importance of H atoms in biological chemistry. Acta Crystallogr. D 2017, 73, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Hannick, S.M.; Kishi, Y. Improved Procedure for the Blaise Reaction—A Short, Practical Route to the Key Intermediates of the Saxitoxin Synthesis. J. Org. Chem. 1983, 48, 3833–3835. [Google Scholar] [CrossRef]
- Kantsadi, A.L.; Bokor, É.; Kun, S.; Stravodimos, G.A.; Chatzileontiadou, D.S.M.; Leonidas, D.D.; Juhász-Tóth, E.; Szakács, A.; Batta, G.; Docsa, T.; et al. Synthetic, enzyme kinetic, and protein crystallographic studies of C-β-d-glucopyranosyl pyrroles and imidazoles reveal and explain low nanomolar inhibition of human liver glycogen phosphorylase. Eur. J. Med. Chem. 2016, 123, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Sondergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pK(a) Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.J.; Reboul, M.; Xiang, J.Y.; Wang, L.L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. 12. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 1972, 56, 2257. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; Defrees, D.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. 23. A Polarization-Type Basis Set for 2nd-Row Elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Ősz, E.; Somsák, L.; Szilágyi, L.; Kovács, L.; Docsa, T.; Tóth, B.; Gergely, P. Efficient inhibition of muscle and liver glycogen phosphorylases by a new glucopyranosylidene-spiro-thiohydantoin. Bioorg. Med. Chem. Lett. 1999, 9, 1385–1390. [Google Scholar] [CrossRef]
