

DFT Studies of Dimethylaminophenyl-Substituted Phthalocyanine and Its Silver Complexes †


Abstract
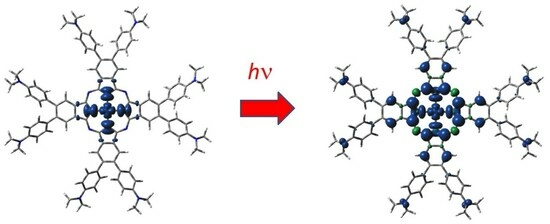
:
1. Introduction
2. Results
2.1. Gibbs Energies
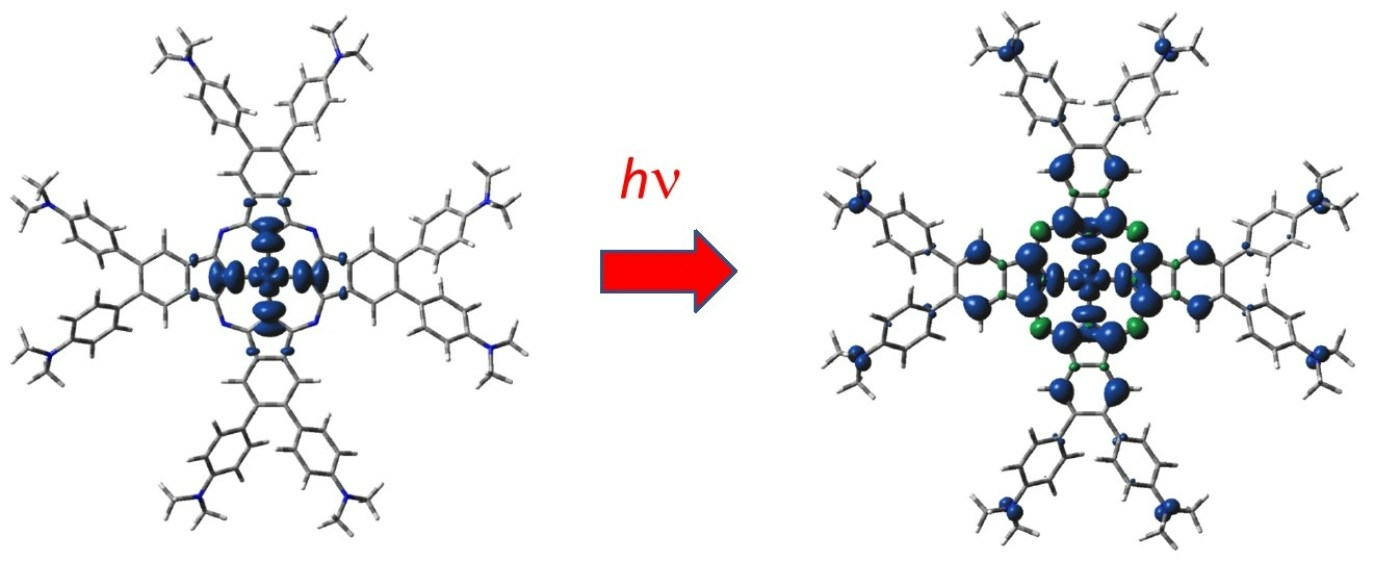
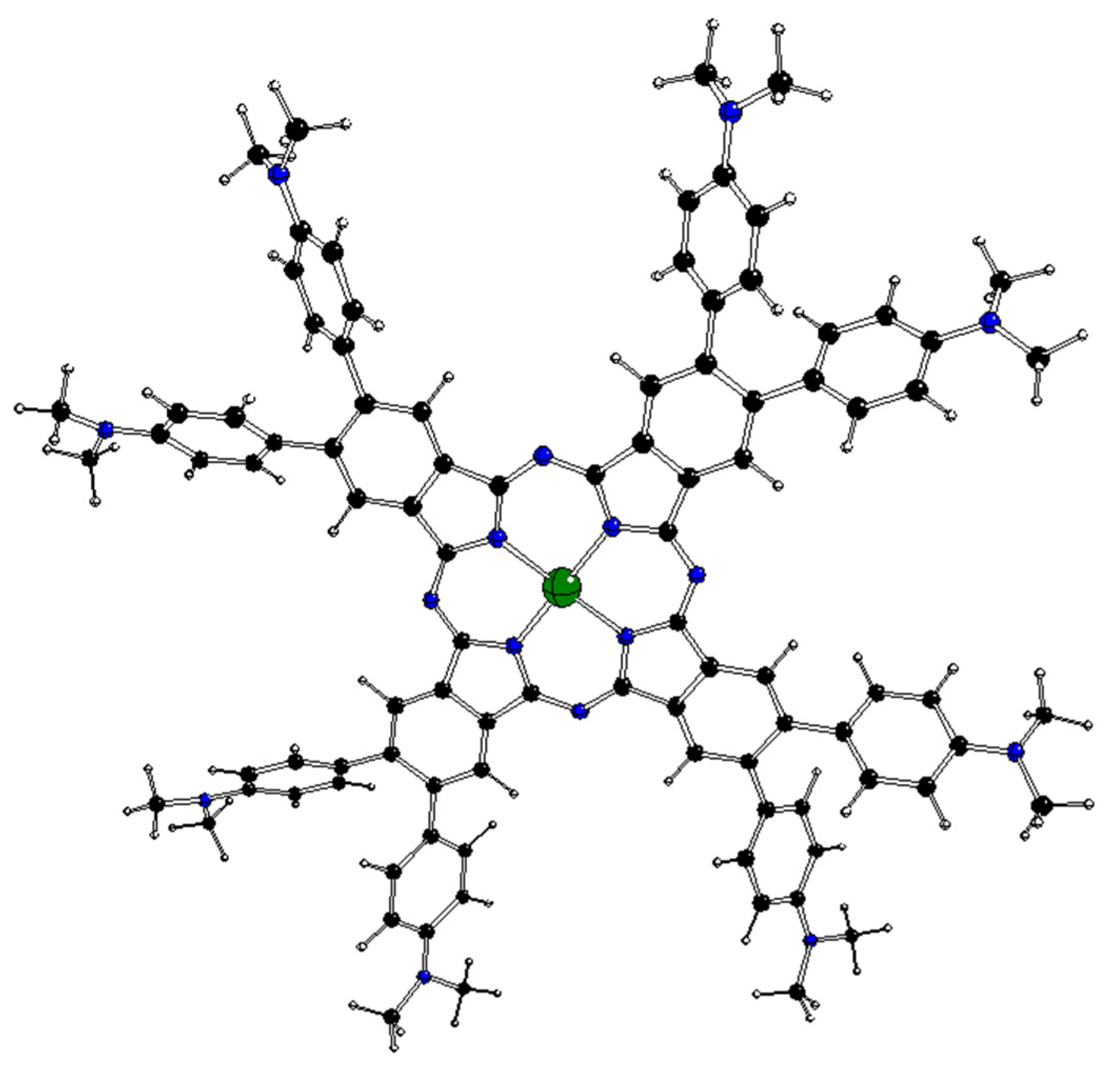
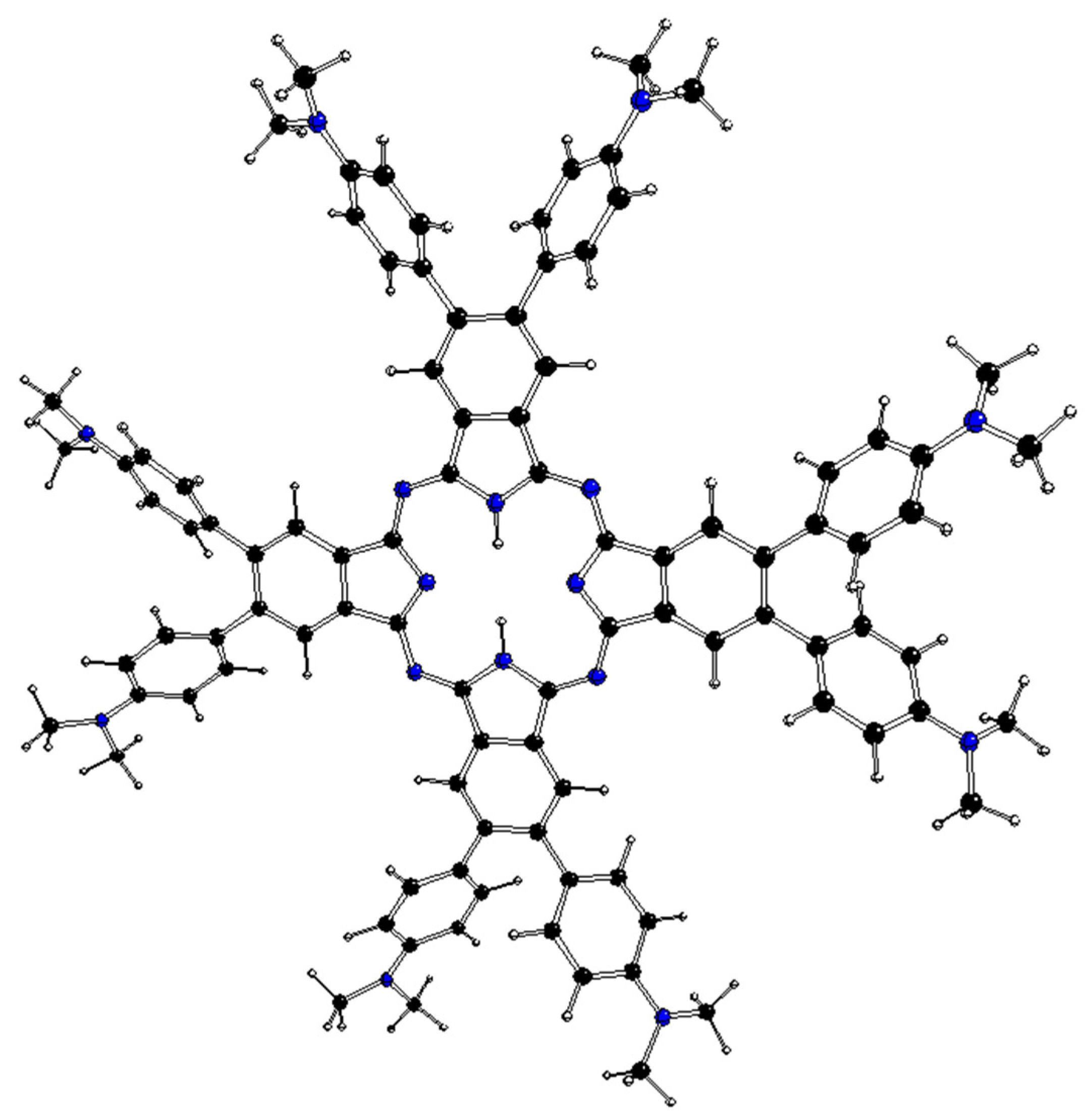
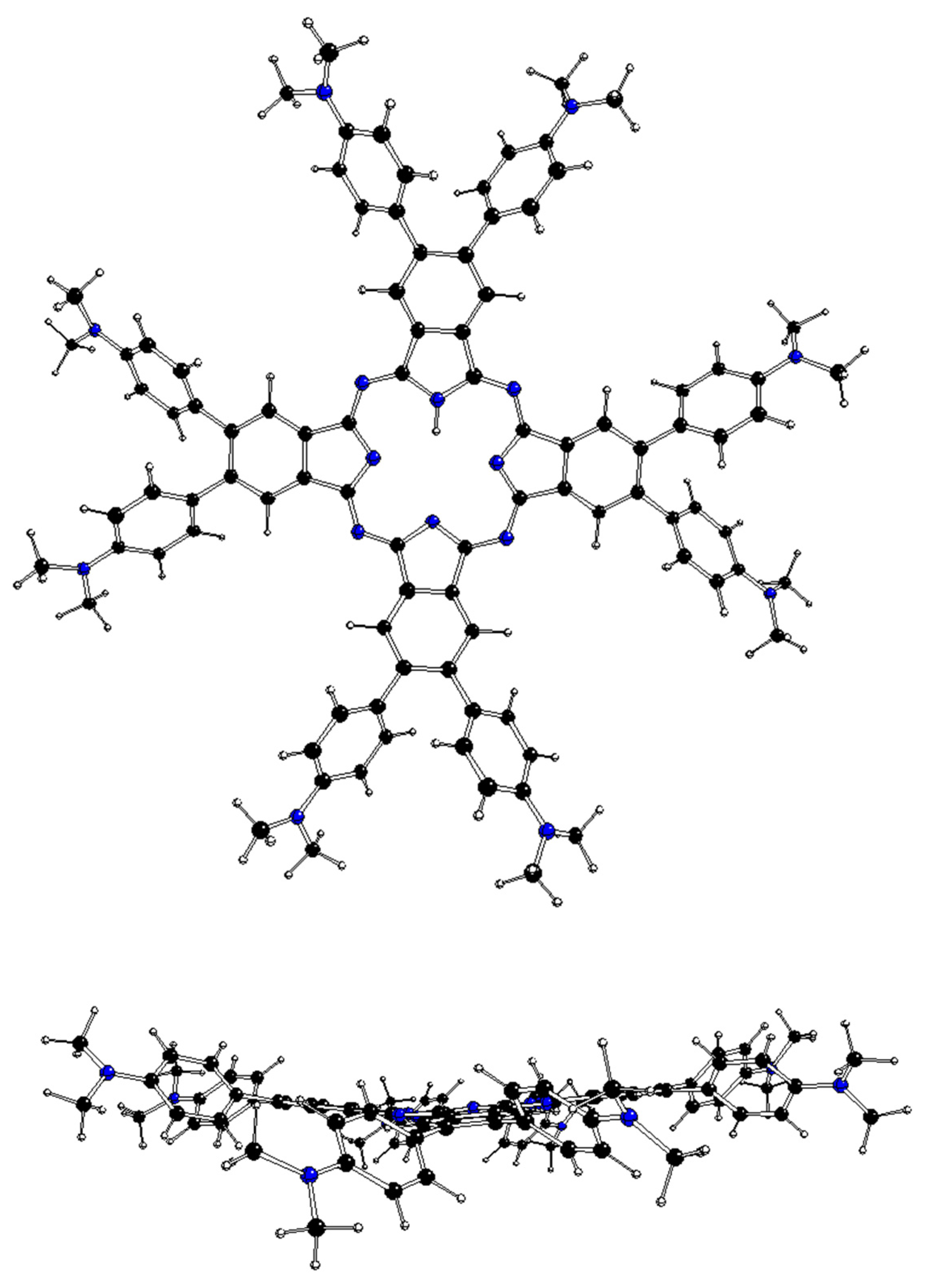
2.2. DFT-Optimized Structures
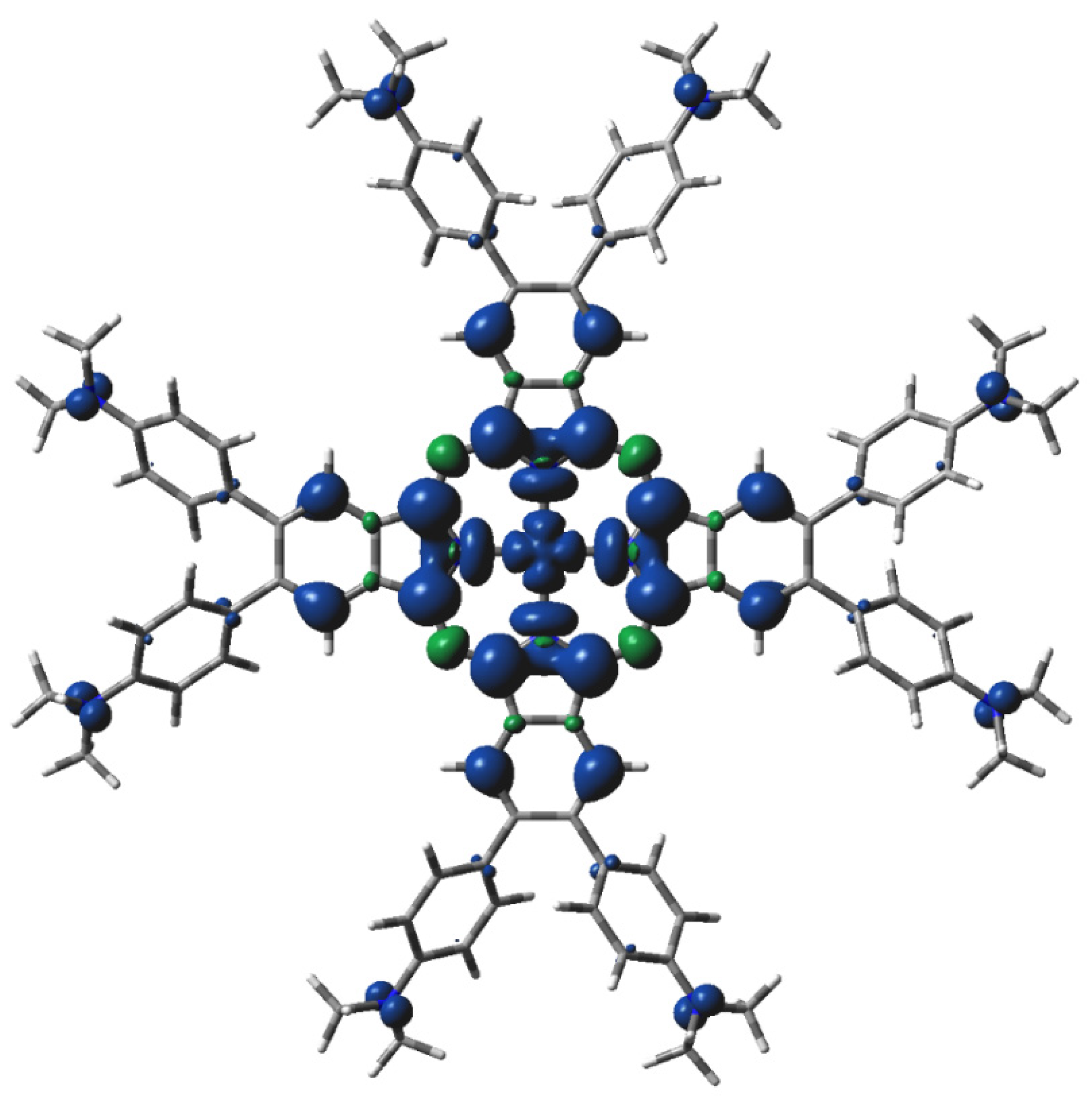
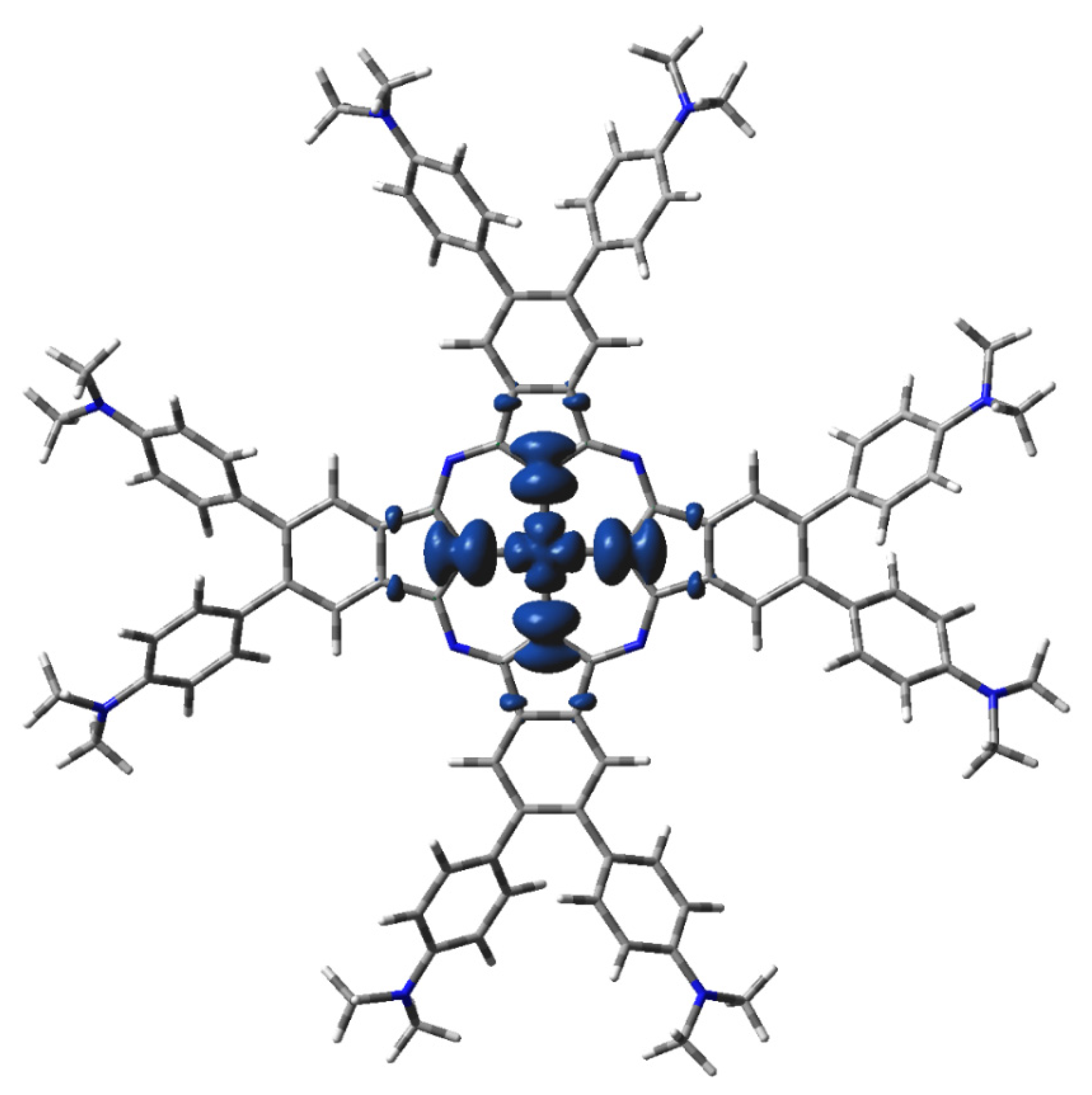
2.3. Electronic Structure Characteristics
2.4. TD-DFT Calculated Electron Transitions
3. Discussion
4. Methods
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Urbani, M.; Ragoussi, M.-E.; Nazeeruddin, M.K.; Torres, T. Phthalocyanines for dye-sensitized solar cells. Coord. Chem. Rev. 2019, 381, 1–64. [Google Scholar] [CrossRef]
- Kaliya, O.L.; Lukyanets, E.A.; Vorozhtsov, G.N. Catalysis and Photocatalysis by Phthalocyanines for Technology, Ecology, and Medicine. J. Porphyr. Phthalocyanines 1999, 3, 592–610. [Google Scholar] [CrossRef]
- Sorokin, A.B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. [Google Scholar] [CrossRef] [PubMed]
- Juzenas, P. Lasers in Therapy of Human Diseases. Trends Cancer Res. 2005, 1, 93–110. [Google Scholar]
- O’Riordan, K.; Akilov, O.E.; Hasan, T. The Potential for Photodynamic Therapy in the Treatment of Localized Infections. Photodiagn. Photodyn. Ther. 2005, 2, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Sekkat, N.; van den Bergh, H.; Nyokong, T.; Lange, N. Like a Bolt from the Blue: Phthalocyanines in Biomedical Optics. Molecules 2012, 17, 98–144. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.M.; Sharman, W.M.; Van Lier, J.E. Current Status of Phthalocyanines in the Photodynamic Therapy of Cancer. J. Porphyr. Phthalocyanines 2001, 5, 161–169. [Google Scholar] [CrossRef]
