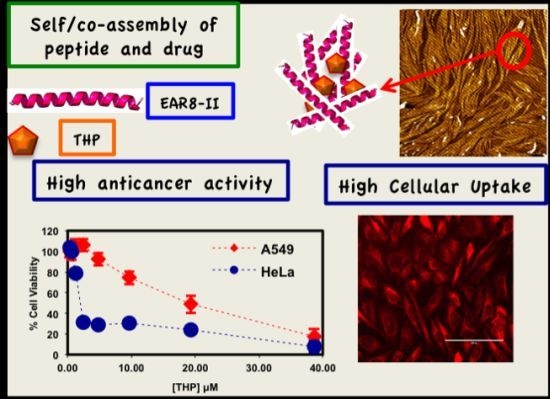
Here, we report details of the complexation between EAR8-II and Pirarubicin (THP), peptide concentration effect on the complex formation, cellular uptake, and anticancer activity of the formulation.
2.1. Effect of Peptide to Drug Ratio on Complex Formation
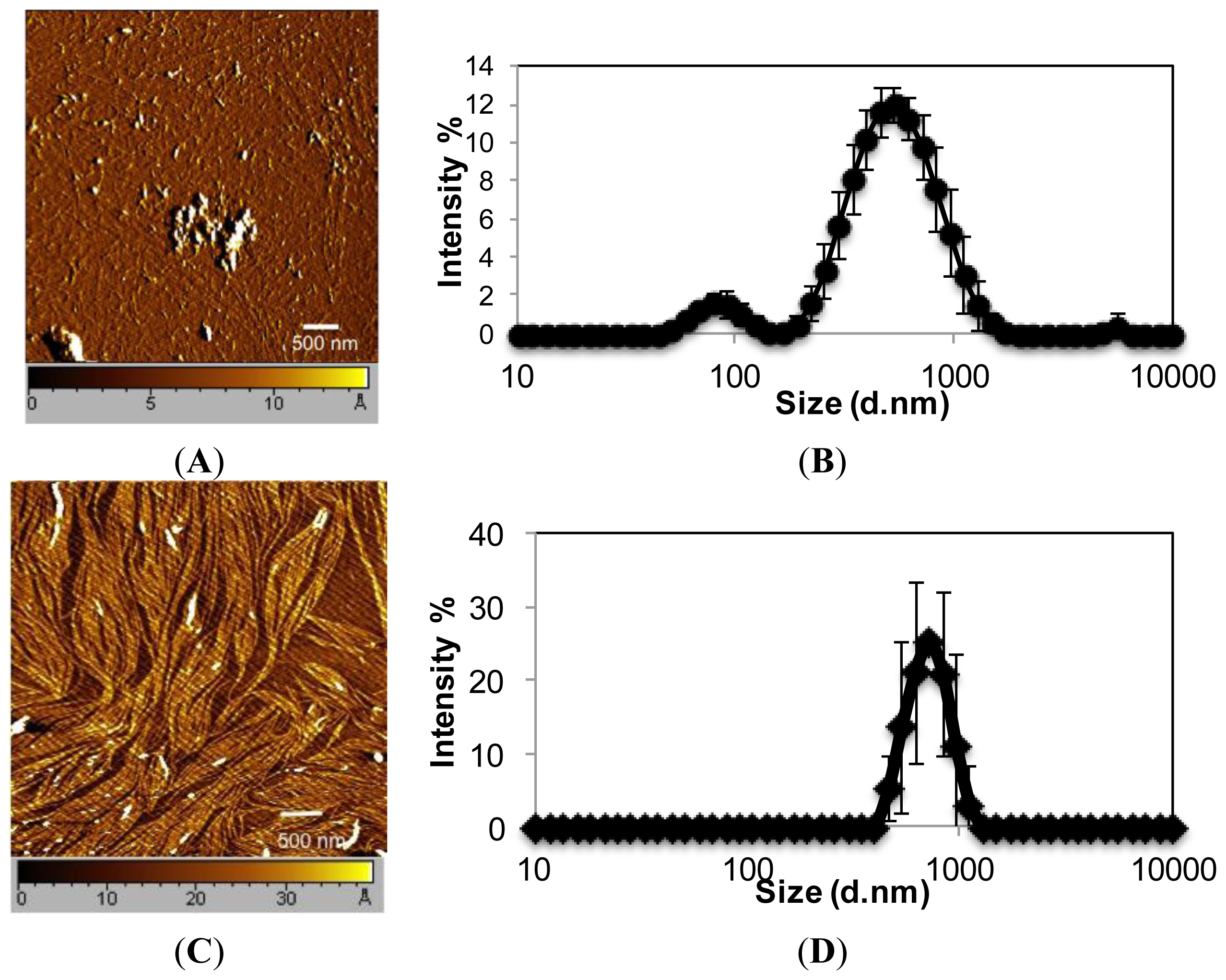
EAR8-II, similar to other ionic-complementary peptides such as EAK16-II, was observed to self-assemble into a dense fibular nanostructure with an average width and height of 10 and 15 nm, respectively. However, there was evidence of large aggregates among the fibers, which occurred due to the high concentration of peptide intermolecular interactions of amino acids. The hydrodynamic diameters of these aggregates were measured at ~500 nm, but fibers were in the range of one or two peptide monomers wide. Interestingly, EAR8-II combined with the hydrophobic drug THP forms a fibular bundle nanostructure with a hydrodynamic diameter of 712.4 ± 17.43 nm, indicating molecular interactions between the peptide and the drug called “co-assembly”. The uniformly shaped fibular structure of the peptide-drug complex provides stability and prevents uncontrollable aggregation in an aqueous solution (
Figure 2).
