
Design, Synthesis, Biological Evaluation and Molecular Docking of Novel F-18-Labeled Focal Adhesion Kinase Inhibitors as Potential Tumor Radiotracers

Abstract
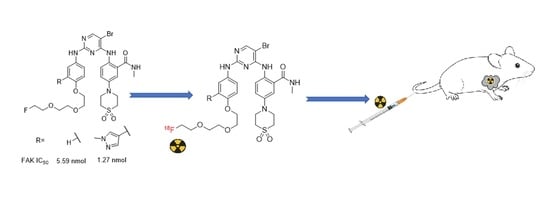
:
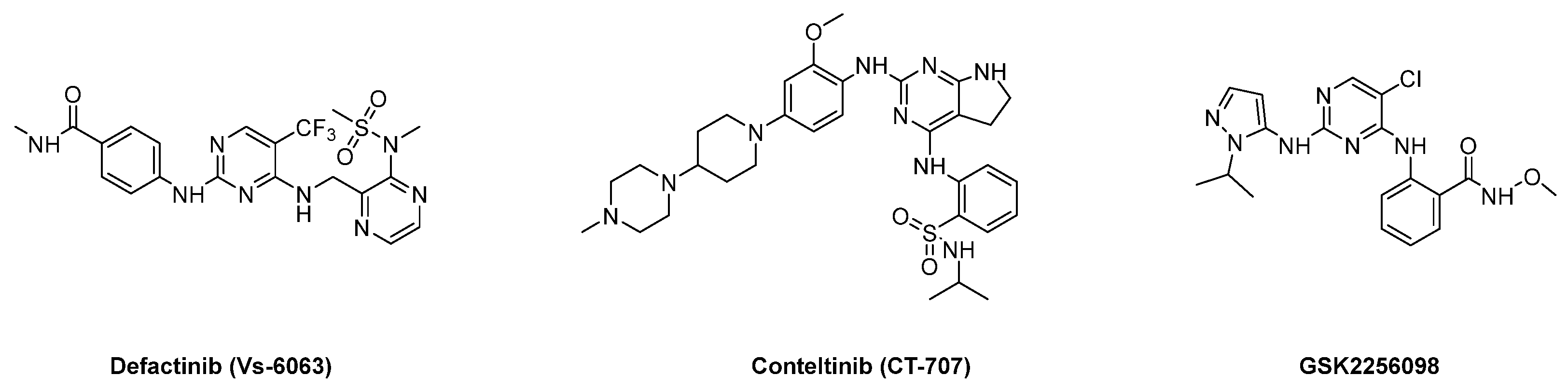
1. Introduction
2. Results
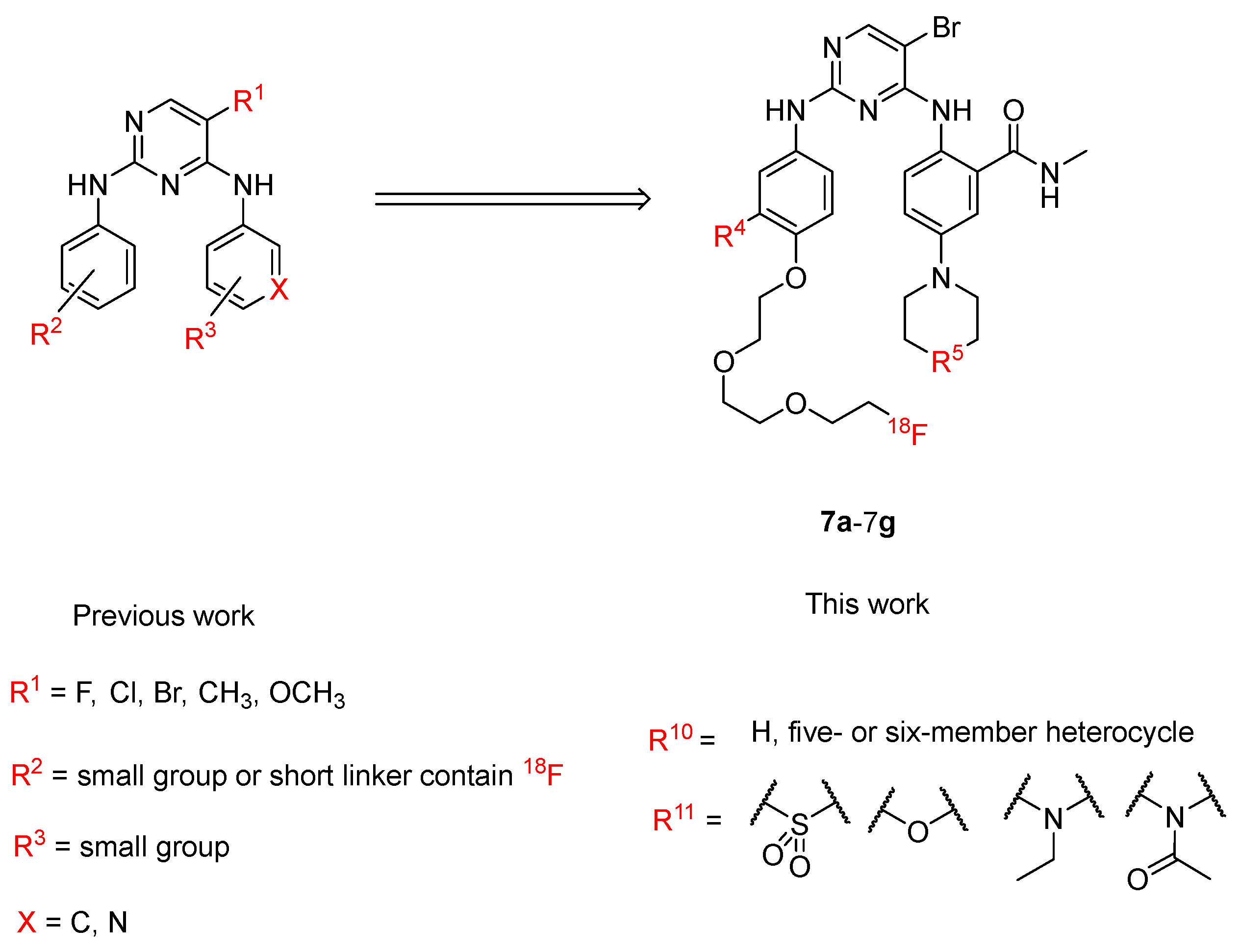
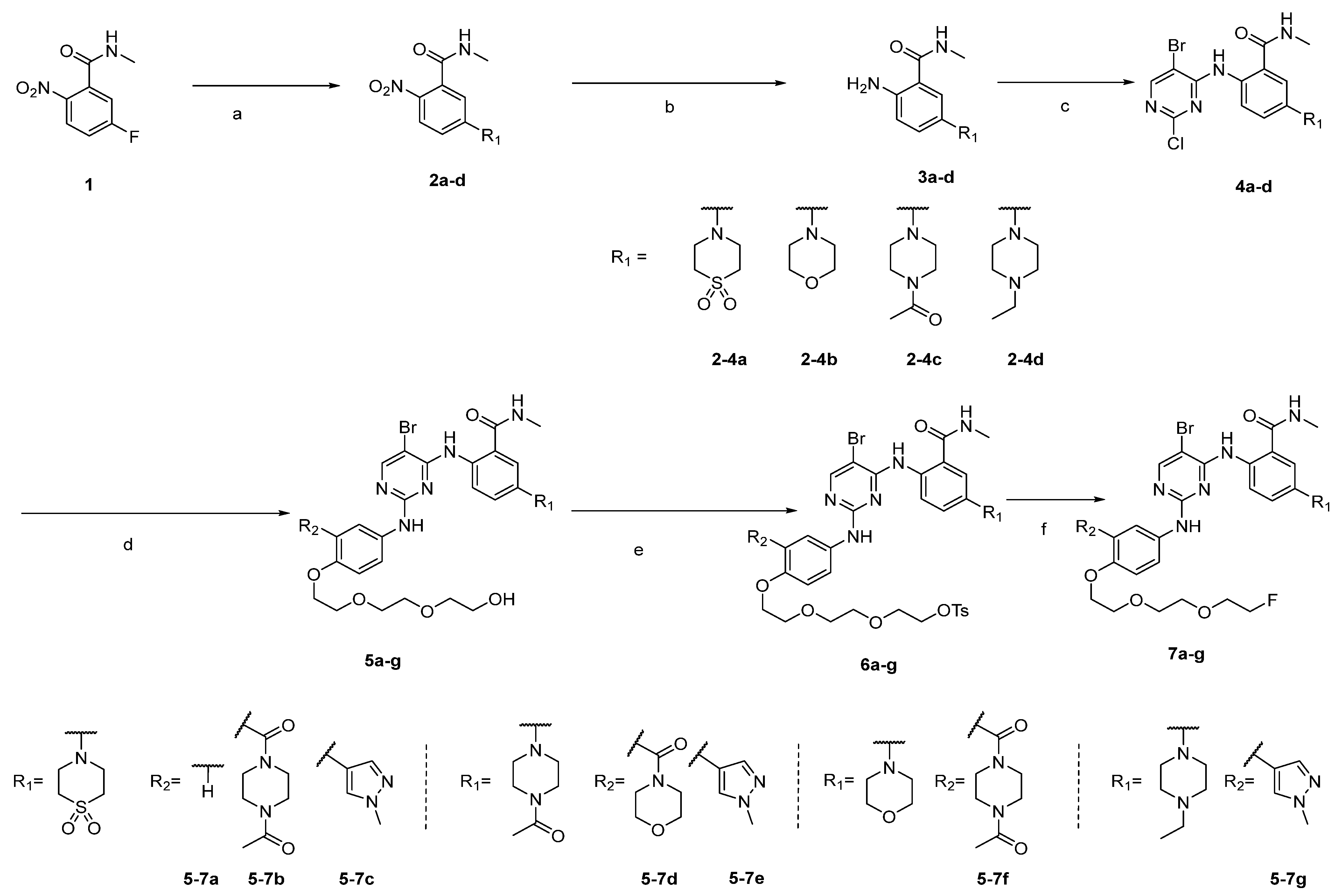
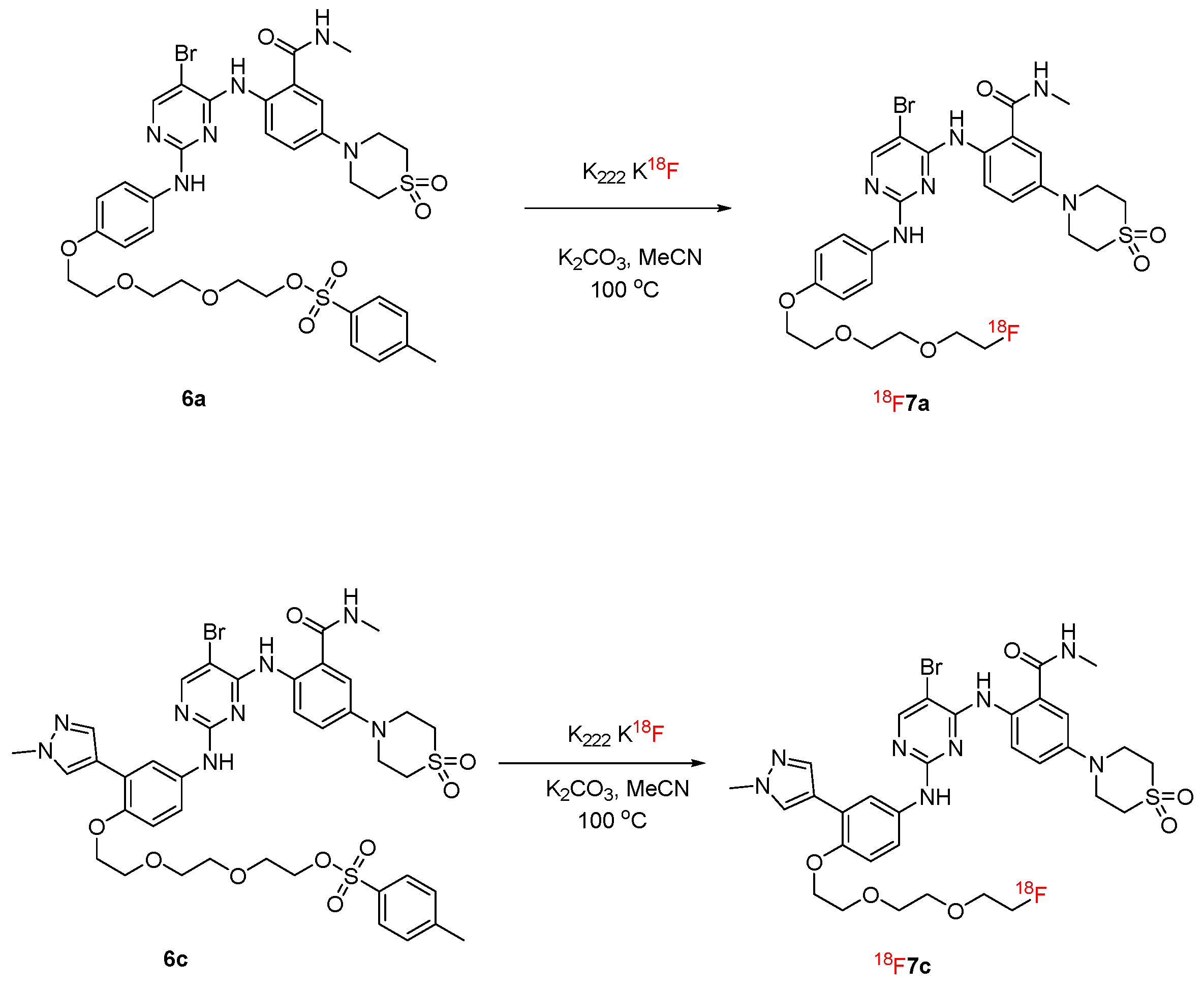
2.1. Synthesis
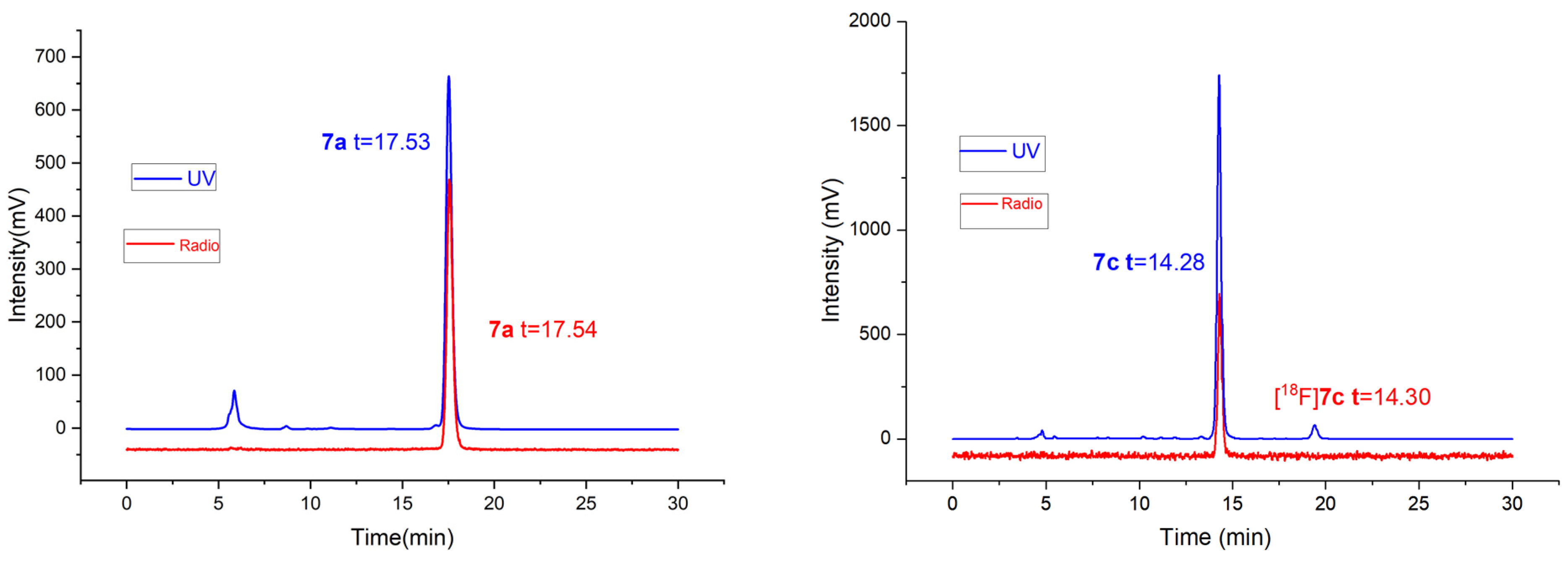
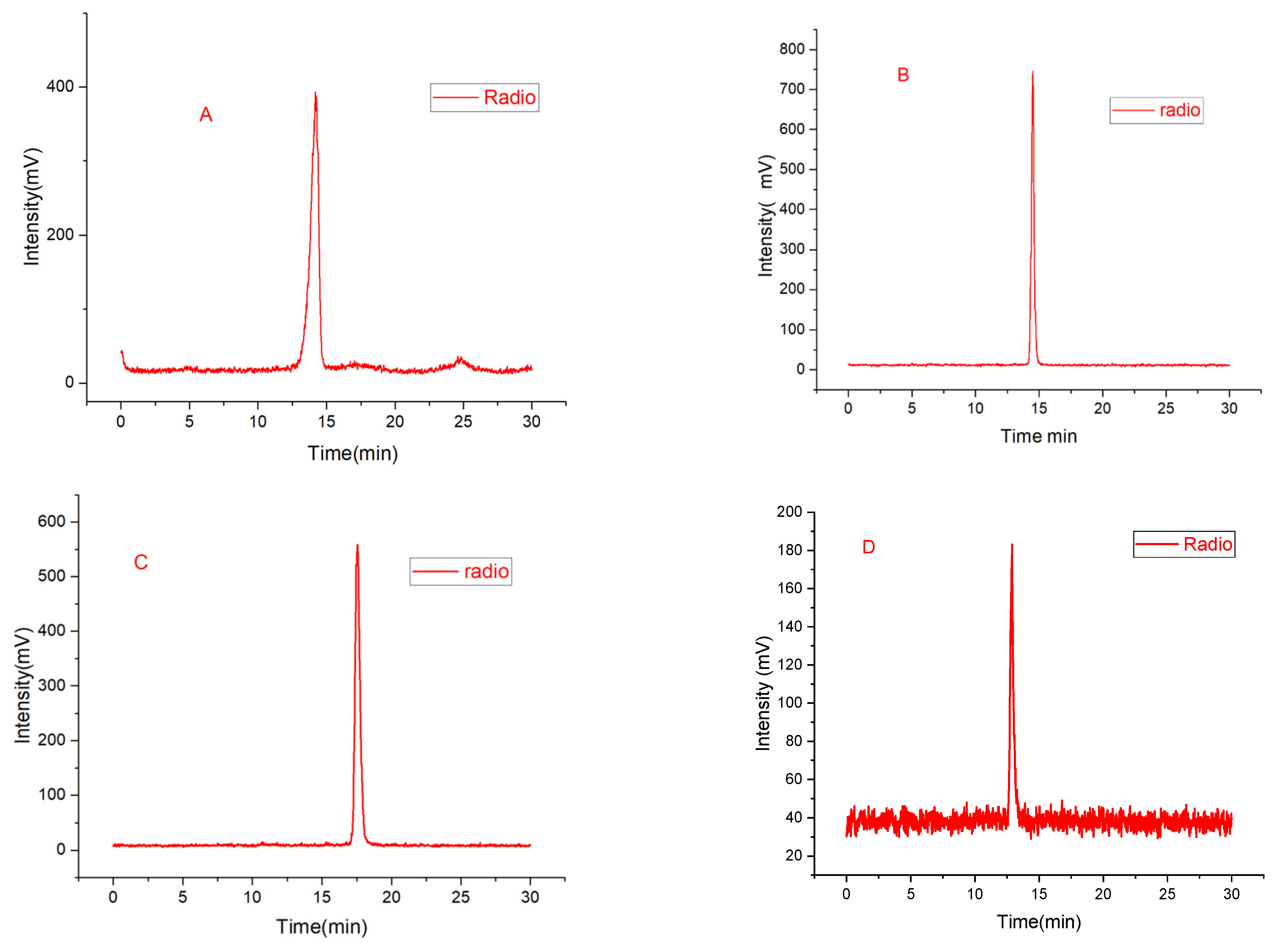
2.2. Radiolabeling and Stability Experiment
2.3. Experiment on Octanol/Water Partition Coefficient Determination
2.4. FAK Inhibitory Assay
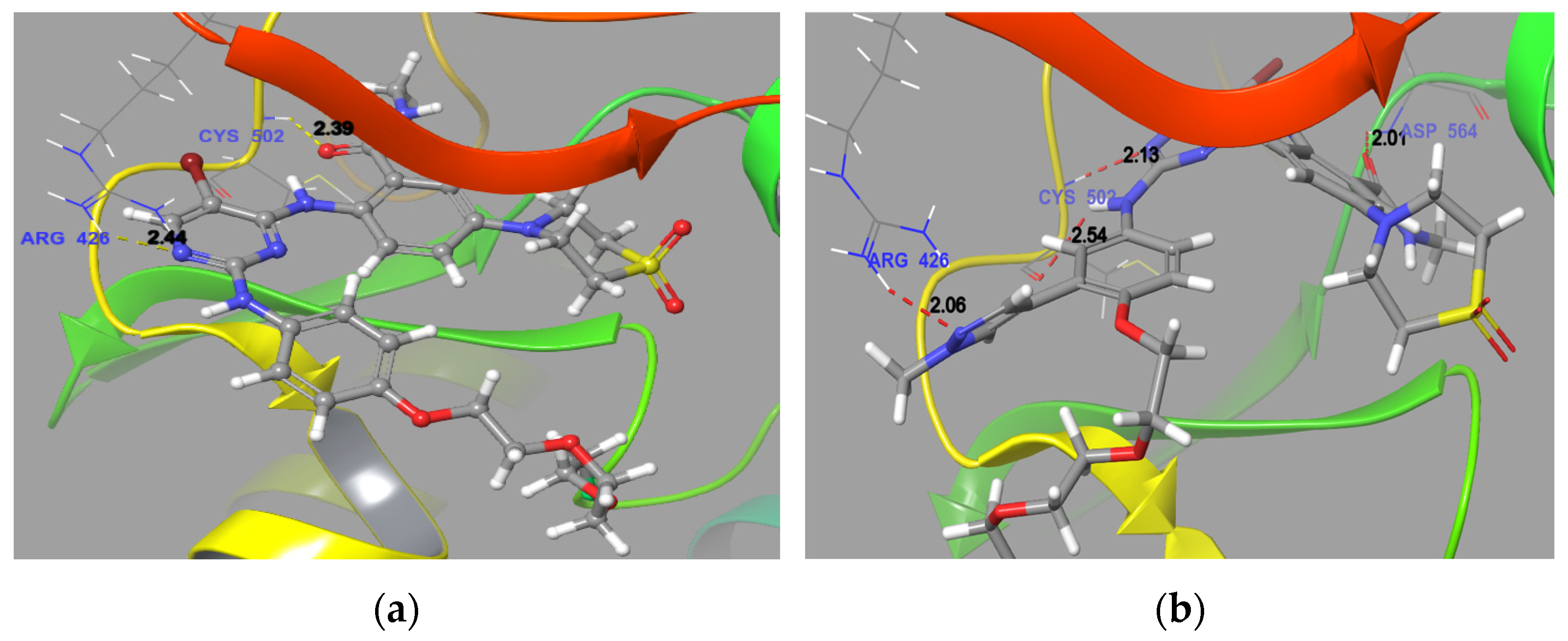
2.5. Molecular Docking
2.6. Biodistributions of [18F]7a and [18F]7c in S-180 Tumor-Bearing Mice
3. Discussion
4. Materials and Methods
4.1. Synthesis
