The Role of Reactive Oxygen Species in Microvascular Remodeling

Abstract
:1. Introduction
2. The Remodeling Process
3. Sources of Reactive Oxygen Species in the Microcirculation
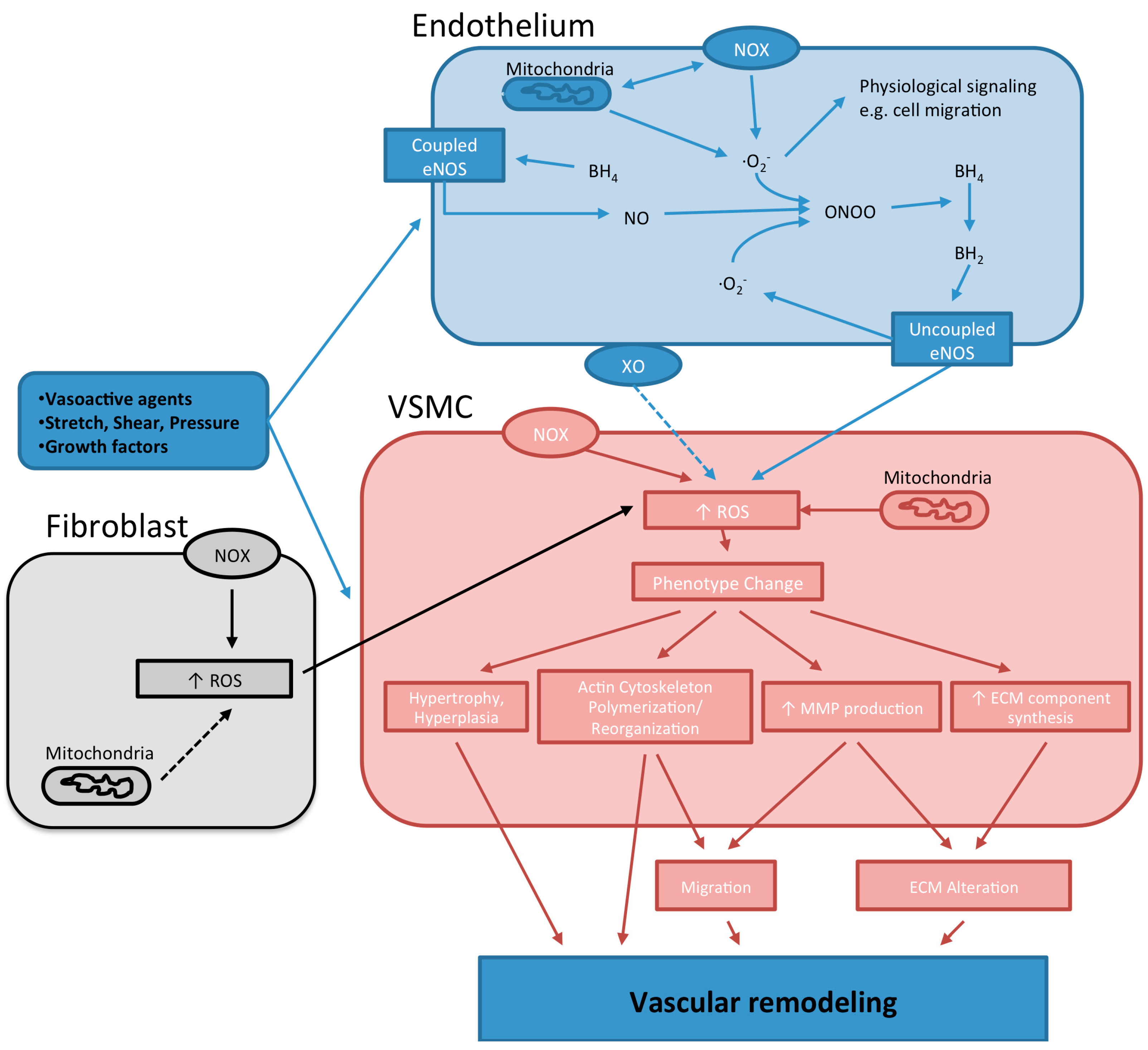
3.1. NADPH Oxidases
3.2. Nitric Oxide Synthase

3.3. Xanthine Oxidase
3.4. Mitochondrial Electron Transport System
4. The Role of ROS in Vascular Remodeling
4.1. ROS in Remodeling of the Microcirculation

{kind=link}
| Vascular Bed | Experimental System | Stimulus | ROS Species | Type of Remodeling | ROS Inhibitor | Ref. |
|---|---|---|---|---|---|---|
| Mouse mesenteric arteriole | PPARγ KO mice | angiotensin II | superoxide, reduced SOD3 expression | Eutrophic remodeling, Hypertrophic remodeling | - | [75] |
| Human subcutaneous arteriole | Human | Cushing syndrome | superoxide | Hypertrophic remodeling | - | [24] |
| Mouse mesenteric arteriole | (NZO) mice | - | superoxide, peroxynitrite | Hypertrophic remodeling | Tempol | [90] |
| Rat mesenteric arteriole | Wistar rats (female) ovareiectomized | high flow | superoxide | Hypertrophic remodeling | - | [39] |
| Rat mesenteric arteriole | Zucker rats | high flow, hyperglycemia | superoxide | Hypertrophic remodeling | Tempol | [132] |
| Mouse mesenteric arteriole | BALB/c male mice | angiotensin II | superoxide | Hypertrophic remodeling | Apocynin | [77] |
| Rat mesenteric arteriole | Wistar rats | angiotensin II | superoxide | Inward eutrophic remodeling | Atorvastatin ** | [79] |
| Mouse basilary artery | PPAR-gamma KO mice | - | superoxide | Inward hypertrophic remodeling | Tempol | [32] |
| Rat cremasteric arteriole | Sprague-Dawley rat | norepinephrine, angiotensin II | superoxide, hydrogen peroxide | Inward remodeling | Tempol, Apocynin | [62] |
| Rat middle cerebral artery | SPSHR rats | serotonin | superoxide | Inward remodeling | Tempol | [130] |
| Rat mesenteric arteriole | Wistar rats | low flow | superoxide | Inward remodeling | Tempol, Apocynin | [133] |
| Rat mesenteric arteriole | Sprague-Dawley rat | angiotensin II | superoxide | Inward remodeling | - | [71] |
| Mouse aferent arteriole | SOD1 tg, SOD1 KO mice | angiotensin II | superoxide | Inward remodeling | Tempol | [76] |
| Rat middle cerebral artery, basilary artery | SHR | - | superoxide | Inward remodeling, Hypertrophic remodeling | Telmisartan # (ARB) | [128] |
| Rat mesenteric arteriole | Wistar rats | low flow, high flow | superoxide | Inward remodeling, Outward remodeling | Tempol | [134] |
| Rat/Mouse mesenteric arteriole | Wistar rats, eNOS KO mice | low flow, high flow | superoxide, hydrogen peroxide | Inward remodeling, Outward remodeling | Apocynin, Catalase | [135] |
| Rat mesenteric arteriole | Wistar rats | high flow | superoxide | Outward hypertrophic remodeling | Tempol, Perindopril *, Candesartan # | [136] |
| Rat mesenteric arteriole | Zucker rats | high flow | superoxide | Outward hypertrophic remodeling | Tempol, Catalase, SOD | [137] |
| Rat mesenteric arteriole | Wistar rats | high flow | superoxide | Outward remodeling | Tempol, Apocynin | [138] |
