Impact of the 237th Residue on the Folding of Human Carbonic Anhydrase II


Abstract
:1. Introduction
2. Results and Discussion
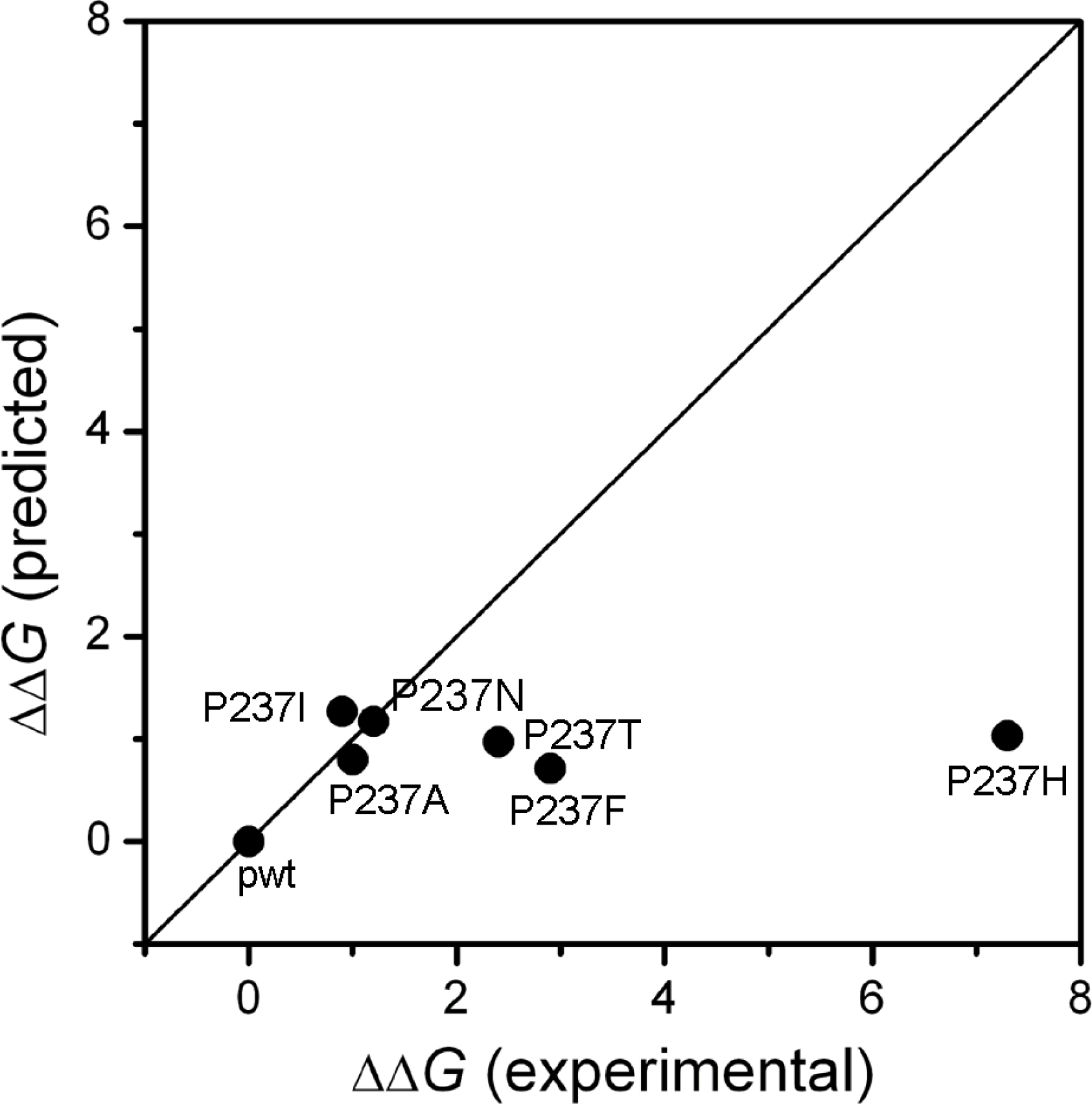
2.1. Stability Changes by Mutations of the 237th Residue Predicted by FoldX
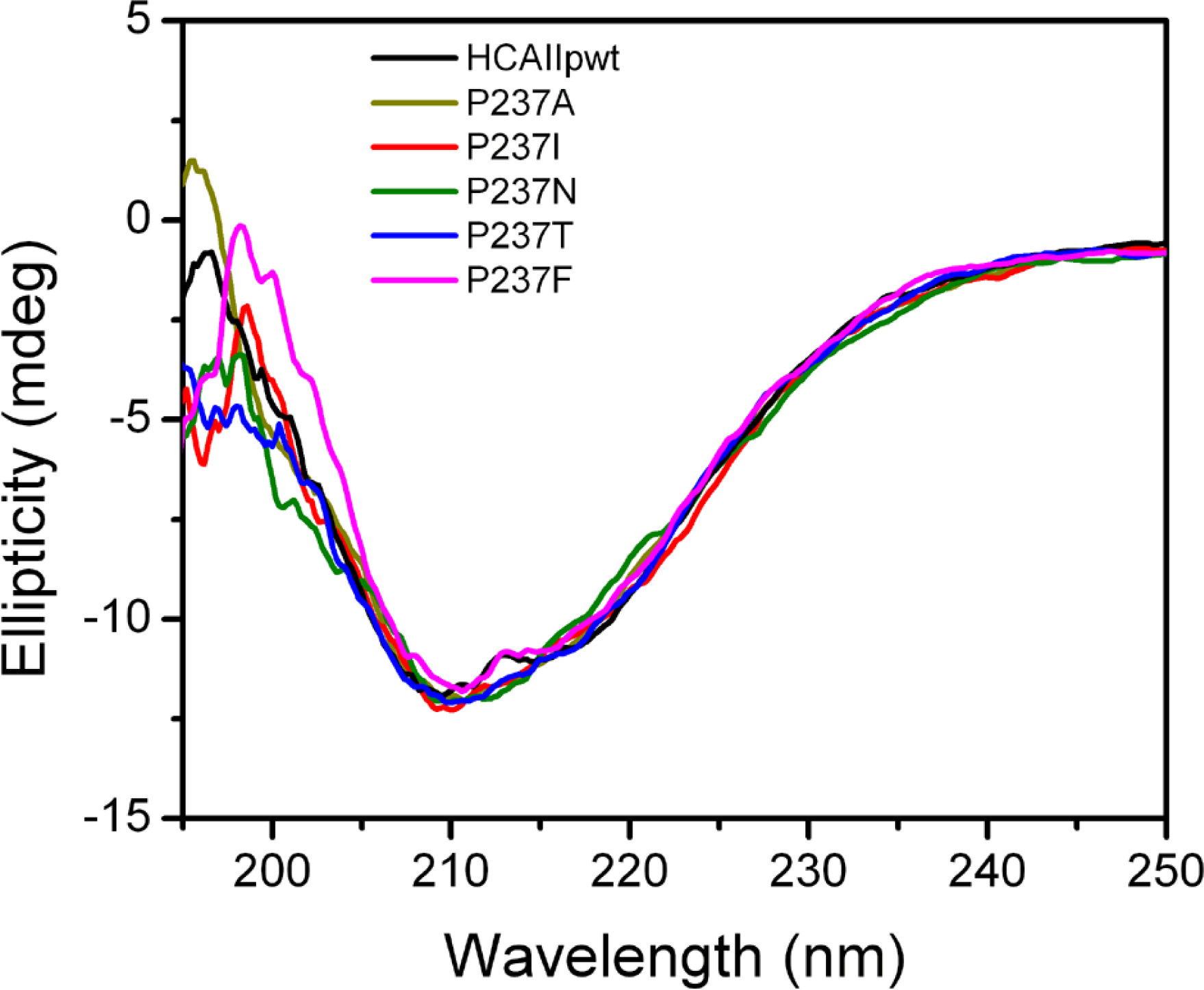
2.2. Characterization of the Mutants
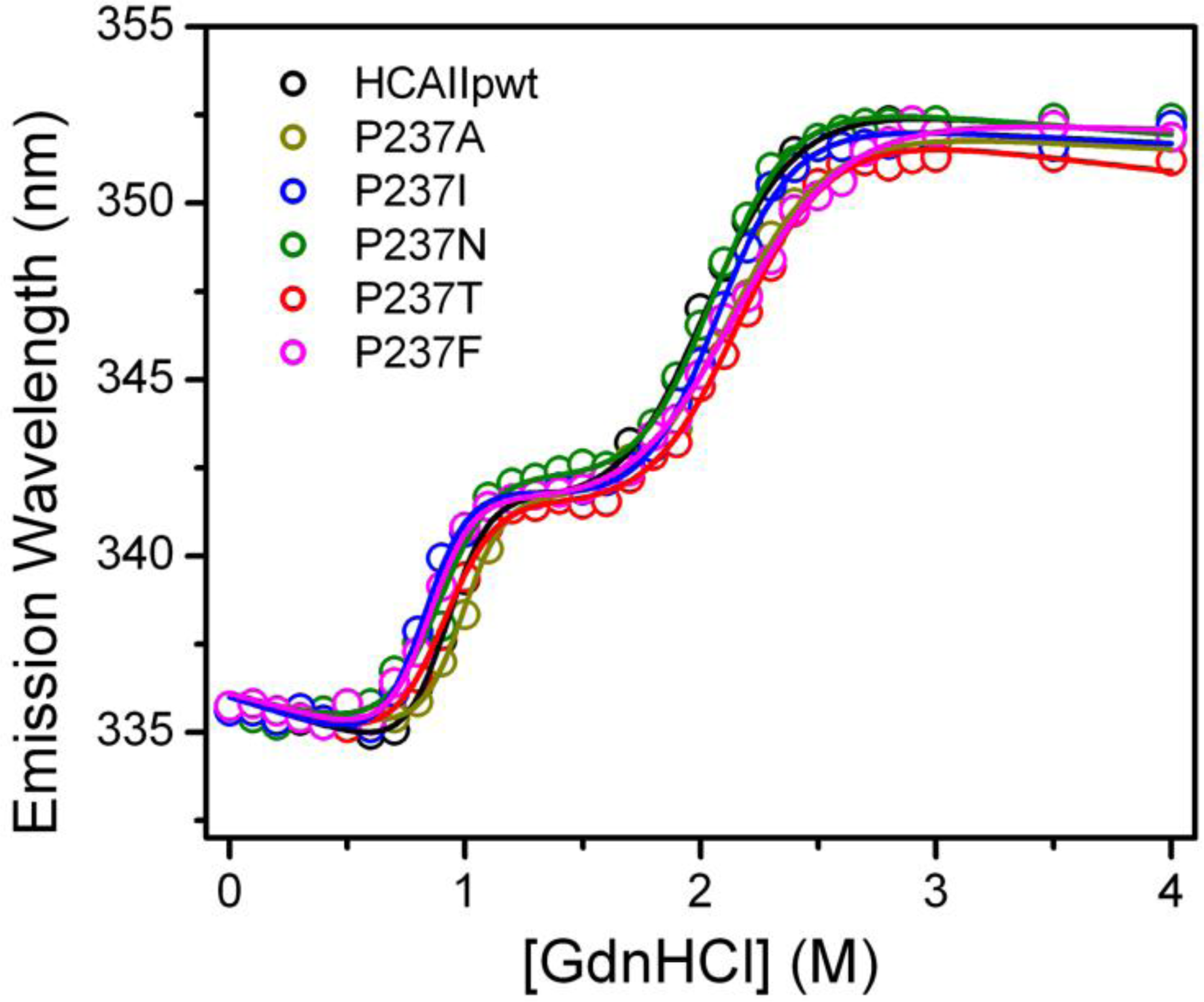
2.3. Evaluation of the Stability Changes by GdnHCl-Induced Folding Experiments
3. Experimental Section
3.1. Materials
3.2. Site-Directed Mutagenesis
- HCAIIP237I-For, 5′-CAATGGGGAGGGTGAAATCGAAGAACTGATG-3′;
- HCAIIP237I-Rev, 5′-CATCAGTTCTTCGATTTCACCCTCCCCATTG-3′;
- HCAIIP237N-For, 5′-CAATGGGGAGGGTGAAAACGAAGAACTGATG-3′;
- HCAIIP237I-Rev, 5′-CATCAGTTCTTCGTTTTCACCCTCCCCATTG-3′;
- HCAIIP237F-For, 5′-CAATGGGGAGGGTGAATTCGAAGAACTGATG-3′;
- HCAIIP237F-Rev, 5′-CATCAGTTCTTCGAATTCACCCTCCCCATTG-3′;
- HCAIIP237A-For, 5′-CAATGGGGAGGGTGAAGCCGAAGAACTGATG-3′;
- HCAIIP237A-Rev, 5′-CATCAGTTCTTCGGCTTCACCCTCCCCATTG-3′;
- HCAIIP237T-For, 5′-CAATGGGGAGGGTGAAACCGAAGAACTGATG-3′;
- HCAIIP237T-Rev, 5′-CATCAGTTCTTCGGTTTCACCCTCCCCATTG-3′.
3.3. Protein Expression and Purification
3.4. Activity Assay
3.5. Protein Folding Experiments
3.6. Spectroscopic Measurements
3.7. Changes in Gibbs Free Energy Calculated by FoldX
4. Conclusions
Acknowledgments
References
- Dobson, CM. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar]
- Crowther, DC. Familial conformational diseases and dementias. Hum. Mutat 2002, 20, 1–14. [Google Scholar]
- Feng, S; Zhao, TJ; Zhou, HM; Yan, YB. Effects of the single point genetic mutation D54G on muscle creatine kinase activity, structure and stability. Int. J. Biochem. Cell. Biol 2007, 39, 392–401. [Google Scholar]