- Oikonomakos, N.G.; Kosmopoulou, M.; Zographos, S.E.; Leonidas, D.D.; Chrysina, E.D.; Somsák, L.; Nagy, V.; Praly, J.P.; Docsa, T.; Tóth, A.; et al. Binding of N-acetyl-N‘-β-d-glucopyranosyl urea and N-benzoyl-N‘-β-d-glucopyranosyl urea to glycogen phosphorylase b—Kinetic and crystallographic studies. Eur. J. Biochem. 2002, 269, 1684–1696. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
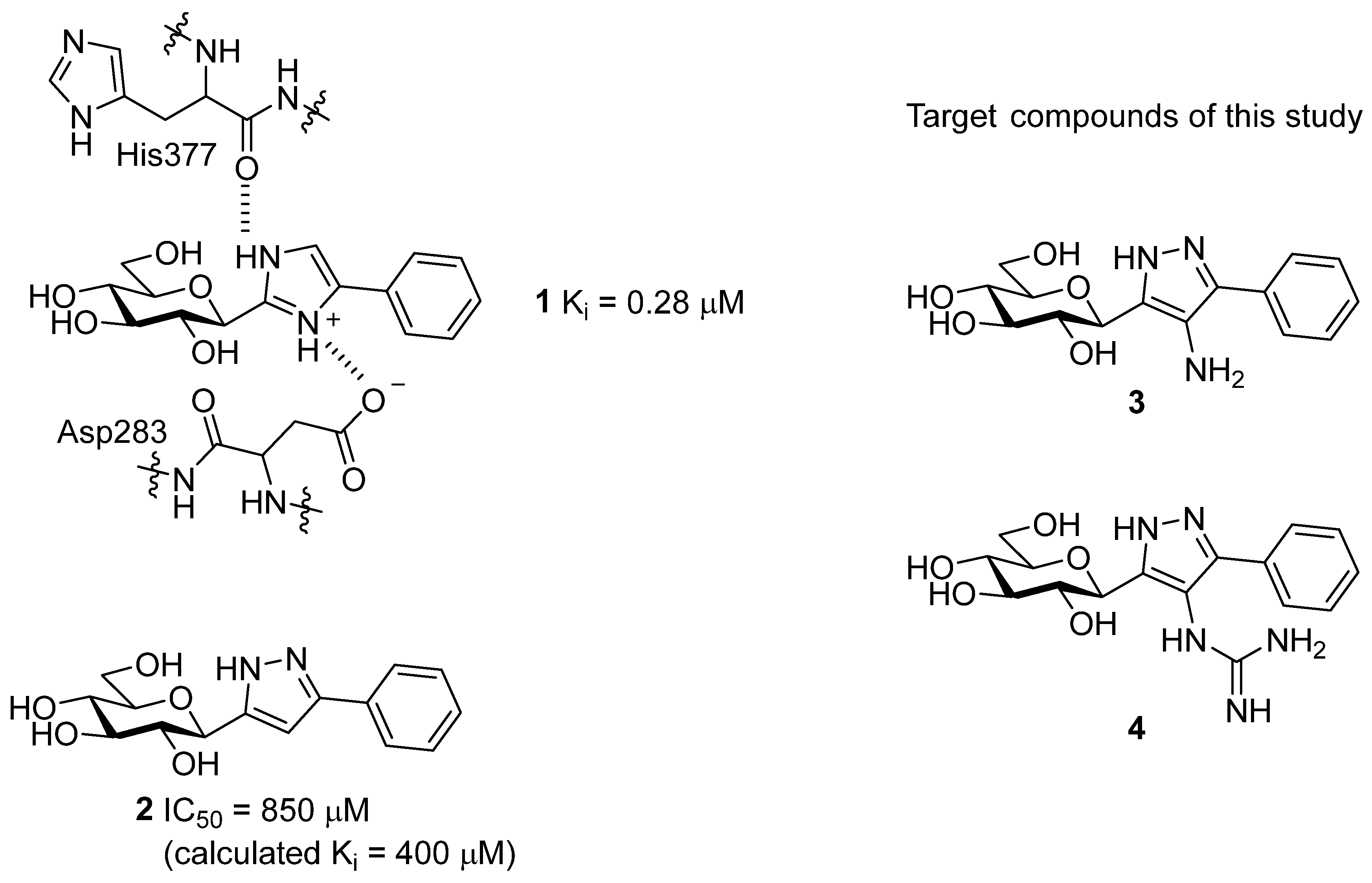
| Compound | IC50 [µM] | |
|---|---|---|
| 1 a |  | 0.28 (Ki) [21] |
| 2 |  | 850 [21] |
| 3 |  | 565 |
| 4 |  | no inhibition at 625 µM |
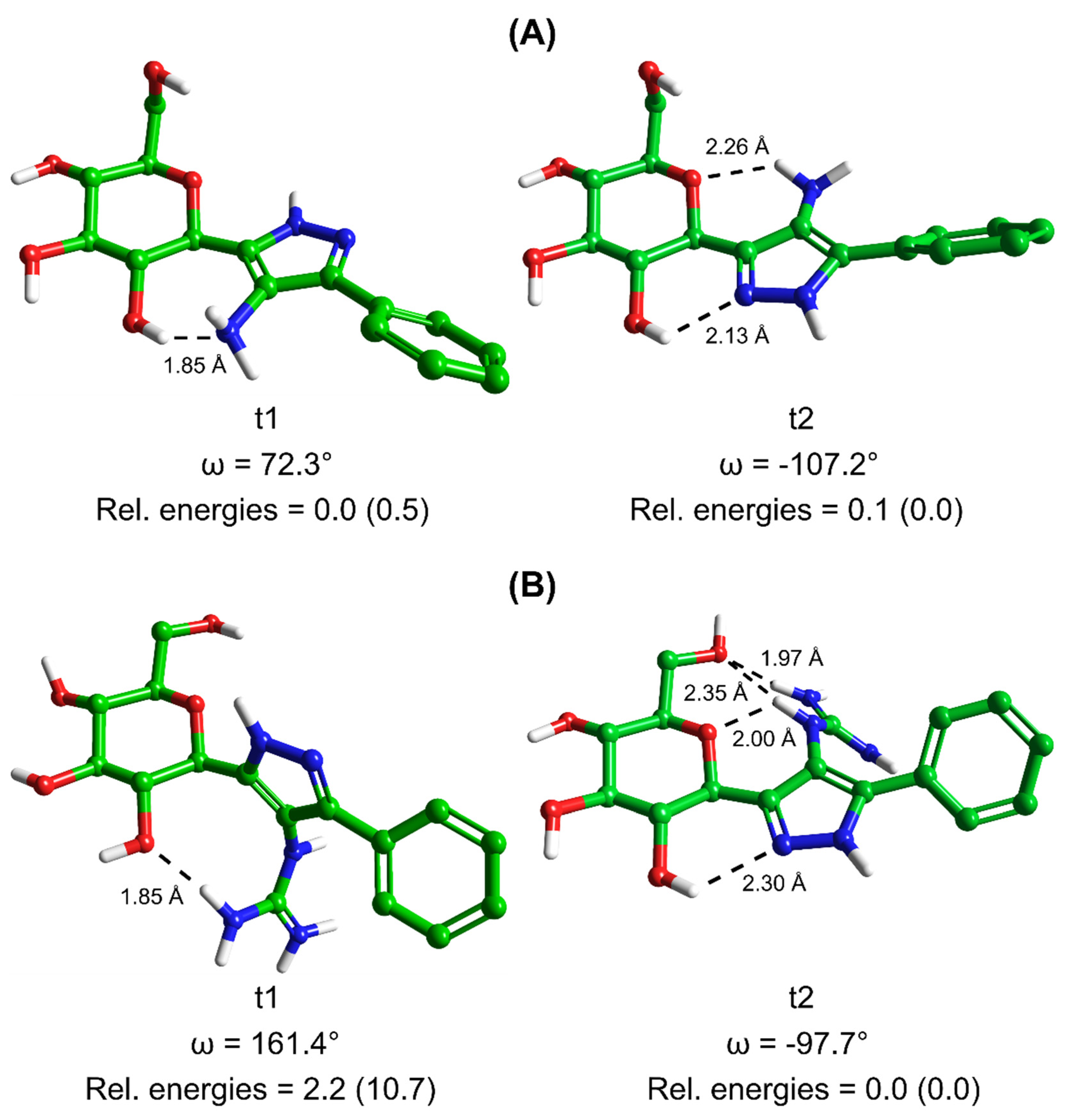
| Conformation a |  3 tautomer 1 (t1) |  3 tautomer 2 (t2) | ||||
|---|---|---|---|---|---|---|
| Dihedral Angle, ω (Degrees) b | Relative Energies (kcal/mol) | Dihedral Angle, ω (Degrees) b | Relative Energies (kcal/mol) | |||
| Gas Phase | Solution Phase c | Gas Phase | Solution Phase c | |||
| 1 | 66.6 | 1.9 | 1.7 | −104.8 | 0.3 | 0.7 |
| 2 | 65.8 | 1.1 | 0.6 | −114.9 | 0.8 | 1.1 |
| 3 | 147.6 | 4.6 | 4.3 | −89.9 | 1.5 | 1.2 |
| 4 | 118.0 | 2.0 | 2.5 | −102.0 | 0.9 | 0.4 |
| 5 | 100.8 | 1.9 | 1.4 | −100.7 | 0.7 | 0.6 |
| 6 | 82.7 | 2.2 | 1.4 | −107.2 | 0.0 | 0.1 |