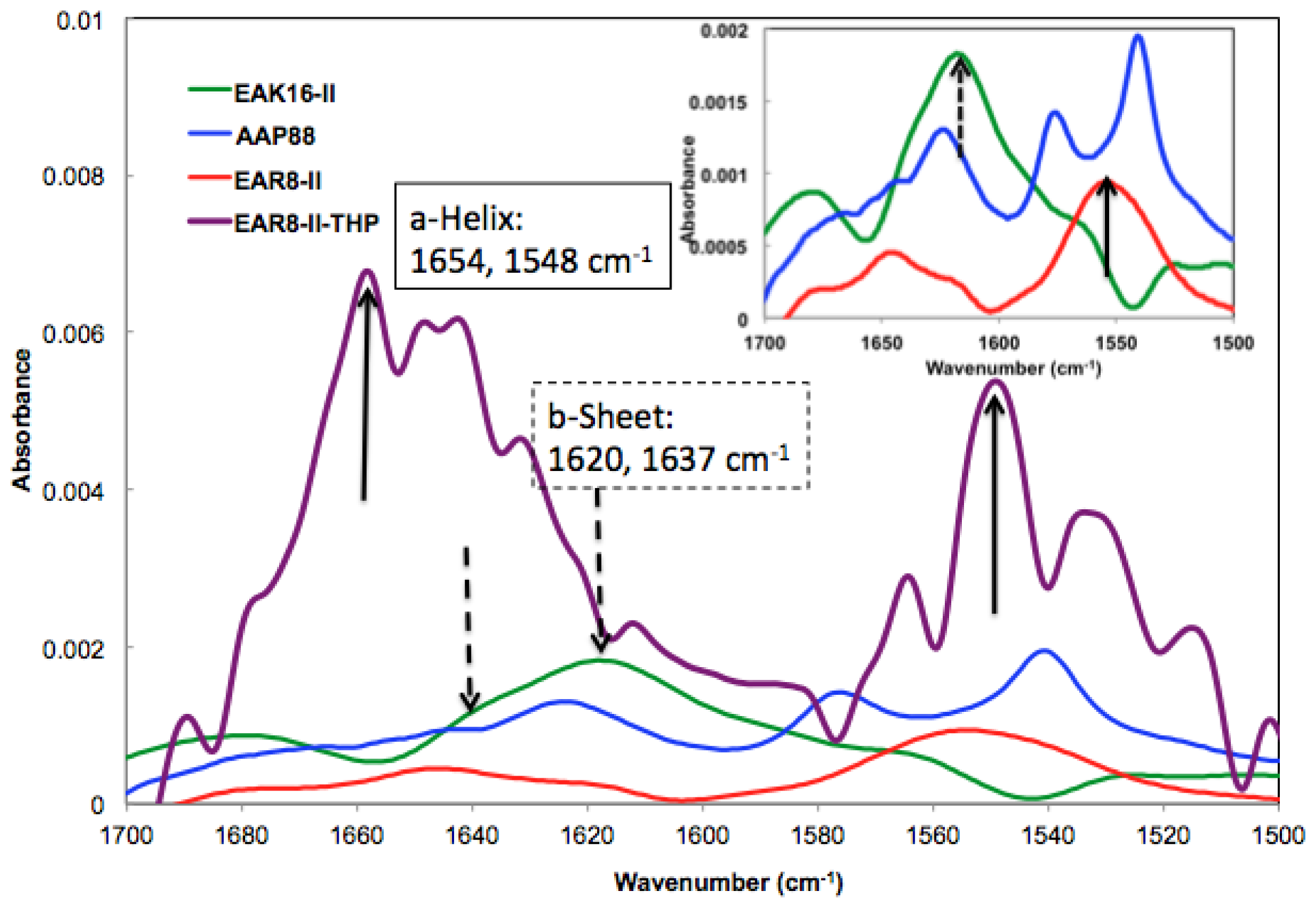
The absorbance peaks from the FT-IR spectrum at 1614–1622 and 1630–1637 cm
−1 correspond to β-sheets, and peaks at 1650–1658 and 1548 cm
−1 correspond to α-helical secondary structures. As shown in
Figure 3, EAK16-II formed β-sheets dominantly, and AAP8 formed a mixture of β-sheets and α-helices. However, EAR8-II showed stronger formation of α-helical structures than the other two self-assembling peptides, probably due to the length of peptide, because intramolecular interactions causing a helical structure in short sequences are stronger than the ones in longer sequences. The FT-IR spectrum collected from the complex containing EAR8-II and THP illustrated a significant increase in absorption intensity at wavenumbers corresponding to α-helical secondary (1654 and 1548 cm
−1) structures, compared to the spectrum collected from the peptide solution. The higher absorption intensity in the FT-IR spectrum implies higher amounts of corresponding secondary structures. The helical fibular bundle structures formed in EAR8-II-THP complexes demonstrate the strong interactions between the peptide and drug to form an organized nanostructure.