- Lukyanets, E.A. Phthalocyanines as Photosensitizers in the Photodynamic Therapy of Cancer. J. Porphyr. Phthalocyanines 1999, 3, 424–432. [Google Scholar] [CrossRef]
- da Silva, R.N.; Cunha, A.; Tomé, A.C. Phthalocyaninesulfonamide Conjugates: Synthesis and Photodynamic Inactivation of Gram-negative and Gram-positive Bacteria. Eur. J. Med. Chem. 2018, 154, 60–67. [Google Scholar] [CrossRef]
- Elemans, J.A.A.W.; van Hameren, R.; Nolte, R.J.M.; Rowan, A.E. Molecular Materials by Self-assembly of Porphyrins, Phthalocyanines, and Perylenes. Adv. Mater. 2006, 18, 1251–1266. [Google Scholar] [CrossRef]
- Gsaenger, M.; Bialas, D.; Huang, L.; Stolte, M.; Wuerthner, F. Organic Semiconductors Based on Dyes and Color Pigments. Adv. Mater. 2016, 28, 3615–3645. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, A.L.; Delile, S.; Brunet, J.; Varenne, C.; Pauly, A. Electrochemical Sensors Based on Screen-printed Electrodes: The Use of Phthalocyanine Derivatives for Application in VFA Detection. Biosensors 2016, 6, 0046. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lv, W.; Luo, X.; He, L.; Xu, K.; Zhao, F.; Huang, F.; Lu, F.; Peng, Y. A Comprehensive Investigation of Organic Active Layer Structures toward High Performance Near-infrared Phototransistors. Synth. Met. 2018, 240, 44–51. [Google Scholar] [CrossRef]
- Woehrle, D.; Schnurpfeil, G.; Makarov, S.G.; Kazarin, A.; Suvorova, O.N. Practical Applications of Phthalocyanines—From Dyes and Pigments to Materials for Optical, Electronic and Photoelectronic Devices. Makrogeterotsikly 2012, 5, 191–202. [Google Scholar]