4.2. Reactive Oxygen Species and the Phenotype of Vascular Smooth Muscle Cells
4.3. ROS-Induced Vascular Smooth Muscle Cell Migration
4.4. Reactive Oxygen Species and the Actin Cytoskeleton
4.5. ROS-Induced Cellular Growth and Apoptosis
4.6. ROS-Induced ECM Reorganization
4.7. Reactive Oxygen Species Contribution to Rarefaction
5. ROS and the Myogenic Response
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Gu, Y.; Dee, C.M.; Shen, J. Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front. Biosci. Schol. Ed. 2011, 3, 1216–1231. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Forstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Forstermann, U. Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol. Sci. 2013, 34, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species, vascular Noxs, and hypertension: Focus on translational and clinical research. Antioxid. Redox Signal. 2014, 20, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Mennuni, S.; Testa, M.; Mennuni, M.; Pierelli, G.; Pagliaro, B.; Gabriele, E.; Coluccia, R.; Autore, C.; Volpe, M. Pathogenesis of chronic cardiorenal syndrome: Is there a role for oxidative stress? Int. J. Mol. Sci. 2013, 14, 23011–23032. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta 2014, 1840, 2709–2729. [Google Scholar] [CrossRef] [PubMed]
- Hafstad, A.D.; Nabeebaccus, A.A.; Shah, A.M. Novel aspects of ROS signalling in heart failure. Basic Res. Cardiol. 2013, 108, 359. [Google Scholar] [CrossRef]
- Satoh, K.; Nigro, P.; Berk, B.C. Oxidative stress and vascular smooth muscle cell growth: A mechanistic linkage by cyclophilin A. Antioxid. Redox Signal. 2010, 12, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases in mechano-transduction: Mechanisms and consequences. Antioxid. Redox Signal. 2014, 20, 887–898. [Google Scholar] [CrossRef] [PubMed]
- San Martin, A.; Griendling, K.K. Redox control of vascular smooth muscle migration. Antioxid. Redox Signal. 2010, 12, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J.; Aalkjaer, C. Structure and function of small arteries. Physiol. Rev. 1990, 70, 921–961. [Google Scholar] [PubMed]
- Martinez-Lemus, L.A. The dynamic structure of arterioles. Basic. Clin. Pharmacol. Toxicol. 2012, 110, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lemus, L.A.; Hill, M.A.; Meininger, G.A. The plastic nature of the vascular wall: A continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology (Bethesda) 2009, 24, 45–57. [Google Scholar] [CrossRef]
- Mulvany, M.J.; Baumbach, G.L.; Aalkjaer, C.; Heagerty, A.M.; Korsgaard, N.; Schiffrin, E.L.; Heistad, D.D. Vascular remodeling. Hypertension 1996, 28, 505–506. [Google Scholar] [PubMed]
- Staiculescu, M.C.; Galinanes, E.L.; Zhao, G.; Ulloa, U.; Jin, M.; Beig, M.I.; Meininger, G.A.; Martinez-Lemus, L.A. Prolonged vasoconstriction of resistance arteries involves vascular smooth muscle actin polymerization leading to inward remodelling. Cardiovasc. Res. 2013, 98, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Vital signs: Prevalence, treatment, and control of hypertension—United States, 1999–2002 and 2005–2008. MMWR Morb. Mortal. Wkly Rep. 2011, 60, 103–108. [Google Scholar]