- 2,2,3,3-tetramethyl-4,7,10-trioxa-3-siladodecan-12-ol (9). To a stirring solution of triethylene glycol (10.00 g, 66.59 mmol, 1.00 eq) and imidazole (2.72 g, 39.95 mmol, 0.60 eq) in DCM (150 mL), a solution of t-Butyldimethylsilyl chloride (5.02 g, 33.30 mmol, 0.50 eq) in DCM (50 mL) was added slowly at 0 °C. The temperature was allowed to cool to r.t., and the solution then reacted overnight. After the reaction was completed, the mixture was extracted using H2O and DCM. The organic phase was collected and concentrated under medium pressure. The crude product was purified with a flash silica gel column (petroleum ether/ethyl acetate = 3:1 to 1:1) to produce 9 (11.62 g, yellow solid, yield 66.10%). 1H NMR (600 MHz, Chloroform-d) δ 3.77–3.71 (m, 2H), 3.71–3.66 (m, 2H), 3.59–3.55 (m, 2H), 3.55–3.50 (m, 2H), 2.84–2.67 (m, 1H), 0.85 (d, J = 1.6 Hz, 9H), 0.04–0.01 (m, 6H). 13C NMR (101 MHz, Chloroform-d) δ 72.71, 72.60, 70.82, 70.49, 62.73, 61.75, 25.96, 18.40, −5.26. ESI-MS m/z 287.1626 [M + Na]+.