| 7 | 95.4 | 0.7 | 0.7 | −88.7 | 2.0 | 1.8 |
| 8 | 72.3 | 0.5 | 0.0 | −100.1 | 0.8 | 0.8 |
| 9 | 141.3 | 7.3 | 5.6 | −135.9 | 8.4 | 5.1 |
| 10 | 84.3 | 2.2 | 1.5 | −164.5 | 8.9 | 4.8 |
| Ligand and State | Dihedral Angle, ω C1H-C1-C2-N3 (Degrees) a | Post-Docking MM-GBSA (kcal/mol) b | |||
|---|---|---|---|---|---|
| 1 c | |||||
| t1 | −167.5 | −64.8 | 10.1 | 18.8 | −35.9 |
| t2 | −146.5 | −51.7 | 10.7 | 18.9 | −22.1 |
| +1 charged | −163.7 | −81.4 | 10.2 | 18.1 | −53.1 |
| 3 | |||||
| t1 | −173.0 | −67.1 | 7.9 | 18.3 | −40.9 |
| t2 | −170.3 | −52.8 | 11.4 | 18.0 | −23.4 |
| 4 | |||||
| t1 | −166.2 | −82.5 | 18.3 | 20.7 | −43.5 |
| t2 | −177.1 | −67.9 | 17.7 | 22.0 | −28.2 |
| Conformation a |  4 tautomer 1 (t1) |  4 tautomer 2 (t2) | ||||
|---|---|---|---|---|---|---|
| Dihedral Angle, ω (Degrees) b | Relative Energies (kcal/mol) | Dihedral Angle, ω (Degrees) b | Relative Energies (kcal/mol) | |||
| Gas Phase | Solution Phase c | Gas Phase | Solution Phase c | |||
| 1 | 160.9 | 10.7 | 2.4 | −141.7 | 3.9 | 0.9 |
| 2 | 9.8 | 17.0 | 5.0 | −120.6 | 10.2 | 4.0 |
| 3 | 131.7 | 8.3 | 3.0 | −97.7 | 0.0 | 0.0 |
| 4 | 132.6 | 11.7 | 5.7 | −140.9 | 3.7 | 0.4 |
| 5 | −154.2 | 9.7 | 4.0 | −32.9 | 6.4 | 3.2 |
| 6 | 161.4 | 10.7 | 2.2 | −178.2 | 13.2 | 2.1 |
| 7 | −155.2 | 9.8 | 4.3 | −144.1 | 5.5 | 2.7 |
| 8 | 158.8 | 10.7 | 3.4 | 175.8 | 10.0 | 1.5 |