- de la Torre, G.; Vazquez, P.; Agullo-Lopez, F.; Torres, T. Phthalocyanines and Related Compounds: Organic Targets for Nonlinear Optical Applications. J. Mater. Chem. 1998, 8, 1671–1683. [Google Scholar] [CrossRef]
- Chen, Y.; Hanack, M.; Blau, W.J.; Dini, D.; Liu, Y.; Lin, Y.; Bai, J. Soluble Axially Substituted Phthalocyanines: Synthesis and Nonlinear Optical Response. J. Mater. Sci. 2006, 41, 2169–2185. [Google Scholar] [CrossRef]
- Wrobel, D.; Dudkowiak, A. Porphyrins and Phthalocyanines—Functional Molecular Materials for Optoelectronics and Medicine. Mol. Cryst. Liq. Cryst. 2006, 448, 15–38. [Google Scholar] [CrossRef]
- Wu, Z.; Jung, K.; Boyer, C. Effective Utilization of NIR Wavelengths for Photo-Controlled Polymerization: Penetration through Thick Barriers and Parallel Solar Syntheses. Angew. Chem. 2020, 59, 2013–2017. [Google Scholar] [CrossRef]
- Corrigan, N.; Xu, J.; Boyer, C. A Photoinitiation System for Conventional and Controlled Radical Polymerization at Visible and NIR Wavelengths. Macromolecules 2016, 49, 3274–3285. [Google Scholar] [CrossRef]
- Korkut, S.E.; Temel, G.; Balta, D.K.; Arsu, N.; Şener, M.K. Type II photoinitiator substituted zinc phthalocyanine: Synthesis, photophysical and photopolymerization studies. J. Lumin. 2013, 136, 389–394. [Google Scholar] [CrossRef]
- Breloy, L.; Brezová, V.; Blacha-Grzechnik, A.; Presset, M.; Yildirim, M.S.; Yilmaz, I.; Yagci, Y.; Versace, D.-L. Visible Light Anthraquinone Functional Phthalocyanine Photoinitiator for Free-Radical and Cationic Polymerizations. Macromolecules 2020, 53, 112–124. [Google Scholar] [CrossRef]
