Dysfunction of Endocytic Kinase AAK1 in ALS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
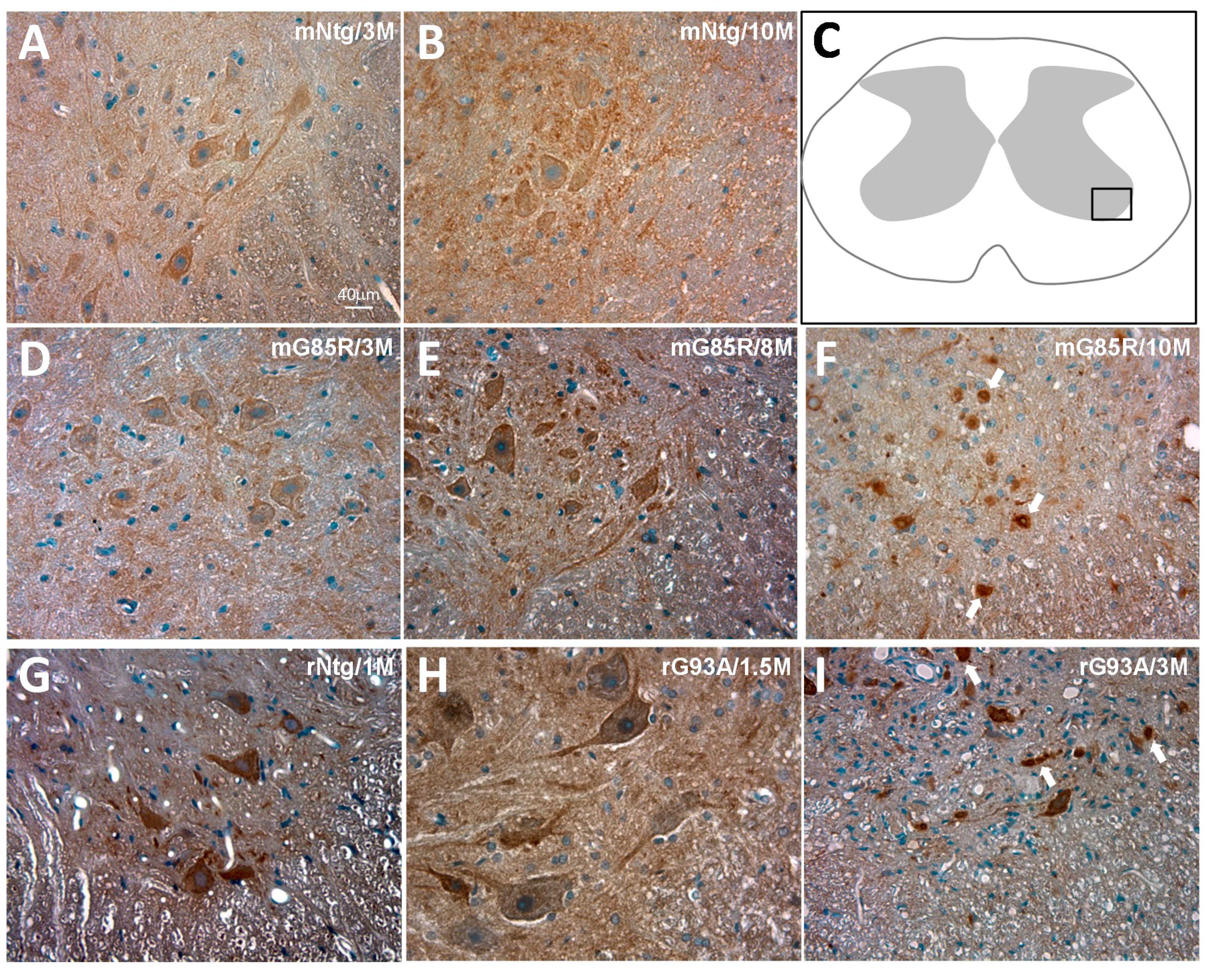
2.1. AAK1 Is Expressed in the Rodent Spinal Cord and Its Patterns of Expression Are Altered in ALS Pathology

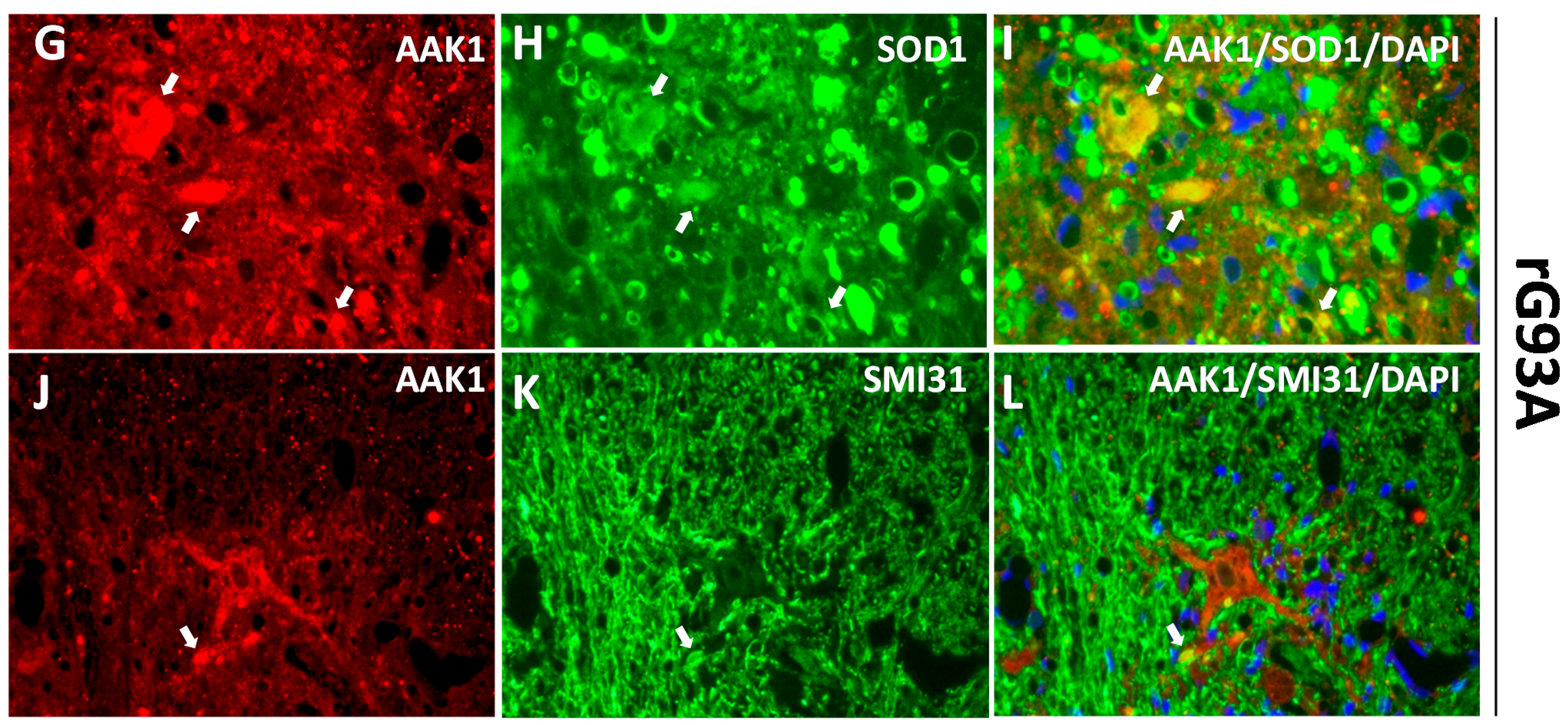
2.2. AAK1 Is Partially Colocalized with the Endosomal and Presynaptic Protein Markers

2.3. AAK1-Containing Aggregates Are Heterogeneous


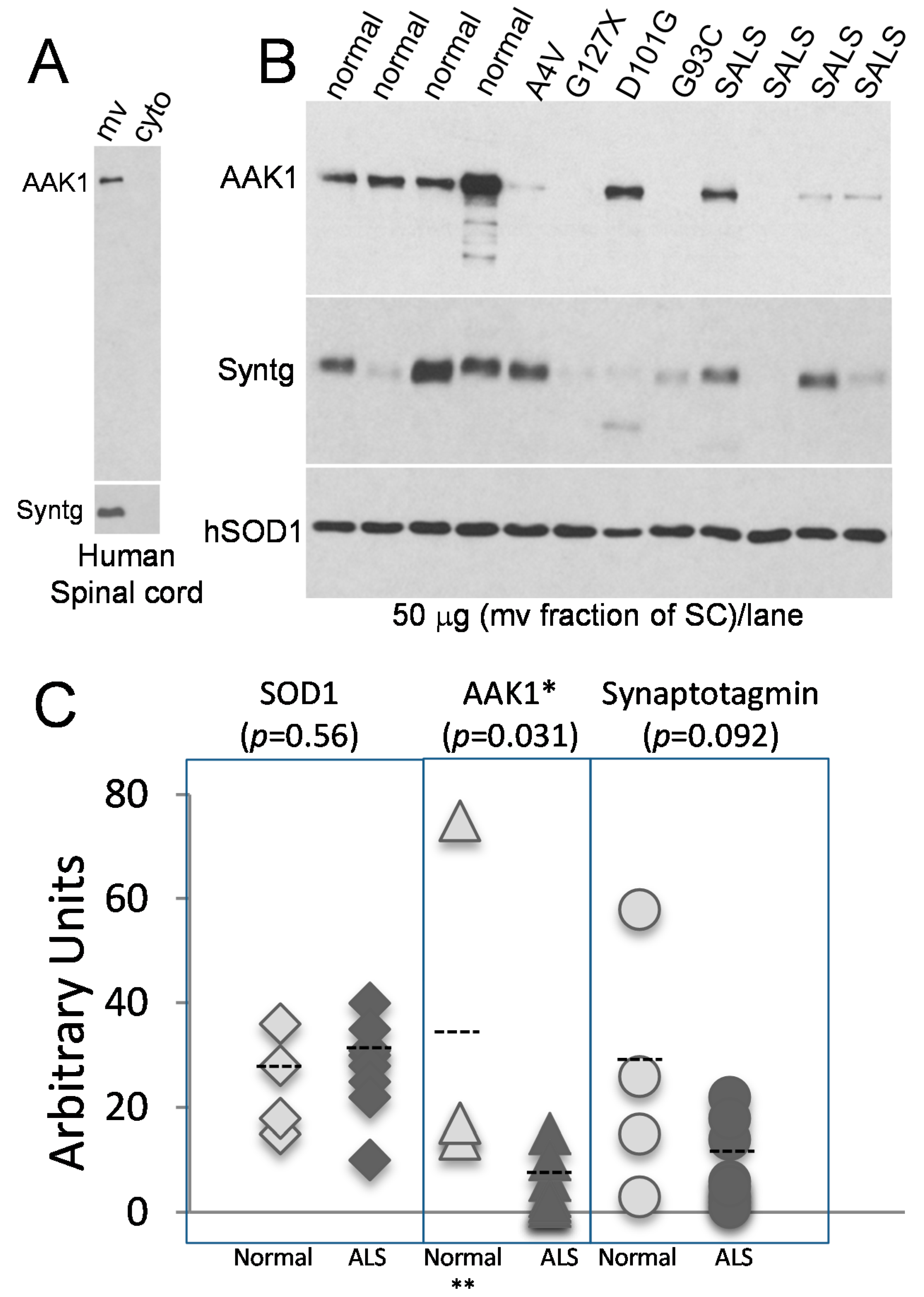
2.4. The AAK1 Protein Levels Are Decreased in Human ALS Spinal Cords

3. Discussion
3.1. AAK1 and Aggregate Pathology in ALS
3.2. AAK1 and Presynaptic Dysfunction in ALS
3.3. AAK1 and Cell Death in ALS
4. Materials and Methods
4.1. Animals
4.2. Human Tissues
4.3. Dual Bait Yeast Two-Hybrid Screening
4.4. Immunohistochemistry and Immunofluorescent Labeling
4.5. Immunoblot Analysis
4.6. Antibodies
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.F.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H., Jr.; et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar]
- Urushitani, M.; Sik, A.; Sakurai, T.; Nukina, N.; Takahashi, R.; Julien, J.P. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci. 2006, 9, 108–118. [Google Scholar]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guegan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M., II; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. USA 2006, 103, 6025–6030. [Google Scholar]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar]