- 2,2,3,3-tetramethyl-4,7,10-trioxa-3-siladodecan-12-yl 4-methylbenzenesulfonate (10). To a stirring solution of 10 (10.00 g, 37.73 mmol, 1.00 eq) and triethylamine (5.72 g, 56.60 mmol, 1.5 eq) in DCM (100 mL), a solution of p-Toluenesulfonyl chloride (8.64 g, 45.24 mmol, 1.2 eq) was added. The mixture was then stirred at room temperature overnight. The reactant solution was concentrated and purified with a flash silica gel column (petroleum ether/ethyl acetate = 10:1 to 3:1) to produce 10 (13.40 g, yellow solid, yield 91.63%). 1H NMR (600 MHz, Chloroform-d) δ 7.78 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 7.7 Hz, 2H), 4.20–4.12 (m, 2H), 3.73–3.71 (m, 2H), 3.71–3.64 (m, 2H), 3.58–3.54 (m, 4H), 3.50 (t, J = 5.4 Hz, 2H), 2.43 (s, 3H), 0.87 (s, 9H), 0.04 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 144.84, 133.01, 129.87, 128.03, 72.73, 70.82, 70.70, 69.31, 68.71, 62.74, 25.97, 25.74, 21.68, 18.39, −5.23. ESI-MS m/z 411.1654 [M + Na]+.
- 2,2,3,3-tetramethyl-12-(4-nitrophenoxy)-4,7,10-trioxa-3-siladodecane (11a). A mixture of 10 (500 mg, 1.29 mmol, 1.00 eq), 4-nitrophenol (269 mg, 1.94 mmol, 1.50 eq), potassium carbonate (359 mg, 2.58 mmol, 2.0 eq) and DMF (10 mL) was stirred at 70 °C overnight [21]. The reaction system was cooled to r.t. and extracted using H2O (100 mL) and ethyl acetate (100 mL). The organic phase was combined, washed with brine, dried by anhydrous sodium sulfate and concentrated under medium pressure to obtain 11a (372 mg, yellow solid, 74.90%). The crude product was used for the next step without further purification. 1H NMR (600 MHz, Chloroform-d) δ 8.19–8.14 (m, 2H), 6.98–6.94 (m, 2H), 4.23–4.18 (m, 2H), 3.90–3.87 (m, 2H), 3.78–3.73 (m, 2H), 3.72–3.69 (m, 2H), 3.69–3.66 (m, 2H), 3.57–3.53 (m, 2H), 0.87 (d, J = 3.6 Hz, 9H), 0.04 (d, J = 3.5 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 163.96, 141.61, 125.88, 125.86, 114.64, 72.77, 71.02, 70.81, 69.41, 68.29, 62.76, 25.94, 18.37, −5.24. ESI-MS m/z 424.3035 [M + Na]+.
- 4-((2,2,3,3-tetramethyl-4,7,10-trioxa-3-siladodecan-12-yl)oxy)aniline (12a). To a mixture solvent of THF (5 mL), MeOH (5 mL) and H2O (2 mL), 11a (300 mg, 0.78 mmol, 1.00 eq), iron powder (218 mg, 3.89 mmol, 5.00 eq) and ammonium chloride (210 mg, 3.89 mmol, 5.00 eq) were added [10]. The mixture was stirred overnight at 70 °C. After the reaction was completed, the mixture was filtered and washed with methanol (3 × 10 mL). The organic phase was collected, concentrated under reduced pressure, and purified with a flash silica gel column (petroleum ether/ethyl acetate = 5:1 to 1:1) to produce 12a (230 mg, brown oil, 83.06%). 1H NMR (600 MHz, Chloroform-d) δ 6.76 (d, J = 8.1 Hz, 2H), 6.63 (d, J = 8.2 Hz, 2H), 4.09–4.02 (m, 2H), 3.85–3.80 (m, 2H), 3.76 (dt, J = 5.6, 2.8 Hz, 2H), 3.72–3.63 (m, 4H), 3.59–3.53 (m, 2H), 0.89 (s, 9H), 0.06 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 151.74, 139.93, 116.12, 115.64, 72.48, 70.61, 70.56, 69.71, 67.91, 62.50, 25.73, 18.17, −5.47. ESI-MS m/z 356.2208 [M + H]+.
- 5-(1,1-dioxidothiomorpholino)-N-methyl-2-nitrobenzamide (2a). The method described for 2b was used to obtain 2a. 1H NMR (600 MHz, DMSO-d6) δ 8.36 (q, J = 4.7 Hz, 1H), 7.98 (d, J = 9.3 Hz, 1H), 7.15 (dd, J = 9.3, 2.9 Hz, 1H), 7.02 (d, J = 2.9 Hz, 1H), 4.03–3.98 (m, 4H), 3.19–3.13 (m, 4H), 2.75 (d, J = 4.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.23, 151.66, 136.72, 136.46, 127.32, 114.17, 113.64, 50.81, 46.00, 26.58. ESI-MS m/z 314.0780 [M + H]+.
- 2-amino-5-(1,1-dioxidothiomorpholino)-N-methylbenzamide (3a). The method described for 12a was used to obtain 3a. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (d, J = 4.7 Hz, 1H), 7.07 (d, J = 2.8 Hz, 1H), 6.96 (dd, J = 8.8, 2.8 Hz, 1H), 6.64 (d, J = 8.8 Hz, 1H), 6.02 (s, 2H), 3.61–3.40 (m, 4H), 3.23–3.08 (m, 4H), 2.72 (d, J = 4.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 169.58, 139.57, 123.47, 118.49, 117.31, 50.82, 49.47, 26.43. ESI-MS m/z 284.1038 [M + H]+.
- 2-((5-bromo-2-chloropyrimidin-4-yl)amino)-5-(1,1-dioxidothiomorpholino)-N-methylbenzamide (4a). The method described in 4b was used to obtain 4a. 1H NMR (600 MHz, DMSO-d6) δ 11.52 (s, 1H), 8.83–8.79 (m, 1H), 8.47 (s, 1H), 8.29 (d, J = 9.0 Hz, 1H), 7.30–7.25 (m, 2H), 3.84 (t, J = 5.3 Hz, 4H), 3.17 (t, J = 5.3 Hz, 4H), 2.81 (d, J = 4.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.15, 158.35, 158.09, 157.35, 143.87, 130.34, 123.69, 123.18, 119.40, 115.28, 105.08, 50.32, 47.24, 26.80. ESI-MS m/z 473.9926 [M + H]+.
- 2-((5-bromo-2-((4-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)phenyl)amino)pyrimidin-4-yl)amino)-5-(1,1-dioxidothiomorpholino)-N-methylbenzamide (5a). A solution of 4a (133 mg, 0.28 mmol, 1.00 eq), 12a (120 mg, 0.34 mmol, 1.20 eq) and 4-Toluenesulfonic acid (7 mg, 0.03 mmol, 0.10 eq) in 1,4-dioxane was stirred at 80 °C for 48 h. After the reaction was completed, the mixture was purified with a flash silica gel column (dichloromethane/methanol = 20:1 to 10:1) to produce 5a (110 mg, brown solid, 57.77%). 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 9.20 (s, 1H), 8.71 (d, J = 4.6 Hz, 1H), 8.39 (d, J = 8.8 Hz, 1H), 8.18 (s, 1H), 7.51 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 2.8 Hz, 1H), 7.17 (dd, J = 9.2, 2.9 Hz, 1H), 6.94–6.79 (m, 2H), 4.59 (t, J = 5.5 Hz, 1H), 4.05 (t, J = 4.6 Hz, 2H), 3.85–3.79 (m, 4H), 3.73 (t, J = 4.7 Hz, 2H), 3.61–3.57 (m, 2H), 3.57–3.53 (m, 2H), 3.51–3.47 (m, 2H), 3.45–3.41 (m, 2H), 3.21–3.16 (m, 4H), 2.80 (d, J = 4.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 168.79, 158.47, 157.14, 155.67, 153.70, 142.70, 133.43, 131.24, 123.54, 123.06, 121.53, 118.90, 115.02, 114.34, 93.38, 72.39, 70.00, 69.82, 69.10, 67.34, 60.26, 49.88, 47.16, 26.26. ESI-MS m/z 679.1574 [M + H]+.
- 2-(2-(2-(4-((5-bromo-4-((4-(1,1-dioxidothiomorpholino)-2-(methylcarbamoyl)phenyl)amino)pyrimidin-2-yl)amino)phenoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (6a). The method described for 10 was used to produce 6a (77 mg, white solid, 62.90% yield). 1HNMR (600 MHz, DMSO-d6) δ 10.82 (s, 1H), 9.21 (s, 1H), 8.72 (q, J = 4.8 Hz, 1H), 8.46–8.36 (m, 1H), 8.18 (s, 1H), 7.81–7.75 (m, 2H), 7.51 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.24 (d, J = 3.0 Hz, 1H), 7.18–7.12 (m, 1H), 6.89–6.83 (m, 2H), 4.12–4.09 (m, 2H), 4.05–4.01 (m, 2H), 3.83–3.79 (m, 4H), 3.72–3.68 (m, 2H), 3.59–3.56 (m, 2H), 3.55–3.51 (m, 2H), 3.49–3.46 (m, 2H), 3.21–3.16 (m, 4H), 2.80 (d, J = 4.7 Hz, 3H), 2.39 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.28, 158.91, 157.58, 156.15, 154.15, 145.44, 143.13, 133.92, 132.91, 131.72, 130.67, 128.17, 124.00, 123.48, 121.95, 119.35, 115.51, 114.79, 93.87, 70.51, 70.33, 70.25, 69.59, 68.45, 67.75, 50.33, 47.61, 26.76, 21.61. ESI-MS m/z 833.1060 [M + H]+.