- Almstedt, K; Lundqvist, M; Carlsson, J; Karlsson, M; Persson, B; Jonsson, BH; Carlsson, U; Hammarström, P. Unfolding a folding disease: Folding, misfolding and aggregation of the marble brain syndrome-associated mutant H107Y of human carbonic anhydrase II. J. Mol. Biol 2004, 342, 619–633. [Google Scholar]
- Jiang, Y; Su, JT; Zhang, J; Wei, X; Yan, YB; Zhou, HM. Reshaping the folding energy landscape of human carbonic anhydrase II by a single point genetic mutation Pro237His. Int. J. Biochem. Cell Biol 2008, 40, 776–788. [Google Scholar]
- Hernández-Santoyo, A; del Pozo Yauner, L; Fuentes-Silva, D; Ortiz, E; Rudiño-Piñera, E; Sánchez-López, R; Horjales, E; Becerril, B; Rodríguez-Romero, A. A single mutation at the sheet switch region results in conformational changes favoring λ6 light-chain fibrillogenesis. J. Mol. Biol 2010, 396, 280–292. [Google Scholar]
- Pang, M; Su, J-T; Feng, S; Tang, Z-W; Gu, F; Zhang, M; Ma, X; Yan, Y-B. Effects of congenital cataract mutation R116H on αA-crystallin structure, function and stability. Biochim. Biophys. Acta Proteins Proteomics 2010, 1804, 948–956. [Google Scholar]
- Knaupp, AS; Levina, V; Robertson, AL; Pearce, MC; Bottomley, SP. Kinetic instability of the serpin Zα1-antitrypsin promotes aggregation. J. Mol. Biol 2010, 396, 375–383. [Google Scholar]
- Bordner, AJ; Abagyan, RA. Large-scale prediction of protein geometry and stability changes for arbitrary single point mutations. Proteins 2004, 57, 400–413. [Google Scholar]