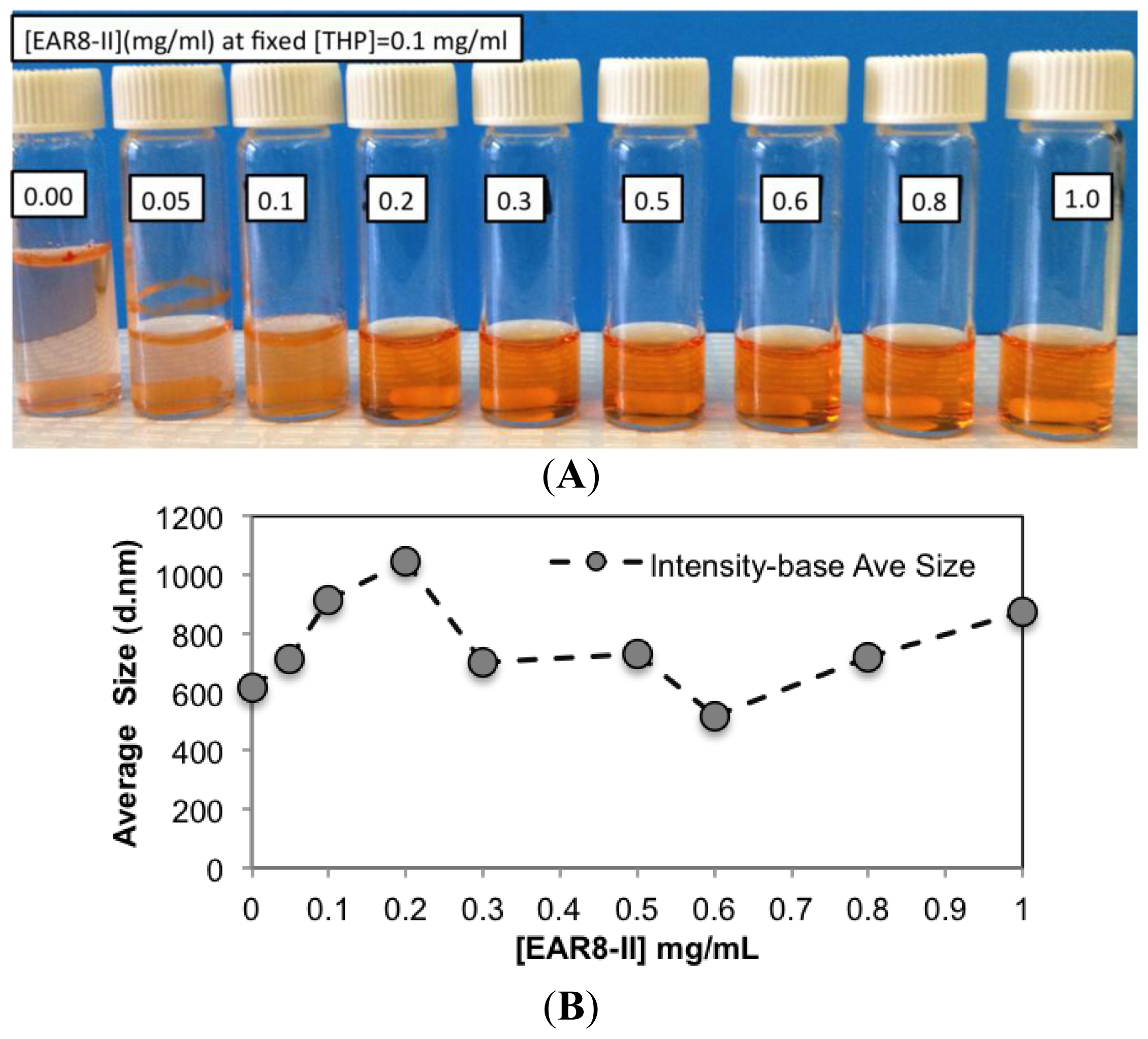
To determine a suitable concentration of required EAR8-II for THP encapsulation, different concentrations of EAR8-II, in a range of 0.05–1.0 mg/mL, were prepared and combined with THP at 0.1 mg/mL concentration in aqueous solution. Solubility of THP in water is very low, whereas it can be dissolved and stabilized in an EAR8-II solution at a minimum concentration of 0.2 mg/mL. As illustrated in
Figure 4A, at lower EAR8-II concentrations (0.05–0.1 mg/mL), THP molecules were not fully dissolved, and crystalline THP particles floated in the solution. However, at higher EAR8-II concentrations (0.2–0.6 mg/mL), THP molecules were fully dissolved, and consequently, a clear-transparent solution of the peptide-drug complexes formed. At much higher peptide concentrations (above 0.6 mg/mL), THP molecules were still fully soluble, but the excess amount of EAR8-II produced a cloudier solution, with clearly a higher particle size distribution. Note that amphiphilic features of EAR8-II, including the ionic and hydrophobic residues, stimulated binding with hydrophobic molecules of THP through protonation of NH
2 to NH
3+ in THP molecules and hydrophobic interactions between alanine residues and aromatic rings in the THP structure.
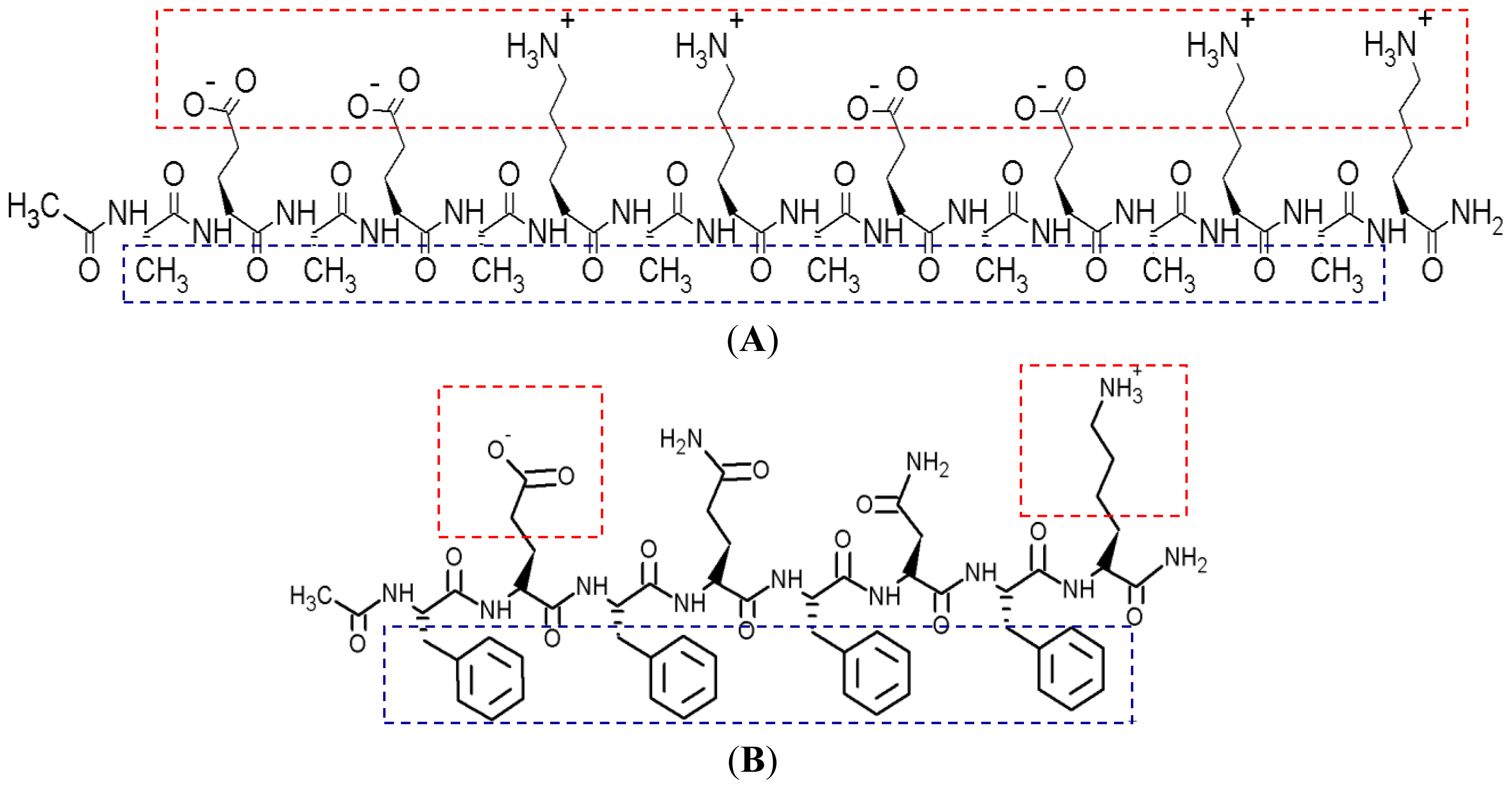
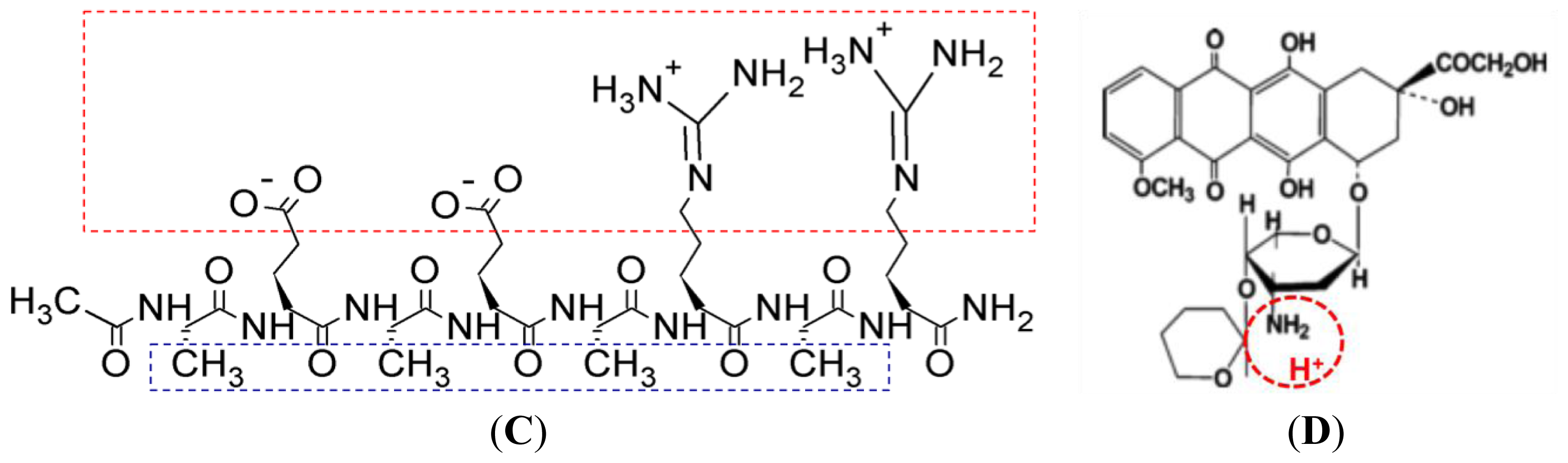