- Wang, Y.; Han, B.; Shi, R.; Pan, L.; Zhang, H.; Shen, Y.; Li, C.; Huang, F.; Xie, A. Preparation and characterization of a novel hybrid hydrogel shell for localized photodynamic therapy. J. Mater. Chem. B 2013, 1, 6411–6417. [Google Scholar] [CrossRef]
- Breloy, L.; Alcay, Y.; Yilmaz, I.; Breza, M.; Bourgon, J.; Brezová, V.; Yagci, Y.; Versace, D.-L. Dimethyl amino phenyl substituted silver phthalocyanine as a UV- and visible-light absorbing photoinitiator: In situ preparation of silver/polymer nanocomposites. Polym. Chem. 2021, 12, 1273–1285. [Google Scholar] [CrossRef]
- Breza, M. On the Jahn–Teller Effect in Silver Complexes of Dimethyl Amino Phenyl Substituted Phthalocyanine. Molecules 2023, 28, 7019. [Google Scholar] [CrossRef] [PubMed]
- Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimer. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural Hybrid Orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | q | m | G298 (Hartree) | ΔG298 (kJ/mol) |
|---|---|---|---|---|
| 1[dmaphPcAg]+ | +1 | 1 | −4733.24222 | 453.4 |
| 3[dmaphPcAg]+ | +1 | 3 | −4733.25119 | 429.9 |
| 2[dmaphPcAg]0 | 0 | 2 | −4733.41492 | 0.0 |
| 4[dmaphPcAg]0 | 0 | 4 | −4733.38011 | 91.4 |
| 1[dmaphPcAg]− | −1 | 1 | −4733.51744 | −269.2 |
| 3[dmaphPcAg]− | −1 | 3 | −4733.51527 | −263.5 |
| 2[dmaphPcAg]2− | −2 | 2 | −4733.59008 | −459.9 |
| 4[dmaphPcAg]2− | −2 | 4 | −4733.57970 | −432.6 |
| 1[dmaphPcH2]0 | 0 | 1 | −4587.58495 | 0.0 |
| 1[dmaphPcH]− | −1 | 1 | −4587.08582 | 1310.5 |
| 2[dmaphPcH]0 | 0 | 2 | −4586.94941 | 1668.6 |
| 1[dmaphPc]2− | −2 | 1 | −4586.54403 | 2732.9 |
| 2[dmaphPc]− | −1 | 2 | −4586.43712 | 3013.6 |