- 2-((5-bromo-2-((4-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)phenyl)amino)pyrimidin-4-yl)amino)-5-(1,1-dioxidothiomorpholino)-N-methylbenzamide (7a). To a solution of 6a (50 mg, 0.06 mmol, 1.00 eq) in THF (1 mL), tetrabutylammonium fluoride (1 mol/L in THF, 0.30 mL, 5.00 eq) was added slowly, and then reacted for 2 h at 50 °C. After the reaction completed, the mixture was extracted using ethyl acetate and water. The organic phase was combined, washed with brine, dried using anhydrous sodium sulfate, concentrated under medium pressure and purified with a flash silica gel column (dichloromethane/methanol = 20:1 to 10:1) to produce 7a (28 mg, white solid, 68.53%) [22]. Rf (MeOH/DCM = 1/10) = 0.58. 1H NMR (600 MHz, DMSO-d6) δ 10.83 (s, 1H), 9.23 (s, 1H), 8.71 (q, J = 4.6 Hz, 1H), 8.38 (s, 1H), 8.18 (s, 1H), 7.50 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 2.9 Hz, 1H), 7.16 (dd, J = 9.2, 3.0 Hz, 1H), 6.87 (d, J = 8.6 Hz, 2H), 4.52 (dt, J = 48.0, 4.0 Hz, 2H), 4.05 (t, J = 4.6 Hz, 2H), 3.82 (t, J = 5.1 Hz, 4H), 3.74 (t, J = 4.6 Hz, 2H), 3.70–3.67 (m, 1H), 3.64–3.62 (m, 1H), 3.62–3.58 (m, 4H), 3.21–3.16 (m, 4H), 2.80 (d, J = 4.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 169.27, 158.95, 157.62, 156.16, 154.16, 143.14, 133.95, 131.75, 124.00, 123.48, 121.98, 119.35, 115.55, 114.81, 93.85, 84.40, 82.75, 70.45, 70.38, 70.33, 70.14, 69.61, 67.80, 50.37, 47.64, 26.74. The ESI-HRMS m/z calculated for C28H35BrFN6O6S+ 681.1501 was found to be 681.1498. IR for 7a (KBr, cm−1): 3230 (vw), 2860 (vw), 1570 (m), 1510 (s), 1420 (2), 1320 (w), 1280 (m), 1230 (m), 1180 (m), 1120 (w), 1010 (w), 928 (w), 865 (w), 773 (m), 539 (m).
- morpholino(5-nitro-2-((2,2,3,3-tetramethyl-4,7,10-trioxa-3-siladodecan-12-yl)oxy)phenyl)methanone (11d). The method described for 11a was used to produce 11d (yellow solid, 71.23%). 1H NMR (600 MHz, Chloroform-d) δ 8.25 (dd, J = 9.1, 2.8 Hz, 1H), 8.19 (d, J = 2.8 Hz, 1H), 7.01 (d, J = 9.1 Hz, 1H), 4.38–4.29 (m, 1H), 4.25–4.18 (m, 1H), 3.95–3.87 (m, 1H), 3.86–3.83 (m, 1H), 3.82–3.78 (m, 2H), 3.77–3.74 (m, 4H), 3.73–3.70 (m, 1H), 3.69–3.64 (m, 4H), 3.60–3.56 (m, 1H), 3.54 (t, J = 5.2 Hz, 2H), 3.40–3.29 (m, 1H), 3.27–3.15 (m, 1H), 0.88 (s, 9H), 0.05 (s, 6H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 165.28, 159.42, 141.78, 126.74, 124.61, 111.78, 72.83, 71.05, 69.28, 68.95, 66.80, 62.76, 61.83, 60.48, 47.28, 42.40, 25.99, 25.72, −3.51, −5.20. ESI-MS m/z 499.2460 [M + H]+.
- (5-amino-2-((2,2,3,3-tetramethyl-4,7,10-trioxa-3-siladodecan-12-yl)oxy)phenyl)(morpholino)methanone (12d). The method described for 12a was used to produce 12d (brown oil, 85.21% yield). 1H NMR (600 MHz, Chloroform-d) δ 6.76–6.72 (m, 1H), 6.69–6.65 (m, 1H), 6.64–6.62 (m, 1H), 4.15–4.06 (m, 1H), 4.03–3.97 (m, 1H), 3.83–3.79 (m, 1H), 3.79–3.75 (m, 4H), 3.74–3.70 (m, 4H), 3.67–3.64 (m, 4H), 3.58–3.50 (m, 3H), 3.42–3.35 (m, 1H), 3.26–3.16 (m, 1H), 0.89–0.86 (m, 9H), 0.07–0.03 (m, 6H). 13C NMR (151 MHz, Chloroform-d) δ 167.81, 147.16, 140.93, 126.68, 117.04, 114.95, 114.57, 72.65, 70.75, 70.68, 69.79, 69.00, 66.91, 66.71, 62.65, 47.24, 42.11, 25.91, 18.32, −5.28. ESI-MS m/z 469.2664 [M + H]+.
- 5-(4-acetylpiperazin-1-yl)-2-((5-bromo-2-((4-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)-3-(morpholine-4-carbonyl)phenyl)amino)pyrimidin-4-yl)amino)-N-methylbenzamide (5d). The method described for 5a was used to produce 5d (white solid, yield 53.18%). 1H NMR (600 MHz, DMSO-d6) δ 9.24 (s, 1H), 9.10 (q, J = 4.8 Hz, 1H), 8.68 (s, 1H), 8.18 (s, 1H), 7.83 (d, J = 2.7 Hz, 1H), 7.74 (s, 1H), 7.67 (dd, J = 9.1, 2.7 Hz, 1H), 7.34–7.24 (m, 1H), 7.20 (d, J = 8.7 Hz, 1H), 6.83 (d, J = 9.0 Hz, 1H), 4.58 (t, J = 5.5 Hz, 1H), 4.10–4.00 (m, 2H), 3.75–3.68 (m, 2H), 3.66–3.61 (m, 4H), 3.61–3.55 (m, 8H), 3.55–3.53 (m, 2H), 3.49–3.47 (m, 2H), 3.41 (t, J = 5.1 Hz, 2H), 3.23–3.03 (m, 2H), 2.96–2.91 (m, 2H), 2.87 (t, J = 5.2 Hz, 2H), 2.85 (d, J = 4.8 Hz, 3H), 2.05 (s, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm):168.24, 166.35, 166.16, 157.88, 157.25, 156.50, 148.47, 146.21, 134.28, 133.67, 128.70, 126.19, 125.14, 124.97, 120.91, 119.93, 118.36, 112.18, 92.27, 72.16, 69.82, 69.59, 68.82, 67.70, 66.00, 65.84, 60.00, 52.63, 52.07, 46.46, 45.63, 41.40, 40.89, 25.74, 21.03. ESI-MS m/z 785.2104 [M + H]+.
- 2-(2-(2-(4-((4-((4-(4-acetylpiperazin-1-yl)-2-(methylcarbamoyl)phenyl)amino)-5-bromopyrimidin-2-yl)amino)-2-(morpholine-4-carbonyl)phenoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (6d). The method described for 10 was used to produce 6d (white solid, yield 57.2%). 1H NMR (600 MHz, DMSO-d6) δ 9.25 (s, 1H), 9.10 (q, J = 4.7 Hz, 1H), 8.68 (s, 1H), 8.19 (s, 1H), 7.83 (d, J = 2.8 Hz, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.74 (s, 1H), 7.67 (dd, J = 9.0, 2.7 Hz, 1H), 7.46 (d, J = 8.1 Hz, 2H), 7.26 (s, 1H), 7.19 (d, J = 8.7 Hz, 1H), 6.82 (d, J = 8.9 Hz, 1H), 4.17–4.08 (m, 2H), 4.03 (t, J = 5.3 Hz, 2H), 3.68 (t, J = 4.8 Hz, 2H), 3.64–3.60 (m, 4H), 3.60–3.54 (m, 8H), 3.53–3.49 (m, 2H), 3.50–3.45 (m, 2H), 3.21–3.02 (m, 2H), 2.93 (t, J = 5.5 Hz, 2H), 2.87 (t, J = 5.0 Hz, 2H), 2.84 (d, J = 4.7 Hz, 3H), 2.39 (s, 3H), 2.04 (s, 3H).13C NMR (151 MHz, DMSO-d6) δ 168.40, 166.52, 166.34, 158.08, 156.69, 148.64, 146.39, 144.88, 134.48, 133.88, 132.40, 130.11, 128.89, 127.59, 125.33, 125.18, 121.09, 120.11, 118.54, 112.34, 69.94, 69.86, 69.70, 69.00, 67.93, 67.85, 66.16, 66.01, 52.82, 52.27, 46.63, 45.92, 41.57, 41.07, 25.93, 21.22, 21.05. ESI-MS m/z 939.2494 [M + H]+.
- 5-(4-acetylpiperazin-1-yl)-2-((5-bromo-2-((4-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)-3-(morpholine-4-carbonyl)phenyl)amino)pyrimidin-4-yl)amino)-N-methylbenzamide (7d). The method described for 7a was used to produce 7d (white solid, yield 65.4%). Rf (MeOH/DCM = 1/10) = 0.47. 1H NMR (600 MHz, DMSO-d6) δ 9.26 (s, 1H), 9.10 (q, J = 4.7 Hz, 1H), 8.70 (s, 1H), 8.19 (s, 1H), 7.83 (d, J = 2.7 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.67 (dd, J = 9.0, 2.7 Hz, 1H), 7.26 (s, 1H), 7.20 (d, J = 8.7 Hz, 1H), 6.83 (d, J = 9.1 Hz, 1H), 4.58–4.53 (m, 1H), 4.52–4.45 (m, 1H), 4.09–4.01 (m, 2H), 3.72 (td, J = 5.6, 3.0 Hz, 2H), 3.68–3.66 (m, 1H), 3.65–3.64 (m, 1H), 3.63–3.60 (m, 4H), 3.60–3.55 (m, 8H), 3.46–3.41 (m, 2H), 3.22–3.17 (m, 1H), 3.11–3.05 (m, 1H), 2.93 (t, J = 5.3 Hz, 2H), 2.87 (t, J = 5.1 Hz, 2H), 2.85 (d, J = 4.7 Hz, 3H), 2.05 (s, 3H).13C NMR (100 MHz, DMSO-d6, δ ppm): 168.03, 166.13, 165.94, 167.55, 156.33, 148.29, 146.33, 134.04, 133.39, 128.47, 126.05, 124.92, 124.83, 120.76, 129.73, 128.20, 111.93, 83.45, 81.81, 69.59, 69.45, 69.41, 69.22, 69.63, 67.45, 65.80, 65.64, 52.43, 51.88, 46.25, 45.52, 41.18, 40.67, 25.55, 20.84. The ESI-HRMS m/z calculated for C35H45BrFN8O7+ 787.2573 was found to be 787.2573. IR for 7d (KBr, cm−1): 3280 (w), 2850 (m), 1630 (s), 1640 (s), 1490 (m), 1420 (vs), 1240 (s), 1110 (s), 1030 (m), 999 (m), 896 (m), 810 (m), 771 (m), 698 (w), 643 (w), 578 (w), 554 (s), 524 (m).
- N-methyl-5-morpholino-2-nitrobenzamide (2b). To a stirring mixture of 5-fluoro-N-methyl-2-nitrobenzamide (2.0 g, 10.1 mmol, 1.0 eq), K2CO3 (2.8 g, 20.2 mmol, 2.0 eq) and acetonitrile (20 mL), morpholine (0.97 g, 11.1 mmol, 1.1 eq) was added. Then, the reaction continued to react overnight at 50 °C. After the reaction was completed, the mixture was poured into water, the solid was filtered under a vacuum, washed with water (50 mL, 3 times) and dried naturally to obtained 2b (yellow solid, 1.9 g, yield 71.2%). 1H NMR (600 MHz, DMSO-d6) δ 8.31 (d, J = 4.7 Hz, 1H), 7.99 (d, J = 9.3 Hz, 1H), 7.03 (dd, J = 9.4, 2.9 Hz, 1H), 6.88 (d, J = 2.9 Hz, 1H), 3.71 (t, 4H), 3.41 (t, J = 5.0 Hz, 4H), 2.74 (d, J = 4.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.06, 153.90, 136.27, 135.09, 126.70, 112.64, 112.19, 65.74, 46.50, 26.07. ESI-MS m/z 266.1110 [M + H]+.
- 2-amino-N-methyl-5-morpholinobenzamide (3b). The method described for 12a was used to produce 3b (brown solid, yield 82.1%). 1H NMR (600 MHz, Chloroform-d) δ 6.99–6.84 (m, 2H), 6.67 (d, J = 8.2 Hz, 1H), 6.15 (s, 1H), 3.85 (t, J = 4.7 Hz, 4H), 3.01 (s, 4H), 2.96 (d, J = 4.9 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.26, 143.46, 141.11, 121.69, 117.23, 115.32, 66.13, 50.43, 25.77. ESI-MS m/z 236.0715 [M + H]+.