- Capriotti, E; Fariselli, P; Rossi, I; Casadio, R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinform 2008, 9. [Google Scholar] [CrossRef]
- Cheng, J; Randall, A; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 2006, 62, 1125–1132. [Google Scholar]
- Pey, AL; Stricher, F; Serrano, L; Martinez, A. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am. J. Hum. Genet 2007, 81, 1006–1024. [Google Scholar]
- Reumers, J; Schymkowitz, J; Rousseau, F. Using structural bioinformatics to investigate the impact of non synonymous SNPs and disease mutations: Scope and limitations. BMC Bioinform 2009, 10. [Google Scholar] [CrossRef]
- Tan, Y; Luo, R. Structural and functional implications of p53 missense cancer mutations. PMC Biophys 2009, 2. [Google Scholar] [CrossRef]
- Lindskog, S. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther 1997, 74, 1–20. [Google Scholar]
- Sly, WS; Hu, PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Ann. Rev. Biochem 1995, 64, 375–401. [Google Scholar]
- Sly, WS; Hewett-Emmett, D; Whyte, MP; Yu, YS; Tashian, RE. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc. Natl. Acad. Sci. USA 1983, 80, 2752–2756. [Google Scholar]
- Hu, PY; Lim, EJ; Ciccolella, J; Strisciuglio, P; Sly, WS. Seven novel mutations in carbonic anhydrase II deficiency syndrome identified by SSCP and direct sequencing analysis. Hum. Mutat 1997, 9, 383–387. [Google Scholar]
- Roth, DE; Venta, PJ; Tashian, RE; Sly, WS. Molecular basis of human carbonic anhydrase II deficiency. Proc. Natl. Acad. Sci. USA 1992, 89, 1804–1808. [Google Scholar]