| q | +1 | +1 | 0 | 0 | −1 | −1 | −2 | −2 |
| m | 1 | 3 | 2 | 4 | 1 | 3 | 2 | 4 |
| Bond length (Å) | ||||||||
| Ag-Npy | 2.000(4×) | 2.056(4×) | 2.059(4×) | 2.058(2×) 2.062(2×) | 2.067(2×) 2.133(2×) | 2.063(2×) 2.067(2×) | 2.072(4×) | 2.071(4×) |
| Ag—plane distance (Å) | ||||||||
| Npy plane | 0.001 | 0.019 | 0.007 | 0.000 | 0.005 | 0.000 | 0.000 | 0.000 |
| Bond angle (deg) | ||||||||
| (Npy-Ag-Npy)adj | 90.0(4×) | 90.0(4×) | 90.0(4×) | 90.0(4×) | 89.9(2×) 90.1(2×) | 90.0(4×) | 90.0(4×) | 90.0(4×) |
| (Npy-Ag-Npy)op | 179.9(2×) | 178.9(2×) | 179.7(2×) | 180.0(2×) | 179.2 179.5 | 180.0(2×) | 180.0(2×) | 180.0(2×) |
| Bond order | ||||||||
| Ag-Npy | 0.448(4×) | 0.377(4×) | 0.379(4×) | 0.379(2×) 0.377(2×) | 0.363(2×) 0.330(2×) | 0.382(2×) 0.381(2×) | 0.383(4×) | 0.384(4×) |
| Charge | ||||||||
| Ag | 1.023 | 0.857 | 0.843 | 0.833 | 0.713 | 0.814 | 0.787 | 0.785 |
| Npy | −0.574(4×) | −0.623(4×) | −0.614(4×) | −0.613(2×) −0.662(2×) | −0.637(2×) −0.614(2×) | −0.603(2×) −0.647(2×) | −0.633(2×) −0.637(2×) | −0.634(4×) |
| Nbr | −0.533(4×) | −0.553(4×) | −0.550(4×) | −0.587(4×) | −0.566(4×) | −0.579(4×) | −0.617(4×) | −0.610(4×) |
| Cα | 0.497(8×) | 0.528(8×) | 0.485(8×) | 0.484(4×) 0.532(4×) | 0.464(4×) 0.435(4×) | 0.422(4×) 0.476(4×) | 0.422(8×) | 0.418(8×) |
| Cβ | −0.083(8×) | −0.092(8×) | −0.085(8×) | −0.101(4×) −0.086(4×) | −0.083(4×) −0.091(4×) | −0.100(4×) −0.081(4×) | −0.101(8×) | −0.101(8×) |
| Namin | −0.482(8×) | −0.475(8×) | −0.492(8×) | −0.489(8×) | −0.497(8×) | −0.497(8×) | −0.502(8×) | −0.502(8×) |
| Cmet | −0.418(16×) | −0.418(16×) | −0.418(16×) | −0.418(16×) | −0.418(16×) | −0.418(16×) | −0.418(16×) | −0.418(16×) |