- 2-((5-bromo-2-chloropyrimidin-4-yl)amino)-N-methyl-5-morpholinobenzamide (4b). To a solution of 5-bromo-2,4-dichloropyrimidine (1.0 g, 4.4 mmol, 1.0 eq) in DMF (10 mL), 3b (1.0 g, 4.4 mmol, 1.0 eq) and K2CO3 (917 mg, 6.6 mmol, 1.6 eq) were added slowly. The mixture was allowed to react overnight at 50 °C, and then, it was poured into water and filtered. The filter cake was washed with water (50 mL, 3 times) and dried naturally to obtained 4b (1.3 g, yellow solid, 67.4%) [10]. 1H NMR (600 MHz, DMSO-d6) δ 11.57 (s, 1H), 8.81 (d, J = 4.8 Hz, 1H), 8.46 (s, 1H), 8.27 (d, J = 9.1 Hz, 1H), 7.26 (d, J = 2.9 Hz, 1H), 7.20 (dd, J = 9.2, 2.9 Hz, 1H), 3.76 (t, J = 4.8 Hz, 4H), 3.17 (t, J = 5.8, 3.9 Hz, 4H), 2.80 (d, J = 4.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.73, 157.74, 157.58, 156.82, 146.86, 129.81, 122.72, 122.42, 118.14, 114.14, 104.54, 66.00, 48.43, 26.29.ESI-MS m/z 426.0280 [M + H]+.
- 2-((2-((3-(4-acetylpiperazine-1-carbonyl)-4-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)phenyl)amino)-5-bromopyrimidin-4-yl)amino)-N-methyl-5-morpholinobenzamide (5f). The method described for 5a was used to obtain 5f (white solid, yield 55.7%). 1H NMR (600 MHz, DMSO-d6) δ 10.98 (d, J = 12.8 Hz, 1H), 9.32 (d, J = 11.3 Hz, 1H), 8.77–8.66 (m, 1H), 8.42 (s, 1H), 8.20 (d, J = 4.0 Hz, 1H), 7.64–7.58 (m, 1H), 7.51 (d, J = 26.2 Hz, 1H), 7.24–7.19 (m, 1H), 7.12 (dt, J = 9.3, 3.6 Hz, 1H), 7.01 (dd, J = 9.0, 3.4 Hz, 1H), 4.58–4.55 (m, 1H), 4.17–4.04 (m, 2H), 3.79–3.74 (m, 4H), 3.73–3.68 (m, 2H), 3.57–3.54 (m, 2H), 3.52–3.45 (m, 8H), 3.45–3.37 (m, 6H), 3.15 (t, J = 5.0 Hz, 4H), 2.79 (d, J = 4.5 Hz, 3H), 2.00 (d, J = 40.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.43, 168.87, 167.05, 158.71, 157.47, 156.15, 149.48, 146.53, 145.97, 138.34, 134.43, 131.73, 128.64, 126.06, 123.27, 119.00, 114.78, 113.27, 94.40, 72.89, 70.48, 70.29, 69.57, 68.46, 66.59, 60.72, 49.32, 26.78, 21.32. ESI-MS m/z 785.2112 [M + H]+.
- 2-(2-(2-(2-(4-acetylpiperazine-1-carbonyl)-4-((5-bromo-4-((2-(methylcarbamoyl)-4-morpholinophenyl)amino)pyrimidin-2-yl)amino)phenoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (6f). The method described for 6a was used to obtain 6f (white solid, yield 74.2%) 1H NMR (600 MHz, DMSO-d6) δ 10.99 (d, J = 12.9 Hz, 1H), 9.33 (d, J = 11.4 Hz, 1H), 8.83–8.64 (m, 1H), 8.42 (s, 1H), 8.20 (d, J = 4.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.64–7.59 (m, 1H), 7.55–7.48 (m, 1H), 7.46 (d, J = 7.9 Hz, 2H), 7.22 (s, 1H), 7.15–7.10 (m, 1H), 7.06–6.97 (m, 1H), 4.13–4.05 (m, 4H), 3.78–3.75 (m, 4H), 3.68 (d, J = 4.8 Hz, 4H), 3.54 (t, J = 4.4 Hz, 4H), 3.51–3.47 (m, 4H), 3.45–3.41 (m, 4H), 3.15 (t, J = 4.9 Hz, 4H), 2.79 (d, J = 4.4 Hz, 3H), 2.39 (d, J = 2.6 Hz, 3H), 1.99 (d, J = 41.7 Hz, 3H).13C NMR (101 MHz, DMSO-d6) δ 169.41, 168.86, 167.00, 158.33, 156.20, 149.57, 146.60, 145.44, 134.24, 132.90, 131.62, 130.66, 128.61, 128.16, 126.07, 123.32, 122.81, 122.18, 119.56, 119.22, 118.96, 114.78, 113.22, 94.42, 70.47, 70.32, 70.19, 69.54, 68.44, 66.58, 55.46, 49.28, 26.79, 21.74, 21.60. ESI-MS m/z 939.2455 [M + H]+.
- 3-((2-((3-(4-acetylpiperazine-1-carbonyl)-4-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)phenyl)amino)-5-bromopyrimidin-4-yl)amino)-N-methyl-5-morpholinobenzamide (7f). The method described for 7a was used to obtain 7f (whited solid, yield 62.3%). Rf (MeOH/DCM = 1/10) = 0.42. 1H NMR (600 MHz, DMSO-d6) δ 10.98 (d, J = 13.0 Hz, 1H), 9.31 (d, J = 11.6 Hz, 1H), 8.78–8.59 (m, 1H), 8.42 (s, 1H), 8.20 (d, J = 4.0 Hz, 1H), 7.67–7.56 (m, 1H), 7.51 (d, J = 26.8 Hz, 1H), 7.22 (t, J = 2.2 Hz, 1H), 7.12 (dt, J = 9.1, 3.4 Hz, 1H), 7.01 (dd, J = 9.1, 3.9 Hz, 1H), 4.60–4.41 (m, 2H), 4.16–4.05 (m, 2H), 3.77 (t, J = 4.8 Hz, 4H), 3.72 (q, J = 5.1 Hz, 2H), 3.67–3.62 (m, 2H), 3.62–3.55 (m, 4H), 3.55–3.52 (m, 2H), 3.51–3.38 (m, 4H), 3.28–3.19 (m, 1H), 3.16 (t, J = 4.9 Hz, 4H), 3.12–3.00 (m, 1H), 2.79 (d, J = 4.5 Hz, 3H), 2.00 (d, J = 40.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 169.43, 168.85, 166.97, 158.69, 157.49, 156.16, 149.41, 146.54, 134.49, 131.72, 130.19, 126.13, 123.28, 122.79, 119.24, 119.00, 114.79, 113.33, 94.14, 84.37, 82.72, 70.43, 70.31, 70.12, 69.58, 68.52, 68.43, 66.59, 49.32, 26.77, 21.75. The ESI-HRMS m/z calculated for C35H45BrFN8O7+ 787.2573 was found to be 787.2573. IR for 7f (KBr, cm−1): 3270 (m), 2920 (m), 2850 (m), 1550 (m), 1500 (m), 1410 (s), 1240 (s), 1120 (s), 1050 (m), 997 (m), 957 (m), 878 (m), 818 (m), 774 (m), 613 (w), 584 (w), 540 (w), 512 (w).
- (2-bromo-4-nitrophenoxy)-2,2,3,3-tetramethyl-4,7,10-trioxa-3-siladodecane (11c). The method described for 11a was used to obtain 11c (yellow solid, yield 77.2%).1H NMR (600 MHz, Chloroform-d) δ 8.46 (d, J = 2.7, 0.7 Hz, 1H), 8.18 (dd, J = 9.1, 2.7, 0.8 Hz, 1H), 7.05–6.94 (m, 1H), 4.29 (t, J = 4.8 Hz, 2H), 3.99–3.92 (m, 2H), 3.78–3.75 (m, 4H), 3.70–3.68 (m, 2H), 3.57–3.55 (m, 2H), 0.88 (s, 9H), 0.05 (s, 6H). 13C NMR (100 MHz, Chloroform-d, δ ppm): 160.32, 141.47, 129.02, 124.51, 112.07, 111.78, 72.63, 71.18, 70.71, 69.64, 69.10, 62.62, 25.80, 25.52, −5.39.ESI-MS m/z 464.1007 [M + H]+.
- 1-methyl-4-(5-nitro-2-((2,2,3,3-tetramethyl-4,7,10-trioxa-3-siladodecan-12-yl)oxy)phenyl)-1H-pyrazole (13). In a one-neck bottle, 11c (500 mg, 1.08 mmol), 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (337 mg, 1.62 mmol), 1,1′-Bis (diphenylphosphino) ferrocene-palladium (II) dichloride dichloromethane complex (90 mg, 0.11 mmol), Cs2CO3 (706.3 mg, 2.16 mmol), 1,4-dioxane (5 mL) and H2O (1 mL), the mixture was allowed to react for 16 h under a nitrogen atmosphere. Then, the solvent was removed under a vacuum, and the crude product was purified with a flash silica gel column (dichloromethane/MeOH = 30/1~20/1) to produce 13 (brown oil, 380 mg, 76%) [23]. 1H NMR (600 MHz, Chloroform-d) δ 8.40 (d, J = 2.8 Hz, 1H), 8.07 (ddd, J = 9.0, 2.8, 1.1 Hz, 1H), 8.00 (d, J = 24.1 Hz, 2H), 6.97 (d, J = 9.0 Hz, 1H), 4.31 (t, J = 4.7 Hz, 2H), 3.98–3.95 (m, 5H), 3.77–3.73 (m, 4H), 3.73–3.70 (m, 2H), 3.57 (t, J = 5.3, 1.1 Hz, 2H), 0.87 (s, 9H), 0.04 (s, 6H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 159.66, 141.81, 138.40, 130.46, 123.05, 122.95, 122.63, 116.84, 111.55, 72.88, 70.98, 70.93, 69.41, 68.22, 62.78, 39.18, 26.00, 24.96, −5.20. ESI-MS m/z 466.2317 [M + H]+.
- 3-(1-methyl-1H-pyrazol-4-yl)-4-((2,2,3,3-tetramethyl-4,7,10-trioxa-3-siladodecan-12-yl)oxy)aniline (14). The method described for 12a was used to obtain 14 (brown oil, yield 78.3%). 1H NMR (600 MHz, Chloroform-d) δ 7.96 (s, 1H), 7.84 (s, 1H), 6.93 (d, J = 2.8 Hz, 1H), 6.77 (d, J = 8.6 Hz, 1H), 6.56 (dd, J = 8.6, 2.8 Hz, 1H), 4.12–4.07 (m, 2H), 3.92 (s, 3H), 3.87–3.84 (m, 2H), 3.76 (t, J = 5.4 Hz, 2H), 3.70 (s, 4H), 3.57 (t, J = 5.4 Hz, 2H), 0.88 (s, 9H), 0.05 (s, 6H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 148.25, 140.09, 137.87, 129.95, 122.82, 118.38, 114.36, 113.84, 72.59, 70.68, 70.50, 69.86, 68.26, 62.56, 38.77, 18.22, −5.41. ESI-MS m/z 436.2564 [M + H]+.
- 2-((2-((3-(4-acetylpiperazine-1-carbonyl)-4-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)phenyl)amino)-5-bromopyrimidin-4-yl)amino)-5-(1,1-dioxidothiomorpholino)-N-methylbenzamide (5b). The method described for 5a was used to obtain 5b (white solid, yield 57.7%). 1H NMR (600 MHz, DMSO-d6) δ 10.93 (d, J = 8.4 Hz, 1H), 9.33 (d, J = 6.6 Hz, 1H), 8.79–8.69 (m, 1H), 8.42 (s, 1H), 8.21 (d, J = 2.7 Hz, 1H), 7.61 (dd, J = 9.0, 2.7 Hz, 1H), 7.55–7.47 (m, 1H), 7.25 (d, J = 2.9 Hz, 1H), 7.23–7.15 (m, 1H), 7.02 (dd, J = 9.0, 5.7 Hz, 1H), 4.57 (t, J = 5.5 Hz, 1H), 4.09 (s, 2H), 3.83 (t, J = 5.2 Hz, 4H), 3.72 (q, J = 5.6 Hz, 2H), 3.56 (t, J = 5.3 Hz, 2H), 3.49 (dt, J = 12.8, 5.1 Hz, 8H), 3.43–3.37 (m, 6H), 3.20 (d, J = 5.9 Hz, 4H), 2.80 (d, J = 4.5 Hz, 3H), 2.00 (d, J = 40.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.30, 168.89, 167.05, 158.71, 156.14, 149.50, 143.00, 131.78, 126.10, 123.49, 123.16, 119.76, 119.25, 119.13, 115.91, 115.58, 113.25, 93.88, 72.89, 70.48, 70.28, 70.23, 69.74, 69.57, 68.46, 60.73, 50.35, 47.59, 26.75, 21.76. ESI-MS m/z 833.1687 [M + H]+.