Figure 1 shows the hydrophobic and hydrophilic regions of EAR8-II peptide, as well as protonation of the amine group in THP. Both ionic and hydrophobic interactions are the possible main interactions of EAR8-II and THP,
i.e., the protonation of THP in an acidic EAR8-II solution and hydrophobic interaction between THP and hydrophobic side chains of EAR8-II are the dominant forces in complexation of EAR8-II-THP.
To evaluate the characteristics of the complex formation, the change in size, pH, surface charge, and the THP fluorescence of the peptide-drug complex was monitored at different peptide concentrations. Increasing the concentration of EAR8-II made the THP become more soluble and stable in the solution. The average particle size in these complexes measured by dynamic light scattering (DLS) presented a size increase of up to [EAR8-II] = 0.3 mg/mL, followed by stabilization of the particles in the smaller range of hydrodynamic diameter ~500–700 nm, where at higher [EAR8-II] = 0.8–1 mg/mL, the solution become unstable with larger particles (
Figure 4). At low concentrations of EAR8-II, non-soluble THP molecules were visible by the naked eye, and the solutions were cloudy. At concentrations of EAR8-II above 0.2 mg/mL, a uniformly suspended complex formed, where THP was fully dissolved. At a very high concentration of EAR8-II (1.0 mg/mL), peptide fibers appeared to aggregate, and the diameter measured by DLS showed a significant size increase.