| d electron population | ||||||||
| Ag | 9.23 | 9.43 | 9.43 | 9.44 | 9.58 | 9.44 | 9.45 | 9.45 |
| Spin population | ||||||||
| Ag | - | 0.426 | 0.422 | 0.426 | - | 0.420 | 0.412 | 0.415 |
| Npy | - | 0.119(4×) | 0.148(4×) | 0.080(2×) 0.206(2×) | - | 0.120(2×) 0.234(2×) | 0.259(2×) 0.039(2×) | 0.208(4×) |
| Nbr | - | −0.037(4×) | 0.002(4×) | 0.009(4×) | - | 0.054(4×) | −0.001(4×) | 0.115(4×) |
| Cα | - | 0.086(8×) | −0.007(8×) | 0.237(4×) 0.100(4×) | - | 0.104(4×) −0.016(4×) | −0.106(4×) 0.096(4×) | 0.079(8×) |
| Cβ | - | −0.007(8×) | 0.006(8×) | 0.016(4×) −0.002(4×) | - | 0.037(4×) 0.004(4×) | −0.043(4×) 0.054(4×) | 0.047(8×) |
| Namin | - | 0.016(8×) | 0.000(8×) | 0.005(8×) | - | 0.000(8×) | 0.000(8×) | 0.001(4×) |
| Cmet | - | −0.001(16×) | 0.000(16×) | −0.000(16×) | - | 0.000(16×) | 0.000(16×) | 0.000(16×) |
| Compound | 1[dmaphPcH2]0 | 1[dmaphPcH]− | 2[dmaphPcH]0 | 1[dmaphPc]2− | 2[dmaphPc]− |
|---|---|---|---|---|---|
| q | 0 | −1 | 0 | −2 | −1 |
| m | 1 | 1 | 2 | 1 | 2 |
| Bond length (Å) | |||||
| H-Npy | 1.017(2×) | 1.028 | 1.029 | - | - |
| Bond order | |||||
| H-Npy | 0.659(2×) | 0.648 | 0.645 | - | - |
| Charge | |||||
| Npy | −0.607(2×) −0.656(2×) | −0.577 −0.613 −0.615 −0.611 | −0.579 −0.617(2×) −0.628 | −0.560(4×) | −0.566(4×) |
| Nbr | −0.550(4×) | −0.571(2×) −0.565(2×) | −0.566(2×) −0.571(2×) | −0.588(4×) | −0.592(4×) |
| Cα | 0.488(4×) 0.475(4×) | 0.461(2×) 0.462(4×) 0.450(2×) | 0.513(2×) 0.519(2×) 0.510(2×) 0.529(2×) | 0.434(8×) | 0.500(8×) |
| Cβ | −0.084(4×) −0.090(4×) | −0.087(2×) −0.085(4×) −0.092(2×) | −0.094(2×) −0.089(2×) −0.097(2×) −0.086(2×) | −0.087(8×) | −0.089(8×) |