- Schymkowitz, J; Borg, J; Stricher, F; Nys, R; Rousseau, F; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res 2005, 33, W382–W388. [Google Scholar]
- Reich, L; Becker, M; Seckler, R; Weikl, TR. In vivo folding efficiencies for mutants of the P22 tailspike beta-helix protein correlate with predicted stability changes. Biophys. Chem 2009, 141, 186–192. [Google Scholar]
- Szczepek, M; Brondani, V; Buchel, J; Serrano, L; Segal, DJ; Segal, DJ; Cathomen, T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol 2007, 25, 786–793. [Google Scholar]
- Kiel, C; Serrano, L. Prediction of Ras-effector interactions using position energy matrices. Bioinformatics 2007, 23, 2226–2230. [Google Scholar]
- Encinar, JA; Fernandez-Ballester, G; Sánchez, IE; Hurtado-Gomez, E; Stricher, F; Beltrao, P; Serrano, L. ADAN: A database for prediction of protein-protein interaction of modular domains mediated by linear motifs. Bioinformatics 2009, 25, 2418–2424. [Google Scholar]
- Alibés, A; Nadra, AD; De Masi, F; Bulyk, ML; Serrano, L; Stricher, F. Using protein design algorithms to understand the molecular basis of disease caused by protein-DNA interactions: The Pax6 example. Nucleic Acids Res 2010, 38, 7422–7431. [Google Scholar]
- Tokuriki, N; Stricher, F; Serrano, L; Tawfik, DS. How protein stability and new functions trade off. PLoS Comput Biol 2008, 4. [Google Scholar] [CrossRef]
- Martensson, LG; Jonsson, BH; Freskgard, PO; Kihlgren, A; Svensson, M; Carlsson, U. Characterization of folding intermediates of human carbonic anhydrase II: Probing substructure by chemical labeling of SH groups introduced by site-directed mutagenesis. Biochemistry 1993, 32, 224–231. [Google Scholar]