- 2-(2-(2-(2-(4-acetylpiperazine-1-carbonyl)-4-((5-bromo-4-((4-(1,1-dioxidothiomorpholino)-2-(methylcarbamoyl)phenyl)amino)pyrimidin-2-yl)amino)phenoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (6b). The method described for 6a was used to obtain 6b. (white solid, yield 74.5%). 1H NMR (600 MHz, DMSO-d6) δ 10.90 (d, J = 8.5 Hz, 1H), 9.30 (d, J = 7.1 Hz, 1H), 8.68 (s, 1H), 8.39 (s, 1H), 8.17 (s, 1H), 7.74 (d, J = 7.7 Hz, 2H), 7.57 (d, J = 9.7 Hz, 1H), 7.48–7.44 (m, 1H), 7.42 (d, J = 8.3 Hz, 2H), 7.22 (s, 1H), 7.17 (d, J = 9.0 Hz, 1H), 6.98 (t, J = 7.6 Hz, 1H), 4.09–3.99 (m, 4H), 3.82–3.75 (m, 4H), 3.72–3.60 (m, 4H), 3.56–3.48 (m, 4H), 3.47–3.42 (m, 4H), 3.40–3.37 (m, 2H), 3.37–3.33 (m, 2H), 3.21–3.13 (m, 4H), 2.77 (d, J = 4.6 Hz, 3H), 2.36 (s, 3H), 1.96 (d, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.30, 168.86, 158.69, 156.13, 149.61, 145.44, 142.99, 132.90, 131.77, 128.58, 126.03, 123.11, 119.71, 115.58, 94.92, 70.47, 70.31, 69.55, 68.43, 55.46, 50.32, 49.12, 47.56, 45.95, 26.75, 21.61, 9.00. ESI-MS m/z 987.2181 [M + H]+.
- 2-((2-((3-(4-acetylpiperazine-1-carbonyl)-4-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)phenyl)amino)-5-bromopyrimidin-4-yl)amino)-5-(1,1-dioxidothiomorpholino)-N-methylbenzamide (7b). The method described for 7a was used to obtain 7b (white solid, 71.3%). Rf (MeOH/DCM = 1/10) = 0.42. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (d, J = 5.3 Hz, 1H), 9.30 (d, J = 4.3 Hz, 1H), 8.68 (q, J = 4.7 Hz, 1H), 8.38 (s, 1H), 8.17 (s, 1H), 7.57 (dd, J = 8.9, 2.7 Hz, 1H), 7.45 (d, J = 9.3 Hz, 1H), 7.22 (d, J = 2.9 Hz, 1H), 7.17 (d, J = 9.3 Hz, 1H), 6.98 (dd, J = 9.1, 4.1 Hz, 1H), 4.54–4.51 (m, 1H), 4.42–4.37 (m, 1H), 4.09–4.03 (m, 2H), 3.79 (t, J = 5.1 Hz, 4H), 3.68 (d, J = 4.5 Hz, 3H), 3.62 (t, J = 4.0 Hz, 2H), 3.58–3.52 (m, 4H), 3.52–3.46 (m, 5H), 3.38–3.36 (m, 2H), 3.17–3.13 (m, 4H), 2.77 (d, J = 4.4 Hz, 3H), 1.96 (d, J = 27.0 Hz, 3H).13C NMR (100 MHz, DMSO-d6, δ ppm):168.76, 168.34, 166.43, 158.15, 156.99, 155.61, 149.00, 142.49, 133.81, 131.23, 129.64, 125.52, 122.96, 122.65, 121.65, 119.21, 118.77, 118.62, 115.03, 112.70, 93.79, 83.83, 82.18, 69.89, 69.77, 69.58, 69.04, 68.00, 49.80, 47.04, 28.99, 26.21, 22.08, 21.19, 13.93. The ESI-HRMS m/z calculated for C35H45BrFN8O8S+ 835.2243 was found to be 835.1687. IR for 7b (KBr, cm−1): 3290 (vw), 2920 (s), 2850 (m), 1630 (m), 1560 (s), 1490 (w), 1460 (m), 1410 (s), 1250 (s), 1120 (s), 1050 (m), 995 (m), 949 (w), 856 (w), 818 (w), 774 (m), 717 (w), 646 (w), 519 (m), 539 (m), 594 (w).
- 2-((5-bromo-2-((4-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)-3-(1-methyl-1H-pyrazol-4-yl)phenyl)amino)pyrimidin-4-yl)amino)-5-(1,1-dioxidothiomorpholino)-N-methylbenzamide (5c). The method described for 5a was used to obtain 5c (white solid, 48.6%). 1H NMR (600 MHz, DMSO-d6) δ 10.87 (s, 1H), 9.11 (s, 1H), 8.66 (d, J = 5.1 Hz, 1H), 8.36 (s, 1H), 8.15 (s, 1H), 8.05 (s, 1H), 7.76 (s, 2H), 7.31–7.23 (m, 1H), 7.22–7.14 (m, 1H), 6.93 (d, J = 8.8 Hz, 1H), 6.66 (s, 1H), 4.57 (t, J = 5.6 Hz, 1H), 4.17–4.05 (m, 2H), 3.82 (s, 3H), 3.81–3.79 (m, 2H), 3.71–3.65 (m, 4H), 3.64–3.60 (m, 2H), 3.58–3.55 (m, 2H), 3.47–3.44 (m, 2H), 3.43–3.39 (m, 2H), 3.13–3.07 (m, 4H), 2.76 (d, J = 4.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 169.30, 159.20, 157.73, 156.05, 150.66, 142.82, 138.02, 133.95, 131.86, 130.32, 123.50, 123.03, 121.66, 120.05, 119.14, 118.35, 115.50, 113.38, 93.80, 72.93, 70.42, 70.32, 69.60, 68.14, 60.75, 55.45, 50.30, 47.44, 39.08, 26.75. ESI-MS m/z 731.1633 [M + H]+.
- 2-(2-(2-(4-((5-bromo-4-((4-(1,1-dioxidothiomorpholino)-2-(methylcarbamoyl)phenyl)amino)pyrimidin-2-yl)amino)-2-(1-methyl-1H-pyrazol-4-yl)phenoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (6c). The method described for 6a was used to obtain 6c (white solid, 72.1%). 1H NMR (600 MHz, DMSO-d6) δ 10.88 (s, 1H), 9.13 (s, 1H), 8.68 (d, J = 4.8 Hz, 1H), 8.36 (s, 1H), 8.15 (s, 1H), 8.03 (d, J = 5.8 Hz, 1H), 7.76 (d, J = 2.7 Hz, 1H), 7.75–7.69 (m, 2H), 7.56–7.49 (m, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.31 (d, J = 7.9 Hz, 1H), 7.29–7.22 (m, 1H), 7.18 (d, J = 2.5 Hz, 1H), 6.93 (dd, J = 8.9, 2.8 Hz, 1H), 6.64 (s, 1H), 4.11–4.04 (m, 3H), 3.83–3.75 (m, 5H), 3.74–3.69 (m, 1H), 3.68–3.62 (m, 4H), 3.62–3.47 (m, 6H), 3.09 (d, J = 5.9 Hz, 4H), 2.76 (d, J = 4.5 Hz, 3H), 2.31 (d, J = 32.0 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 168.60, 155.83, 151.10, 145.72, 142.87, 137.60, 137.49, 129.84, 129.43, 128.04, 127.57, 125.49, 124.47, 123.33, 122.99, 117.60, 114.71, 112.92, 93.33, 72.38, 70.53, 69.88, 69.77, 69.09, 60.20, 46.66, 43.51, 38.56, 26.22, 20.76. ESI-MS m/z 897.1796 [M + H]+.
- 2-((5-bromo-2-((4-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)-3-(1-methyl-1H-pyrazol-4-yl)phenyl)amino)pyrimidin-4-yl)amino)-5-(1,1-dioxidothiomorpholino)-N-methylbenzamide (7c). The method described for 7a was used to obtain 7c (white solid, 68.2%). Rf (MeOH/DCM = 1/10) = 0.53. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 9.14 (s, 1H), 8.78–8.68 (m, 1H), 8.46–8.35 (m, 1H), 8.19 (s, 1H), 8.09 (s, 1H), 7.85–7.69 (m, 2H), 7.34–7.26 (m, 1H), 7.27–7.20 (m, 1H), 6.97 (d, J = 8.9 Hz, 1H), 6.70 (s, 1H), 4.57 (t, J = 4.0 Hz, 1H), 4.45 (t, J = 4.0 Hz, 1H), 4.19–4.08 (m, 2H), 3.85 (s, 5H), 3.74–3.69 (m, 4H), 3.69–3.61 (m, 6H), 3.16–3.08 (m, 4H), 2.80 (d, J = 4.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 169.30, 159.21, 157.73, 156.03, 150.06, 142.82, 137.96, 133.94, 131.87, 130.31, 123.50, 123.02, 121.66, 120.35, 120.05, 119.13, 118.35, 115.51, 113.36, 93.79, 84.40, 82.75, 70.47, 70.35, 70.14, 69.69, 68.13, 50.31, 47.44, 39.06, 26.74. The ESI-HRMS m/z calculated for C32H39BrFN8O6S+ 761.1875 was found to be 761.1875. IR for 7c (KBr, cm−1): 3260 (vw), 2920 (vw), 1260 (w), 1520 (s), 1420 (m), 1270 (m), 1120 (s), 1050 (w), 1000 (w), 949 (w), 870 (m), 810 (m), 775 (m), 714 (m), 668 (m), 536 (m).
- 2-((5-bromo-2-((4-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)-3-(1-methyl-1H-pyrazol-4-yl)phenyl)amino)pyrimidin-4-yl)amino)-5-(4-ethylpiperazin-1-yl)-N-methylbenzamide (5g). The method described for 5a was used to obtain 5g. (white solid, yield 73.5%). 1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1H), 9.04 (s, 1H), 8.59 (s, 1H), 8.13 (s, 1H), 8.00 (s, 1H), 7.78–7.70 (m, 2H), 7.67 (d, J = 2.6 Hz, 1H), 7.57 (s, 1H), 7.26 (d, J = 8.9 Hz, 1H), 6.88 (d, J = 8.7 Hz, 1H), 6.81 (d, J = 8.9 Hz, 1H), 4.59 (s, 1H), 4.07–4.03 (m, 2H), 3.82 (s, 3H), 3.80–3.76 (m, 2H), 3.63–3.58 (m, 2H), 3.57–3.52 (m, 2H), 3.49–3.42 (m, 2H), 3.42–3.37 (m, 2H), 3.28–3.25 (m, 2H), 3.13 (d, J = 5.1 Hz, 2H), 2.79 (d, J = 4.7 Hz, 2H), 2.78–2.73 (m, 3H), 2.58–2.48 (m, 2H), 2.43–2.31 (m, 2H), 1.01 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.04, 158.95, 158.16, 157.09, 150.24, 137.79, 134.94, 134.11, 130.29, 126.72, 125.32, 121.54, 120.31, 119.28, 118.31, 113.30, 92.60, 72.94, 70.42, 69.65, 68.06, 60.74, 53.10, 52.03, 39.09, 26.37, 12.50. The ESI-MS m/z calculated for C34H45BrN9O5+ 738.2722 was found to be 738.2655.