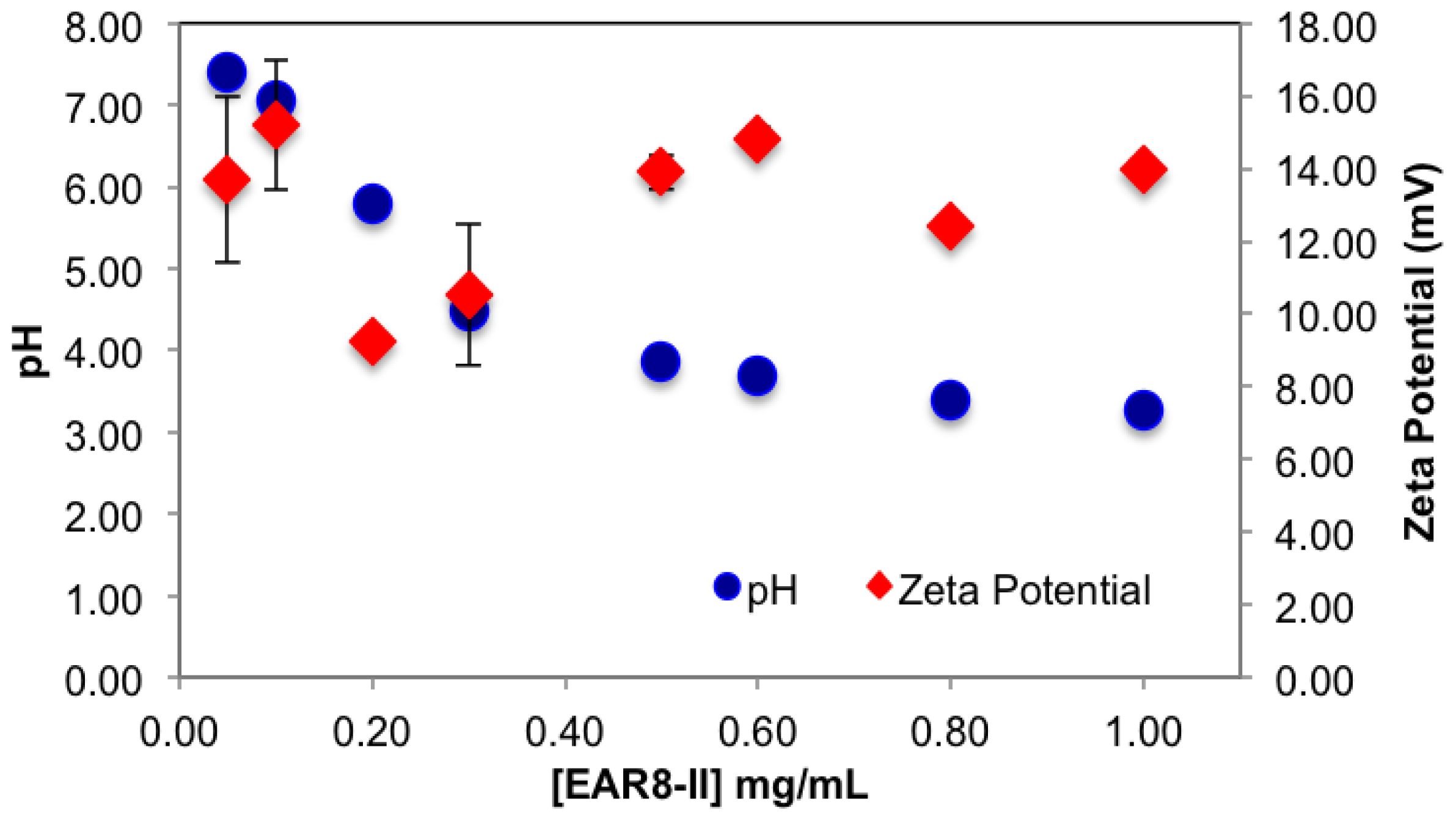
The surface charge and the pH of the complex were also affected by concentration of EAR8-II. EAR8-II has an isoelectric point of ~6.55, in which at lower pH, the net charge of the peptide in aqueous solution is positive. Results showed that when the peptide concentration was increased, the pH of the solution decreased from pH 5.5 to 3.5 at a concentration of 0.05–1 mg/mL, owing to the higher [H
+] at higher peptide concentrations. Since THP has a pKa of ~7.3 (pyridine-like nitrogen) it can be protonated in a weakly acidic environment. Consequently, acidic EAR8-II produced protonation of THP at higher peptide-to-drug ratios and led to more positive zeta potential values (
Figure 5). Basically, an acidic environment provided higher [H
+], and accordingly a higher positive surface charge reflected in zeta potential values. In
Figure 5, the zeta potential values increased with increasing peptide concentration in the complex. Zeta potential values reached a plateau at EAR8-II concentrations above 0.5 mg/mL, indicating saturation of the positive charge on the surface at high peptide concentrations. Note, negatively charged glutamic acid residues in the peptide sequence may also help to stabilize the protonated THP. The high zeta potential values at very low peptide concentrations did not follow the above trend, due to the presence of non-stabilized protonated THP on the surface.