| 9 | −19.1 | 10.4 | 6.2 | −143.4 | 5.1 | 2.1 |
| 10 | 159.6 | 13.6 | 3.5 | −99.0 | 3.2 | 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kun, S.; Mathomes, R.T.; Docsa, T.; Somsák, L.; Hayes, J.M. Design and Synthesis of 3-(β-d-Glucopyranosyl)-4-amino/4-guanidino Pyrazole Derivatives and Analysis of Their Glycogen Phosphorylase Inhibitory Potential. Molecules 2023, 28, 3005. https://doi.org/10.3390/molecules28073005
Kun S, Mathomes RT, Docsa T, Somsák L, Hayes JM. Design and Synthesis of 3-(β-d-Glucopyranosyl)-4-amino/4-guanidino Pyrazole Derivatives and Analysis of Their Glycogen Phosphorylase Inhibitory Potential. Molecules. 2023; 28(7):3005. https://doi.org/10.3390/molecules28073005
Chicago/Turabian StyleKun, Sándor, Rachel T. Mathomes, Tibor Docsa, László Somsák, and Joseph M. Hayes. 2023. "Design and Synthesis of 3-(β-d-Glucopyranosyl)-4-amino/4-guanidino Pyrazole Derivatives and Analysis of Their Glycogen Phosphorylase Inhibitory Potential" Molecules 28, no. 7: 3005. https://doi.org/10.3390/molecules28073005
APA StyleKun, S., Mathomes, R. T., Docsa, T., Somsák, L., & Hayes, J. M. (2023). Design and Synthesis of 3-(β-d-Glucopyranosyl)-4-amino/4-guanidino Pyrazole Derivatives and Analysis of Their Glycogen Phosphorylase Inhibitory Potential. Molecules, 28(7), 3005. https://doi.org/10.3390/molecules28073005