- 2-(2-(2-(4-((5-bromo-4-((4-(4-ethylpiperazin-1-yl)-2-(methylcarbamoyl)phenyl)amino)pyrimidin-2-yl)amino)-2-(1-methyl-1H-pyrazol-4-yl)phenoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (6g). The method described for 6a was used to obtain 6g (white solid, yield 69.8%). 1H NMR (600 MHz, DMSO-d6) δ 9.39–9.19 (m, 1H), 9.09 (s, 1H), 8.64 (s, 1H), 8.17 (s, 1H), 8.02 (s, 1H), 7.84–7.77 (m, 2H), 7.76–7.74 (m, 2H), 7.73–7.70 (m, 1H), 7.60 (s, 1H), 7.45–7.41 (m, 2H), 7.33 (s, 1H), 6.92 (s, 1H), 6.85 (d, J = 8.9 Hz, 1H), 4.13–4.05 (m, 4H), 3.84 (s, 3H), 3.79 (t, J = 4.7 Hz, 2H), 3.62–3.56 (m, 4H), 3.54–3.49 (m, 2H), 3.37–3.34 (m, 2H), 3.33–3.30 (m, 2H), 3.20–3.09 (m, 2H), 2.87–2.75 (m, 5H), 2.55–2.51 (m, 1H), 2.49–2.46 (m, 1H), 2.37 (s, 3H), 1.26–1.17 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.64, 166.96, 158.96, 158.14, 157.09, 150.22, 145.42, 139.19, 137.74, 134.93, 134.12, 132.90, 130.65, 130.23, 128.12, 127.24, 126.76, 126.62, 126.12, 125.35, 121.54, 120.37, 119.27, 118.30, 118.07, 113.28, 70.44, 70.36, 70.21, 69.65, 68.46, 68.03, 55.47, 53.29, 52.93, 52.44, 52.07, 50.76, 26.32, 21.58, 12.53. ESI-MS m/z 892.2792 [M + H]+.
- 2-((5-bromo-2-((4-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)-3-(1-methyl-1H-pyrazol-4-yl)phenyl)amino)pyrimidin-4-yl)amino)-5-(4-ethylpiperazin-1-yl)-N-methylbenzamide (7g). The method described for 7a was used to obtain 7g (white solid, yield 57.6%). Rf (MeOH/DCM = 1/5) = 0.66. 1H NMR (600 MHz, DMSO-d6) δ 9.27 (d, J = 4.7 Hz, 1H), 9.09 (s, 1H), 8.63 (s, 1H), 8.17 (s, 1H), 8.04 (d, J = 2.6 Hz, 1H), 7.81 (d, J = 2.8 Hz, 1H), 7.78 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 2.7 Hz, 1H), 7.60 (s, 1H), 7.31 (s, 1H), 6.93 (s, 1H), 6.85 (d, J = 9.1 Hz, 1H), 4.09 (dd, J = 5.8, 3.5 Hz, 2H), 3.86 (d, J = 3.0 Hz, 3H), 3.85–3.81 (m, 2H), 3.69 (qd, J = 4.1, 1.8 Hz, 3H), 3.66 (dd, J = 6.1, 3.7 Hz, 2H), 3.64–3.61 (m, 2H), 3.21–3.12 (m, 3H), 2.83 (d, J = 4.8 Hz, 3H), 2.80 (d, J = 5.2 Hz, 4H), 2.52 (s, 2H), 2.40 (q, J = 7.2 Hz, 2H), 1.04 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, DMSO-D6) δ 166.96, 158.97, 158.11, 157.09, 150.24, 147.11, 137.75, 134.92, 134.11, 130.24, 128.98, 126.75, 125.35, 121.55, 120.39, 119.30, 118.31, 113.29, 84.11, 83.01, 71.10, 70.48, 70.33, 70.30, 70.18, 69.67, 58.07, 55.46, 53.33, 52.96, 52.10, 44.07, 39.10, 39.06, 23.60, 14.02. The ESI-HRMS m/z calculated for C34H44BrFN9O4+ 740.2678 was found to be 740.2682. IR for 7g (KBr, cm−1): 3400 (m), 2920 (s), 2850 (m), 1650 (s), 1610 (m), 1530 (s), 1490 (s), 1410 (s), 1290 (m), 1110 (s), 1050 (w), 942 (m), 838 (m), 813 (m), 773 (m), 691 (m), 666 (m), 625 (m), 555 (m).
- 5-(4-acetylpiperazin-1-yl)-2-((5-bromo-2-((4-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)-3-(1-methyl-1H-pyrazol-4-yl)phenyl)amino)pyrimidin-4-yl)amino)-N-methylbenzamide (5e). The method described for 5a was used to obtain 5e (white solid, 66.8%). 1H NMR (600 MHz, DMSO-d6) δ 9.09 (s, 1H), 9.02 (d, J = 4.7 Hz, 1H), 8.63 (s, 1H), 8.17 (s, 1H), 8.05 (s, 1H), 7.78 (d, J = 2.3 Hz, 2H), 7.71 (d, J = 2.8 Hz, 1H), 7.59 (s, 1H), 7.35–7.18 (m, 1H), 6.90 (s, 1H), 6.85 (d, J = 8.9 Hz, 1H), 4.61 (t, J = 5.5 Hz, 1H), 4.10–4.06 (m, 2H), 3.85 (s, 3H), 3.83–3.80 (m, 2H), 3.67–3.63 (m, 2H), 3.62–3.57 (m, 6H), 3.50–3.47 (m, 2H), 3.45–3.42 (m, 2H), 2.85–2.82 (m, 3H), 2.82–2.77 (m, 2H), 2.75–2.70 (m, 2H), 2.05 (s, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 168.43, 166.61, 158.42, 157.64, 156.56, 149.70, 146.14, 137.24, 134.48, 133.60, 129.77, 128.97, 126.13, 124.78, 121.00, 119.81, 118.72, 118.66, 117.79, 112.79, 72.40, 69.89, 69.13, 67.56, 60.22, 52.74, 52.07, 45.93, 41.08, 38.55, 25.92, 21.26. ESI-MS m/z 752.2333 [M + H]+.
- 2-(2-(2-(4-((4-((4-(4-acetylpiperazin-1-yl)-2-(methylcarbamoyl)phenyl)amino)-5-bromopyrimidin-2-yl)amino)-2-(1-methyl-1H-pyrazol-4-yl)phenoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (6e). The method described for 6a was used to obtain 6e (white solid, 81.2%). 1H NMR (600 MHz, DMSO-d6) δ 9.10 (s, 1H), 9.02 (d, J = 4.9 Hz, 1H), 8.65 (s, 1H), 8.17 (s, 1H), 8.01 (s, 1H), 7.81–7.76 (m, 2H), 7.75–7.73 (m, 2H), 7.71 (d, J = 2.7 Hz, 1H), 7.60–7.54 (m, 1H), 7.44–7.39 (m, 2H), 7.34–7.27 (m, 1H), 6.88 (s, 1H), 6.84 (d, J = 8.9 Hz, 1H), 4.10–4.06 (m, 4H), 3.82 (s, 3H), 3.80–3.77 (m, 2H), 3.61–3.57 (m, 8H), 3.52–3.49 (m, 2H), 2.83 (d, J = 4.7 Hz, 3H), 2.79 (s, 2H), 2.72 (s, 2H), 2.36 (s, 3H), 2.03 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 168.22, 166.43, 158.26, 157.42, 156.39, 149.54, 145.96, 144.69, 137.05, 134.34, 133.46, 132.24, 129.92, 129.54, 128.81, 127.40, 125.92, 124.62, 120.87, 119.64, 118.54, 118.49, 117.64, 112.63, 91.99, 69.73, 69.67, 69.53, 68.97, 67.77, 67.38, 54.74, 52.56, 51.90, 45.77, 40.92, 38.34, 25.72, 25.62, 21.04, 20.86. ESI-MS m/z 906.2981 [M + H]+.
- 5-(4-acetylpiperazin-1-yl)-2-((5-bromo-2-((4-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)-3-(1-methyl-1H-pyrazol-4-yl)phenyl)amino)pyrimidin-4-yl)amino)-N-methylbenzamide (7e). Rf (MeOH/DCM = 1/10) = 0.51. The method described for 7a was used to obtain 7e (white solid, 58.1%). 1H NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.98 (q, J = 4.8 Hz, 1H), 8.60 (s, 1H), 8.13 (s, 1H), 8.00 (s, 1H), 7.81–7.71 (m, 2H), 7.67 (d, J = 2.7 Hz, 1H), 7.54 (s, 1H), 7.34–7.21 (m, 1H), 6.89–6.83 (m, 1H), 6.81 (d, J = 8.9 Hz, 1H), 4.57–4.48 (m, 1H), 4.43–4.35 (m, 1H), 4.12–4.00 (m, 2H), 3.89–3.74 (m, 5H), 3.68–3.64 (m, 1H), 3.64–3.60 (m, 2H), 3.60–3.52 (m, 7H), 2.80 (d, J = 4.7 Hz, 3H), 2.76 (t, J = 5.0 Hz, 2H), 2.69 (t, J = 5.1 Hz, 2H), 2.01 (s, 3H). 13C NMR (100 MHz, DMSO-d6, δ ppm): 168.39, 116.59, 158.30, 157.38, 156.56, 149.74, 146.13, 137.18, 134.43, 133.50, 129.73, 128.96, 126.10, 124.77, 121.01, 119.80, 118.74, 117.76, 112.78, 92.12, 83.84, 82.19, 69.93, 69.79, 69.59, 69.12, 67.55, 52.72, 52.03, 45.91, 41.06, 38.50, 25.88, 21.20. The ESI-HRMS m/z calculated for C34H42BrFN9O5+ 754.2476 was found to be 754.2477. IR for 7e (KBr, cm−1): 3310 (w), 2870 (w), 1630 (m), 1560 (m), 1520 (s), 1410 (s), 1220 (m), 1130 (m), 1070 (m), 984 (m), 939 (w), 891 (w), 852 (w), 773 (w), 732 (w), 672 (w), 611 (w), 590 (w), 552 (w), 520 (w).