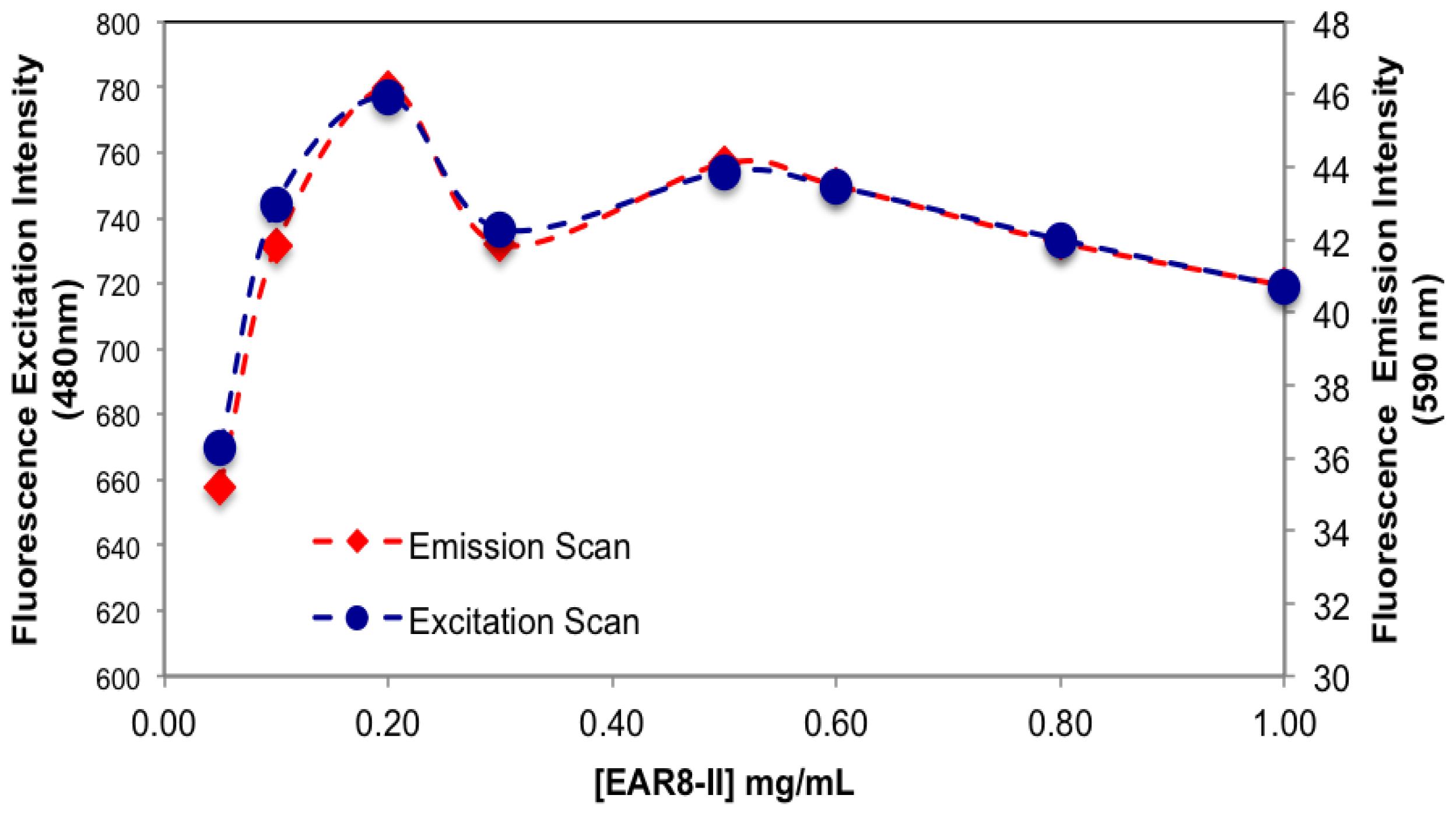
The amount of protonated THP in EAR8-II is also determined by measuring the fluorescence intensity of the excitation and emission by THP at different EAR8-II concentrations. Protonated THP absorbs light at 480 nm, and emits light at 590–600 nm. Fluorescence intensity for both the excitation and emission spectra is proportional to the concentration of protonated THP stabilized in the peptide solution. Although the initial concentration of THP was constant in each complex, the amount of protonated THP varied due to the dependency of the THP encapsulation efficiency on peptide concentration. As shown in
Figure 6, when peptide concentration increased, the THP fluorescence intensities increased, and plateaued at peptide concentrations above 0.5 mg/mL. At lower peptide concentrations, substantial amounts of non-stabilized THP were present in the solutions, and the fluorescence intensities were significantly low, whereas at higher peptide concentrations where all the THP molecules were stabilized in the peptide aqueous environment, higher fluorescence intensities were collected. Our results showed that the minimum concentration of peptides required to fully encapsulate THP was ~0.5 mg/mL.
2.2. Cytotoxicity and Cellular Uptake of the Peptide-Drug Complexes
So far, we have illustrated that EAR8-II can stabilize THP in protonated form in aqueous solutions. It is expected that, due to stability and the peptide-to-drug mass ratio, different anticancer activity against cancer cells will be obtained [
4,
15].
Here, a series of peptide-to-drug mass-ratio complexes at a constant THP concentration of 0.1 mg/mL and EAR8-II at a range of concentrations of 0.05–1 mg/mL were employed to investigate cellular uptake and toxicity against two cancer cell lines (HeLa and A549). Cellular viability profiles for both HeLa and A549 cells at different ratios followed a similar trend, where the cell viability of A549 was relatively higher than that of HeLa, indicating more sensitivity of HeLa cells toward EAR8-II-THP complexes. In the absence of peptide in the complex, THP has been shown to induce non-significant cellular toxicity against both cell lines. Whereas, when EAR8-II stabilized THP in aqueous solution, cellular toxicity of both cells increased significantly. Due to the instability of the THP molecules in aqueous solution they do not penetrate the cell membrane effectively.
As shown in
Figure 7, the complexes with a peptide concentration below 0.2 mg/mL and above 0.6 mg/mL had lower anticancer activity than the complexes with a peptide concentration between 0.2 and 0.6 mg/mL. Cellular viability of the complexes with an EAR8-II concentration beyond 0.2–0.6 mg/mL was significantly high, in the range of cellular viability observed in the THP control sample. This finding indicated effectiveness of EAR8-II in stabilizing THP in certain concentration ranges. As discussed above, the particle size in complexes in the 0.2–0.6 mg/mL ranges were smaller than those in other ratio complexes, which can be correlated to anticancer activity of the complexes. At lower EAR8-II concentrations, not all the THP molecules co-assembled with the peptide molecules, shown in the particle size distribution and in less efficiency toward cancer cells. However, at higher peptide concentrations, the excess presence of peptide suppressed the proficiency of THP by inhibiting the drug release effectively.