- Svensson, M; Jonasson, P; Freskgard, PO; Jonsson, BH; Lindgren, M; Maartensson, L; Gentile, M; Boren, K; Carlsson, U. Mapping the folding intermediate of human carbonic anhydrase II. Probing substructure by chemical reactivity and spin and fluorescence labeling of engineered cysteine residues. Biochemistry 1995, 34, 8606–8620. [Google Scholar]
- Hammarstrom, P; Kalman, B; Jonsson, BH; Carlsson, U. Pyrene excimer fluorescence as a proximity probe for investigation of residual structure in the unfolded state of human carbonic anhydrase II. FEBS Lett 1993, 420, 63–68. [Google Scholar]
- Hammarstrom, P; Persson, M; Carlsson, U. Protein compactness measured by fluorescence resonance energy transfer. Human carbonic anhydrase ii is considerably expanded by the interaction of GroEL. J. Biol. Chem 2001, 276, 21765–21775. [Google Scholar]
- Hammarstrom, P; Persson, M; Freskgard, PO; Martensson, LG; Andersson, D; Jonsson, B; Carlsson, U. Structural mapping of an aggregation nucleation site in a molten globule intermediate. J. Biol. Chem 1999, 274, 32897–32903. [Google Scholar]
- Potapov, V; Cohen, M; Schreiber, G. Assessing computational methods for predicting protein stability upon mutation: Good on average but not in the details. Protein Eng. Des. Sel 2009, 22, 553–560. [Google Scholar]
- Pocker, Y; Stone, JT. Catalytic versatility of erythrocyte carbonic Anhydrase 6 Kinetic Studies of noncompetitive inhibition of enzyme-catalyzed hydrolysis of P-Nitrophenyl acetate. Biochemistry 1968, 7, 2936–2945. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | ΔGNI | ΔGIU | ΔGNU | ΔΔGNU | mNI | mIU | Activity(%) |