| Namin | −0.490(4×) −0.492(4×) | −0.497(6×) −0.495(2×) | −0.490(2×) −0.488(4×) −0.486(2×) | −0.503(8×) | −0.496(8×) |
| Cmet | −0.418(16×) | −0.418(16×) | −0.418(16×) | −0.419(16×) | −0.418(16×) |
| Spin | |||||
| Npy | - | - | −0.050 −0.042(2×) −0.023 | - | −0.049(4×) |
| Nbr | - | - | −0.050(2×) −0.040(2×) | - | −0.054(4×) |
| Cα | 0.125(8×) | 0.140(8×) | |||
| Cβ | −0.007(8×) | −0.002(8×) | |||
| Namin | - | - | 0.004(4×) 0.003(4×) | - | 0.002(8×) |
| Cmet | - | - | 0.000(16×) | - | 0.000(16×) |
| System | λexp (nm) | λcalc (nm) | Compound |
|---|---|---|---|
| 2[dmaphPcAg]0 | 300 m | 306(0.3) | 4[dmaphPcAg]0 |
| 305(0.4) | 1[dmaphPcAg]− | ||
| 350 m | 358(0.7), 359(0.8), 346(0.4), 345(0.5) | 2[dmaphPcAg]0 | |
| 355(0.7), 350(0.8) | 4[dmaphPcAg]0 | ||
| 357(0.3), 355(0.5), 353(0.4), 349(0.5), 342(0.4) | 1[dmaphPcAg]− | ||
| 343(0.8) | 3[dmaphPcAg]− | ||
| 364(1.4), 358(1.0), 349(0.4), 344(0.3), 341(0.5), 333(0.6) | 1[dmaphPcH2]0 | ||
| 325(0.9), 327(0.3), 330(0.5) | 1[dmaphPcH]− | ||
| 334(0.3) | 2[dmaphPc]− | ||
| 660 sh | 668(1.0) | 1[dmaphPcAg]− | |
| 676(1.2), 672(1.2) | 1[dmaphPcH]− | ||
| 660(1.2), 659(1.2) | 1[dmaphPc]2− | ||
| 700 m | 701(0.4) | 4[dmaphPcAg]0 | |
| 687(0.7) | 1[dmaphPcAg]− | ||
| 730 sh | 724(0.8), 723(0.9) | 2[dmaphPcAg]0 | |
| 724(1.3) | 3[dmaphPcAg]− | ||
| 715(0.6) | 4[dmaphPcAg]2− | ||
| 749(1.0), 736(0.8) | 1[dmaphPcH2]0 | ||
| 722(0.5), 721(0.5) | 2[dmaphPc]− | ||
| 1[dmaphPcH2]0 | 340 s | 364(1.4), 358(1.0), 349(0.4), 344(0.3), 341(0.5), 333(0.6) | 1[dmaphPcH2]0 |
| 325(0.9), 327(0.3), 330(0.5) | 1[dmaphPcH]− | ||
| 334(0.3) | 2[dmaphPc]− | ||
| 680 sh | 676(1.2), 672(1.2) | 1[dmaphPcH]− | |
| 660(1.2), 659(1.2) | 1[dmaphPc]2− | ||