- Fujisawa, T.; Homma, K.; Yamaguchi, N.; Kadowaki, H.; Tsuburaya, N.; Naguro, I.; Matsuzawa, A.; Takeda, K.; Takahashi, Y.; Goto, J.; et al. A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann. Neurol. 2012, 72, 739–749. [Google Scholar]
- Gifondorwa, D.J.; Robinson, M.B.; Hayes, C.D.; Taylor, A.R.; Prevette, D.M.; Oppenheim, R.W.; Caress, J.; Milligan, C.E. Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2007, 27, 13173–13180. [Google Scholar]
- Urushitani, M.; Kurisu, J.; Tateno, M.; Hatakeyama, S.; Nakayama, K.; Kato, S.; Takahashi, R. CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem. 2004, 90, 231–244. [Google Scholar]
- Wood, J.D.; Beaujeux, T.P.; Shaw, P.J. Protein aggregation in motor neurone disorders. Neuropathol. Appl. Neurobiol. 2003, 29, 529–545. [Google Scholar]
- Kunst, C.B.; Mezey, E.; Brownstein, M.J.; Patterson, D. Mutations in SOD1 associated with amyotrophic lateral sclerosis cause novel protein interactions. Nat. Genet. 1997, 15, 91–94. [Google Scholar]
- Zhai, J.; Lin, H.; Canete-Soler, R.; Schlaepfer, W.W. HoxB2 binds mutant SOD1 and is altered in transgenic model of ALS. Hum. Mol. Genet. 2005, 14, 2629–2640. [Google Scholar]
- Conner, S.D.; Schmid, S.L. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J. Cell Biol. 2002, 156, 921–929. [Google Scholar]