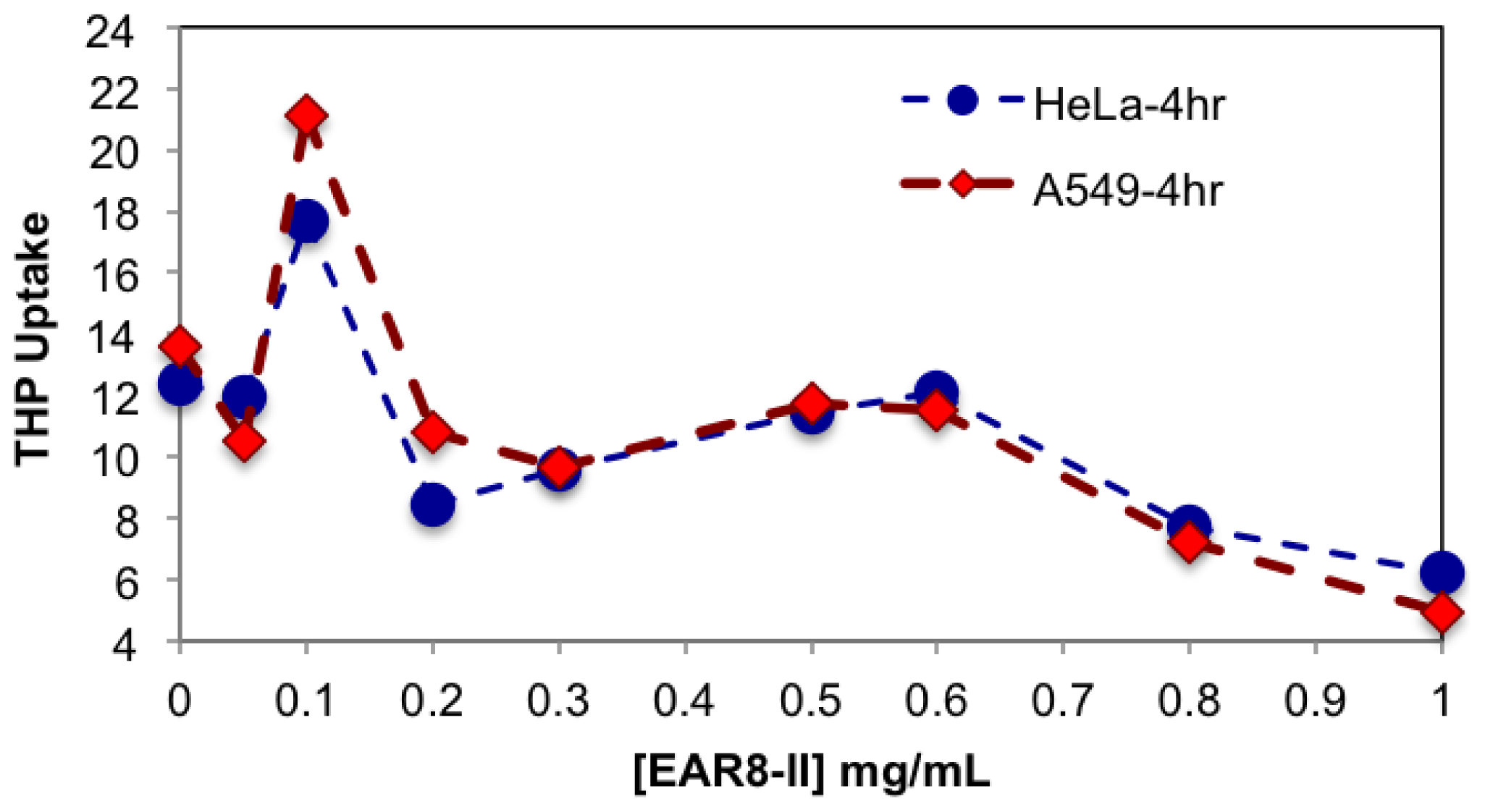
It is speculated that protonated THP molecules were inclined to interact with negatively charged cell membrane and promoted cellular uptake. The hydrophobic properties of THP further stimulated molecules cross the cell membrane and localized in the cytoplasm. As shown in
Figure 8, cellular uptake of complexes with a peptide concentration between 0.3 and 0.6 mg/mL presented higher cellular uptake for both cell lines than the complexes beyond that ratio. The exception was the peptide-to-drug ratio 1 to 1 and the THP control, showing dramatically high uptake due to accumulation of free THP molecules on the cell membrane without them being fully localized in the cytoplasm. On the other hand, the complexes with higher EAR8-II concentrations (0.8–1.0 mg/mL) showed significantly lower cellular uptake due to highly bound EAR8-II to THP and inhibiting release of hydrophobic THP to the cell membranes.
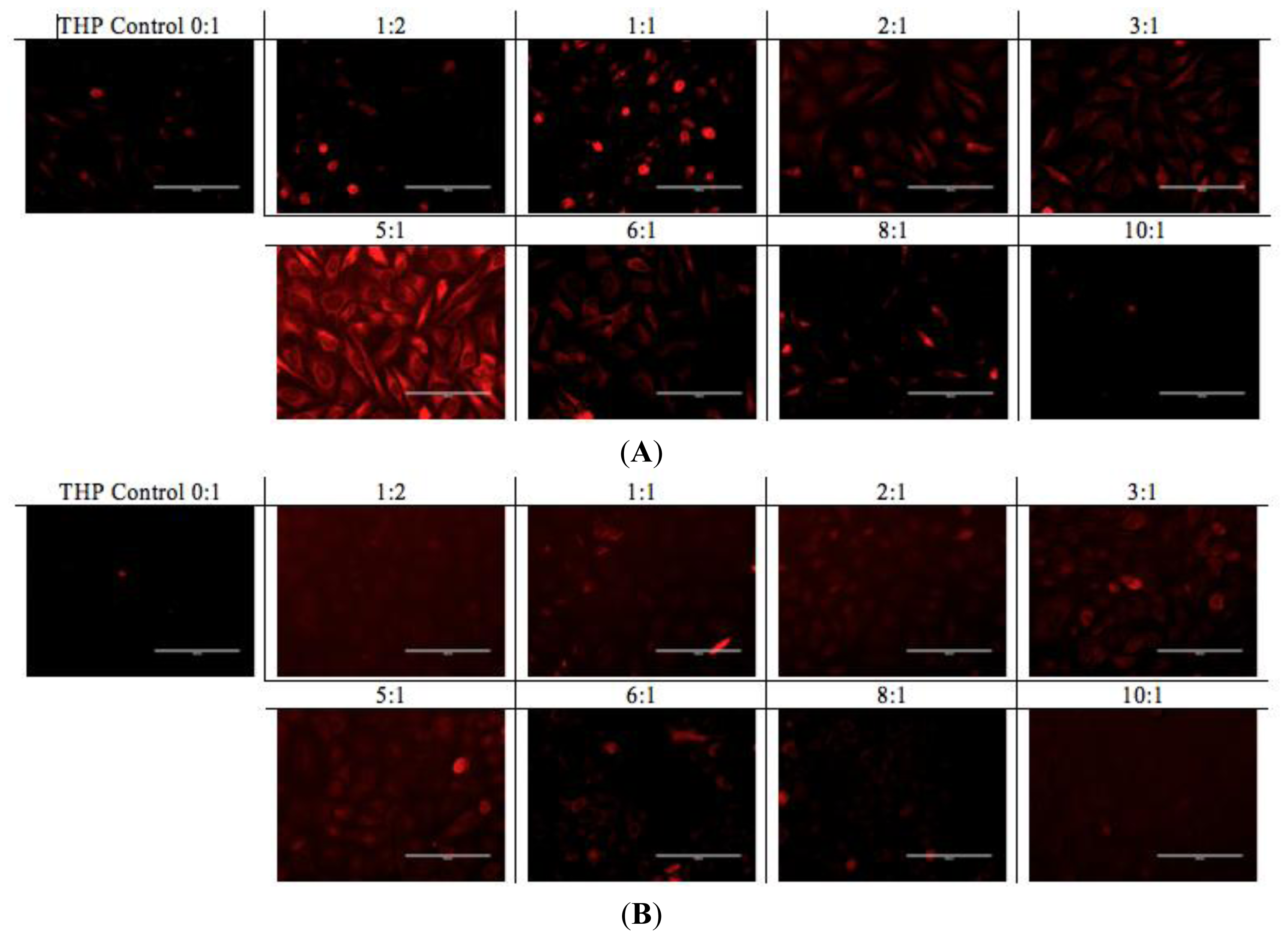
A similar pattern was observed in the fluorescence microscopy images from the cells treated with EAR8-II-THP complexes at different ratios. For both cell lines, complexes with peptide concentrations of 0.3 and 0.5 mg/mL resulted in the highest cellular uptake of THP in cytoplasm. In contrast, complexes at lower and higher concentrations of the peptide showed lower localization of THP in cytoplasm. Note that the bright red fluorescence observed in 1:2 and 1:1 ratio complexes were from non-dissolved THP not localized in cytoplasm. At the ratios of 3:1 and 5:1, the localization of THP in cytoplasm was very uniform, whereas other ratio complexes had THP randomly localized or just accumulated on the membrane (
Figure 9). These results led us to select appropriate formulations to treat different cancer cells. So far, from the cumulative characterization and cellular uptake and toxicity results, the complex with the peptide-to-drug ratio of 5:1 has been shown to be a more stable and efficient solution for drug delivery, because of its high encapsulation efficiency, uniform nanostructure, relatively high cellular toxicity and uptake.
Au
et al. emphasized the importance of drug stability upon dilution during treatment time in clinical trials [
16]. Since, the peptide-drug complex at a 5:1 ratio illustrated appropriate properties such as size, charge and toxicity against cancer cells, cytotoxicity testing was performed for this particular complex, and its serial dilution solutions were tested against two human cancer cell lines, including the non-small lung cancer cell A549, cervical adenocarcinoma HeLa.
Figure 10 represents the viability of the cells treated with the 5:1 ratio complex and its serial dilution in water, where the final concentration of THP is between 0.3 and 38 μM. The IC