|---|---|---|---|---|---|---|---|
| HCAIIpwt | 5.7 ± 0.6 | 7.8 ± 0.6 | 13.5 | 0 | 6.3 ± 0.6 | 3.9 ± 0.2 | 100 |
| HCAIIP237A | 6.5 ± 0.7 | 6.0 ± 0.4 | 12.5 | 1.0 | 6.5 ± 0.7 | 2.8 ± 0.2 | 87 |
| HCAIIP237T | 4.7 ± 0.5 | 6.4 ± 0.4 | 11.1 | 2.4 | 5.2 ± 0.6 | 3.0 ± 0.2 | 99 |
| HCAIIP237N | 5.0 ± 0.7 | 7.3 ± 0.5 | 12.3 | 1.2 | 5.5 ± 0.8 | 3.6 ± 0.3 | 93 |
| HCAIIP237I | 5.1 ± 0.5 | 7.5 ± 0.4 | 12.6 | 0.9 | 6.2 ± 0.6 | 3.7 ± 0.2 | 91 |
| HCAIIP237F | 5.3 ± 0.6 | 5.3 ± 0.3 | 10.6 | 2.9 | 6.3 ± 0.7 | 2.5 ± 0.1 | 91 |
| HCAIIP237H | − | − | − | 7.3 | − | − | − |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, M.-J.; Jiang, Y.; Yan, Y.-B. Impact of the 237th Residue on the Folding of Human Carbonic Anhydrase II. Int. J. Mol. Sci. 2011, 12, 2797-2807. https://doi.org/10.3390/ijms12052797
Wu M-J, Jiang Y, Yan Y-B. Impact of the 237th Residue on the Folding of Human Carbonic Anhydrase II. International Journal of Molecular Sciences. 2011; 12(5):2797-2807. https://doi.org/10.3390/ijms12052797
Chicago/Turabian StyleWu, Ming-Jie, Yan Jiang, and Yong-Bin Yan. 2011. "Impact of the 237th Residue on the Folding of Human Carbonic Anhydrase II" International Journal of Molecular Sciences 12, no. 5: 2797-2807. https://doi.org/10.3390/ijms12052797
APA StyleWu, M. -J., Jiang, Y., & Yan, Y. -B. (2011). Impact of the 237th Residue on the Folding of Human Carbonic Anhydrase II. International Journal of Molecular Sciences, 12(5), 2797-2807. https://doi.org/10.3390/ijms12052797