| 740 s | 749(1.0), 736(0.8) | 1[dmaphPcH2]0 | |
| 722(0.5), 721(0.5) | 2[dmaphPc]− | ||
| Irradiated reaction system | 300 s | 306(0.3) | 4[dmaphPcAg]0 |
| 305(0.4) | 1[dmaphPcAg]− | ||
| 350 s | 358(0.7), 359(0.8), 346(0.4), 345(0.5) | 2[dmaphPcAg]0 | |
| 355(0.7), 350(0.8) | 4[dmaphPcAg]0 | ||
| 357(0.3), 355(0.5), 353(0.4), 349(0.5), 342(0.4) | 1[dmaphPcAg]− | ||
| 343(0.8) | 3[dmaphPcAg]− | ||
| 364(1.4), 358(1.0), 349(0.4), 344(0.3), 341(0.5), 333(0.6) | 1[dmaphPcH2]0 | ||
| 334(0.3) | 2[dmaphPc]− | ||
| 450 sh | - | - | |
| 660 sh | 668(1.0) | 1[dmaphPcAg]− | |
| 676(1.2), 672(1.2) | 1[dmaphPcH]− | ||
| 660(1.2), 659(1.2) | 1[dmaphPc]2− | ||
| 700 s | 701(0.4) | 4[dmaphPcAg]0 | |
| 687(0.7) | 1[dmaphPcAg]− | ||
| 720 sh | 724(0.8), 723(0.9) | 2[dmaphPcAg]0 | |
| 724(1.3) | 3[dmaphPcAg]− | ||
| 715(0.6) | 4[dmaphPcAg]2− | ||
| 722(0.5), 721(0.5) | 2[dmaphPc]− |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breza, M. DFT Studies of Dimethylaminophenyl-Substituted Phthalocyanine and Its Silver Complexes. Molecules 2024, 29, 1344. https://doi.org/10.3390/molecules29061344
Breza M. DFT Studies of Dimethylaminophenyl-Substituted Phthalocyanine and Its Silver Complexes. Molecules. 2024; 29(6):1344. https://doi.org/10.3390/molecules29061344
Chicago/Turabian StyleBreza, Martin. 2024. "DFT Studies of Dimethylaminophenyl-Substituted Phthalocyanine and Its Silver Complexes" Molecules 29, no. 6: 1344. https://doi.org/10.3390/molecules29061344
APA StyleBreza, M. (2024). DFT Studies of Dimethylaminophenyl-Substituted Phthalocyanine and Its Silver Complexes. Molecules, 29(6), 1344. https://doi.org/10.3390/molecules29061344