50 values of the complex for the cancer cells were calculated, and followed the following trend; THP concentration at ~19.3 μM for A549 >~1.8 μM for HeLa cells [
17].
Toxicity results for both serially diluted and variable peptide-to-drug ratio complexes revealed relatively higher cell viability of A549 compared to HeLa cells, denoting that A549 cells are highly resistant towards the EAR8-II-THP complex. Similar results were observed previously for EAK16-II and EPT complexes against MCF-7 cells and A549, in which A549 cells showed higher resistance than other tested cells [
4]. The reason for different levels of toxicity of the same complex towards various cancer cells is not fully clear, but it might arise from different reactions of cancer cells to the EAR8-II-THP complex.
Note that the cell viability of the THP control solution (no peptide) for both types of cell lines was significantly higher than for the respective THP concentrations in the presence of EAR8-II. For THP at 38 μM in water, the cellular viability was 64.9 ± 6.4, 29.58 ± 3.69 (%) for A549, HeLa cells, respectively. These findings clarify the important role of EAR8-II in stabilizing THP in an aqueous environment, and consequently delivering it into cells and causing cellular toxicity.
In the case of a stable complex upon dilution, the increase in cellular viability happens smoothly and gradually due to the lower drug concentration. Based on the observed cell viability results at 24 h incubation time, the viability of A549 increased gradually, when the complex was diluted. In contrast, the viability trend for HeLa cells did not increase as smoothly as for A549 cells. The increase of HeLa cell viability after sixteen times dilution was sharp, indicating instability of the complex at high dilution factors in water, in the presence of a medium. One reason for complex instability is the instability of protonated THP in a highly diluted complex. As discussed, protonated THP is stabilized in peptide environments at low pH. Higher amounts of water lead to increased pH and consequently lower THP stability in solution. This effect might also arise from the stability of the complex in the different culture media used for different cell lines. The content of each culture medium varied, and THP might have distinct properties in each medium. Further experiments are required to confirm this phenomenon. Overall, EAR8-II-THP complexes at a peptide-to-drug ratio of 5:1 demonstrated promising results against cancer cells and stabilized in the cell culture environment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



 ) EAK16-II; (
) EAK16-II; (
 ) AAP8; (
) AAP8; (
 ) EAR8-II; and (
) EAR8-II; and (
 ) EAR8-II-THP complex collected by FT-IR indicating secondary structures.
) EAR8-II-THP complex collected by FT-IR indicating secondary structures.