- Pun, S.; Santos, A.F.; Saxena, S.; Xu, L.; Caroni, P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 2006, 9, 408–419. [Google Scholar]
- Mizusawa, H.; Matsumoto, S.; Yen, S.H.; Hirano, A.; Rojas-Corona, R.R.; Donnenfeld, H. Focal accumulation of phosphorylated neurofilaments within anterior horn cell in familial amyotrophic lateral sclerosis. Acta Neuropathol. 1989, 79, 37–43. [Google Scholar]
- Murayama, S.; Ookawa, Y.; Mori, H.; Nakano, I.; Ihara, Y.; Kuzuhara, S.; Tomonaga, M. Immunocytochemical and ultrastructural study of Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathol. 1989, 78, 143–152. [Google Scholar]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.F.; Teixeira, M.; Valentine, J.S. Superoxide dismutases and superoxide reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar]
- Schiffer, D.; Cordera, S.; Giordana, M.T.; Attanasio, A.; Pezzulo, T. Synaptic vesicle proteins, synaptophysin and chromogranin A in amyotrophic lateral sclerosis. J. Neurol. Sci. 1995, 129, 68–74. [Google Scholar]
- Wang, J.; Farr, G.W.; Hall, D.H.; Li, F.; Furtak, K.; Dreier, L.; Horwich, A.L. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000350. [Google Scholar]
- Cappello, V.; Vezzoli, E.; Righi, M.; Fossati, M.; Mariotti, R.; Crespi, A.; Patruno, M.; Bentivoglio, M.; Pietrini, G.; Francolini, M. Analysis of neuromuscular junctions and effects of anabolic steroid administration in the SOD1G93A mouse model of ALS. Mol. Cell Neurosci. 2012, 51, 12–21. [Google Scholar]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar]