4.2. Radiolabeling
4.2.1. Drying Produce for [18F]-Fluoride
4.2.2. Synthesis of [18F]7a and [18F]7c
4.3. FAK Inhibitory Assay and Partition Coefficient Determination
4.4. Molecular Docking
4.5. In Vitro Stability
4.6. Biodistribution Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Zheng, R.; Zhang, S.; Zeng, H.; Wang, S.; Sun, K.; Chen, R.; Li, L.; Wei, W.; He, J. Cancer incidence and mortality in China, 2016. J. Natl. Cancer Cent. 2022, 2, 1–9. [Google Scholar] [CrossRef]
- Qi, J.; Li, M.; Wang, L.; Hu, Y.; Liu, W.; Long, Z.; Zhou, Z.; Yin, P.; Zhou, M. National and subnational trends in cancer burden in China, 2005–2020: An analysis of national mortality surveillance data. Lancet Public Health 2023, 8, e943–e955. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Yoon, H.; Dehart, J.P.; Murphy, J.M.; Lim, S.S. Understanding the Roles of FAK in Cancer. J. Histochem. Cytochem. 2015, 63, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250–260. [Google Scholar] [CrossRef]
- Pomella, S.; Cassandri, M.; Braghini, M.R.; Marampon, F.; Alisi, A.; Rota, R. New Insights on the Nuclear Functions and Targeting of FAK in Cancer. Int. J. Mol. Sci. 2022, 23, 1998. [Google Scholar] [CrossRef] [PubMed]
- Spallarossa, A.; Tasso, B.; Russo, E.; Villa, C.; Brullo, C. The Development of FAK Inhibitors: A Five-Year Update. Int. J. Mol. Sci. 2022, 23, 6381. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qi, Y.; Fang, Y.; Gao, H.; Zhang, H. Design, Synthesis, and Biological Evaluation of 4-Arylamino Pyrimidine Derivatives as FAK Inhibitors and Tumor Radiotracers. Mol. Pharm. 2022, 19, 2471–2482. [Google Scholar] [CrossRef]
- Hsieh, C.; Giannakoulias, S.; Petersson, E.J.; Mach, R.H. Computational chemistry for the identification of lead compounds for radiotracer development. Pharmaceuticals 2023, 36, 317. [Google Scholar] [CrossRef]
- Salahinejad, M.; Winkler, D.A.; Shiri, F. Discovery and design of radiopharmaceuticals by in silico methods. Curr. Radiopharm. 2022, 15, 271–319. [Google Scholar]
- Lu, Y.; Sun, H. Progress in the development of small molecular inhibitors of Focal Adhesion Kinase (FAK). J. Med. Chem. 2020, 63, 14382–14403. [Google Scholar] [CrossRef]
- Li, M.; Wang, D.; Zhi, Y.; Liu, B.; Yao, Q. Advancing strategies for Proteolysis-Targeting Chimera design. J. Med. Chem. 2023, 66, 2308–2329. [Google Scholar] [CrossRef]
- O’Brien, M.; Konings, L.; Martin, M.; Heap, J. Harnessingopen-sourcetechnologyforlow-costautomationin synthesis:Flow chemical deprotection of silyl ethers using a Homem adeauto sampling system. Tetrahedron Lett. 2017, 58, 2409–2413. [Google Scholar] [CrossRef]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wei, W.; Ehlerding, E.B.; Lan, X.; Luo, Q.; Cai, W. PET and SPECT imaging of melanoma: The state of the art. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 132–150. [Google Scholar] [CrossRef] [PubMed]
- Altine, B.; Gai, Y.; Han, N.; Jiang, Y.; Ji, H.; Fang, H.; Niyonkuru, A.; Bakari, K.H.; Rajab, A.; Maher, M.; et al. Preclinical Evaluation of a Fluorine-18 (18F)-Labeled Phosphatidylinositol 3-Kinase Inhibitor for Breast Cancer Imaging. Mol. Pharm. 2019, 16, 4563–4571. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Rousseau, E.; Kwon, D.; Lin, K.; Bénard, F.; Chen, X. Insight into the Development of PET Radiopharmaceuticals for Oncology. Cancers 2020, 12, 1312. [Google Scholar] [CrossRef] [PubMed]
- Narayanam, M.K.; Tsang, J.E.; Xu, S.; Nathanson, D.A.; Murphy, J.M. 18F-Labeled brain-penetrant EGFR tyrosine kinase inhibitors for PET imaging of glioblastoma. Chem. Sci. 2023, 14, 13825–13831. [Google Scholar] [CrossRef] [PubMed]
- Schibilla, F.; Stegemann, L.; Strassert, C.A.; Rizzo, F.; Ravoo, B.J. Fluorescence quenching in β-cyclodextrin vesicles: Membrane confinement and host-guest interactions. Photochem. Photobiol. Sci. 2016, 15, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Shoda, T.; Kato, M.; Harada, R.; Fujisato, T.; Okuhira, K.; Demizu, Y.; Inoue, H.; Naito, M.; Kurihara, M. Synthesis and evaluation of tamoxifen derivatives with a long alkyl side chain as selective estrogen receptor down-regulators. Bioorg. Med. Chem. 2015, 23, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Kusakabe, K.; Ide, N.; Daigo, Y.; Tachibana, Y.; Itoh, T.; Yamamoto, T.; Hashizume, H.; Hato, Y.; Higashino, K.; Okano, Y.; et al. Indazole-based potent and cell-active mps1 kinase inhibitors: Rational design from pan-kinase inhibitor anthrapyrazolone (SP600125). J. Med. Chem. 2013, 56, 4343–4356. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
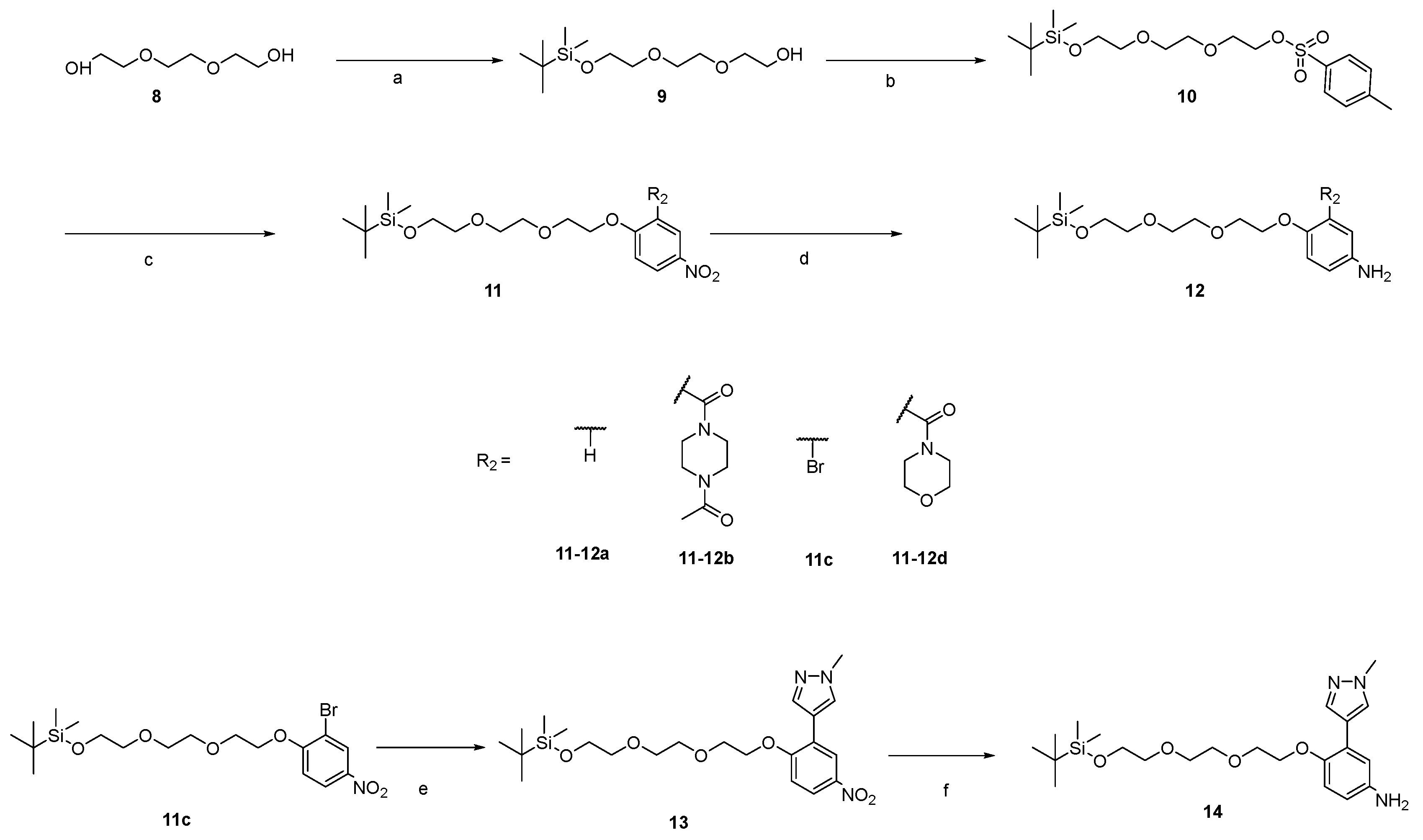{kind=link}
{kind=link}
| Compound ID | Structure | IC50 (nmol) |
|---|---|---|
| 7a |  | 5.59 |
| 7d |  | 1968 |
| 7f |  | 10.02 |
| 7b |  | 24.6 |
| 7c |  | 1.27 |
| 7g |  | 1238 |
| 7e |  | 643 |
| VS-6063 |  | 1.26 |
| Tissue | [18F]7a | [18F]7c | ||||
|---|---|---|---|---|---|---|
| 30 min | 60 min | 120 min | 30 min | 60 min | 120 min | |
| Blood | 0.98 ± 0.26 | 0.96 ± 0.10 | 0.92 ± 0.15 | 2.74 ± 0.11 | 3.43 ± 0.04 | 3.79 ± 0.46 |
| Brine | 0.57 ± 0.08 | 1.08 ± 0.13 | 0.16 ± 0.01 | 2.81 ± 0.06 | 3.12 ± 0.66 | 2.01 ± 0.18 |
| Heart | 3.05 ± 0.29 | 1.12 ± 0.03 | 0.85 ± 0.09 | 3.46 ± 0.65 | 2.53 ± 0.56 | 3.12 ± 0.25 |
| Liver | 23.49 ± 4.29 | 23.16 ± 2.65 | 8.86 ± 3.14 | 17.64 ± 4.45 | 19.21 ± 3.81 | 28.36 ± 5.74 |
| Lung | 33.55 ± 5.59 | 28.77 ± 5.72 | 11.63 ± 1.25 | 120.47 ± 15.08 | 128.13 ± 6.74 | 125.29 ± 16.25 |
| Kidney | 3.02 ± 1.70 | 1.47 ± 0.18 | 1.16 ± 0.06 | 4.00 ± 0.25 | 3.82 ± 0.44 | 3.50 ± 0.37 |
| Spleen | 5.43 ± 1.12 | 2.53 ± 0.21 | 5.58 ± 1.06 | 16.32 ± 1.26 | 8.48 ± 0.10 | 24.70 ± 4.31 |
| Stomach | 0.92 ± 0.28 | 0.80 ± 0.21 | 1.16 ± 0.40 | 5.81 ± 1.02 | 1.14 ± 0.16 | 3.32 ± 0.31 |
| Bone | 2.32 ± 0.45 | 2.96 ± 0.43 | 1.27 ± 0.42 | 4.12 ± 0.39 | 4.86 ± 0.30 | 10.70 ± 1.01 |
| Muscle | 1.63 ± 0.34 | 2.50 ± 0.59 | 0.20 ± 0.03 | 3.50 ± 0.18 | 2.80 ± 0.49 | 2.04 ± 0.12 |
| Small intestine | 2.95 ± 0.17 | 1.58 ± 0.35 | 0.96 ± 0.29 | 13.02 ± 1.93 | 5.73 ± 0.82 | 7.49 ± 1.46 |
| Large intestine | 1.46 ± 0.13 | 1.36 ± 0.36 | 4.5 ± 1.91 | 6.18 ± 0.60 | 4.47 ± 0.44 | 48.07 ± 9.85 |
| Tumor | 1.31 ± 0.07 | 1.39 ± 0.30 | 0.86 ± 0.34 | 6.58 ± 0.46 | 5.85 ± 0.28 | 5.18 ± 0.68 |
| Tumor/blood | 1.34 | 1.45 | 0.89 | 2.40 | 1.71 | 1.37 |
| Tumor/muscle | 0.80 | 0.56 | 4.30 | 1.88 | 2.09 | 2.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Li, Y.; Liang, H.; Cui, C.; Gan, L.; Zhang, H. Design, Synthesis, Biological Evaluation and Molecular Docking of Novel F-18-Labeled Focal Adhesion Kinase Inhibitors as Potential Tumor Radiotracers. Molecules 2024, 29, 1224. https://doi.org/10.3390/molecules29061224
Yang H, Li Y, Liang H, Cui C, Gan L, Zhang H. Design, Synthesis, Biological Evaluation and Molecular Docking of Novel F-18-Labeled Focal Adhesion Kinase Inhibitors as Potential Tumor Radiotracers. Molecules. 2024; 29(6):1224. https://doi.org/10.3390/molecules29061224
Chicago/Turabian StyleYang, Hailong, Ye Li, Huaju Liang, Chun Cui, Lu Gan, and Huabei Zhang. 2024. "Design, Synthesis, Biological Evaluation and Molecular Docking of Novel F-18-Labeled Focal Adhesion Kinase Inhibitors as Potential Tumor Radiotracers" Molecules 29, no. 6: 1224. https://doi.org/10.3390/molecules29061224
APA StyleYang, H., Li, Y., Liang, H., Cui, C., Gan, L., & Zhang, H. (2024). Design, Synthesis, Biological Evaluation and Molecular Docking of Novel F-18-Labeled Focal Adhesion Kinase Inhibitors as Potential Tumor Radiotracers. Molecules, 29(6), 1224. https://doi.org/10.3390/molecules29061224