- Fasana, E.; Fossati, M.; Ruggiano, A.; Brambillasca, S.; Hoogenraad, C.C.; Navone, F.; Francolini, M.; Borgese, N. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 2010, 24, 1419–1430. [Google Scholar]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar]
- Ito, Y.; Yamada, M.; Tanaka, H.; Aida, K.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Inuzuka, T.; Takahashi, H.; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 2009, 36, 470–476. [Google Scholar]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar]
- Conner, S.D.; Schmid, S.L. Differential requirements for AP-2 in clathrin-mediated endocytosis. J. Cell Biol. 2003, 162, 773–779. [Google Scholar]
- Conner, S.D.; Schroter, T.; Schmid, S.L. AAK1-mediated micro2 phosphorylation is stimulated by assembled clathrin. Traffic 2003, 4, 885–890. [Google Scholar]
- Sorensen, E.B.; Conner, S.D. AAK1 regulates Numb function at an early step in clathrin-mediated endocytosis. Traffic 2008, 9, 1791–1800. [Google Scholar]
- Watanabe, S.; Trimbuch, T.; Camacho-Perez, M.; Rost, B.R.; Brokowski, B.; Sohl-Kielczynski, B.; Felies, A.; Davis, M.W.; Rosenmund, C.; Jorgensen, E.M. Clathrin regenerates synaptic vesicles from endosomes. Nature 2014, 515, 228–233. [Google Scholar]
- Baldwin, A.; Li, W.; Grace, M.; Pearlberg, J.; Harlow, E.; Munger, K.; Grueneberg, D.A. Kinase requirements in human cells: II. Genetic interaction screens identify kinase requirements following HPV16 E7 expression in cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16478–16483. [Google Scholar]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am. J. Med. Genet. B 2005, 137B, 5–16. [Google Scholar]
- Latourelle, J.C.; Pankratz, N.; Dumitriu, A.; Wilk, J.B.; Goldwurm, S.; Pezzoli, G.; Mariani, C.B.; DeStefano, A.L.; Halter, C.; Gusella, J.F.; et al. Genomewide association study for onset age in Parkinson disease. BMC Med. Genet. 2009, 10, 98. [Google Scholar]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, B.; Conner, S.D.; Liu, J. Dysfunction of Endocytic Kinase AAK1 in ALS. Int. J. Mol. Sci. 2014, 15, 22918-22932. https://doi.org/10.3390/ijms151222918
Shi B, Conner SD, Liu J. Dysfunction of Endocytic Kinase AAK1 in ALS. International Journal of Molecular Sciences. 2014; 15(12):22918-22932. https://doi.org/10.3390/ijms151222918
Chicago/Turabian StyleShi, Bingxing, Sean D. Conner, and Jian Liu. 2014. "Dysfunction of Endocytic Kinase AAK1 in ALS" International Journal of Molecular Sciences 15, no. 12: 22918-22932. https://doi.org/10.3390/ijms151222918
APA StyleShi, B., Conner, S. D., & Liu, J. (2014). Dysfunction of Endocytic Kinase AAK1 in ALS. International Journal of Molecular Sciences, 15(12), 22918-22932. https://doi.org/10.3390/ijms151222918