- Feihl, F.; Liaudet, L.; Levy, B.I.; Waeber, B. Hypertension and microvascular remodelling. Cardiovasc. Res. 2008, 78, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Agabiti-Rosei, E.; Heagerty, A.M.; Rizzoni, D. Effects of antihypertensive treatment on small artery remodelling. J. Hypertens. 2009, 27, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, F.; Vincent, J.M.; Limiñana, P.; Chillon, J.M.; Capdeville-Atkinson, C.; Atkinson, J. Effects of suboptimal doses of the AT1 receptor blocker, telmisartan, with the angiotensin-converting enzyme inhibitor, ramipril, on cerebral arterioles in spontaneously hypertensive rat. J. Hypertens. 2010, 28, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Hassona, M.D. H.; Abouelnaga, Z.A.; Elnakish, M.T.; Awad, M.M.; Alhaj, M.; Goldschmidt-Clermont, P.J.; Hassanain, H.H. Vascular hypertrophy-associated hypertension of profilin1 transgenic mouse model leads to functional remodeling of peripheral arteries. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2112–H2120. [Google Scholar] [CrossRef] [PubMed]
- Izzard, A.S.; Horton, S.; Heerkens, E.H.; Shaw, L.; Heagerty, A.M. Middle cerebral artery structure and distensibility during developing and established phases of hypertension in the spontaneously hypertensive rat. J. Hypertens. 2006, 24, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Louis, H.; Kakou, A.; Regnault, V.; Labat, C.; Bressenot, A.; Gao-Li, J.; Gardner, H.; Thornton, S.N.; Challande, P.; Li, Z.; Lacolley, P. Role of α 1β 1-integrin in arterial stiffness and angiotensin-induced arterial wall hypertrophy in mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2597–H2604. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.S.; Baker, P.N.; Mayhew, T.M.; Dunn, W.R. Remodeling of myometrial radial arteries in preeclampsia. Am. J. Obstet. Gynecol. 2005, 192, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Porteri, E.; de Ciuceis, C.; Rodella, L.F.; Paiardi, S.; Rizzardi, N.; Platto, C.; Boari, G.E.M.; Pilu, A.; Tiberio, G.A.M.; et al. Hypertrophic remodeling of subcutaneous small resistance arteries in patients with Cushing's syndrome. J. Clin. Endocrinol. Metab. 2009, 94, 5010–5018. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Porteri, E.; Giustina, A.; de Ciuceis, C.; Sleiman, I.; Boari, G.E.; Castellano, M.; Muiesan, M.L.; Bonadonna, S.; Burattin, A.; et al. Acromegalic patients show the presence of hypertrophic remodeling of subcutaneous small resistance arteries. Hypertension 2004, 43, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Rossi, G.P.; Porteri, E.; Sticchi, D.; Rodella, L.; Rezzani, R.; Sleiman, I.; de Ciuceis, C.; Paiardi, S.; Bianchi, R.; et al. Bradykinin and matrix metalloproteinases are involved the structural alterations of rat small resistance arteries with inhibition of ACE and NEP. J. Hypertens. 2004, 22, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Porteri, E.; Guefi, D.; Piccoli, A.; Castellano, M.; Pasini, G.; Muiesan, M.L.; Mulvany, M.J.; Rosei, E.A. Cellular hypertrophy in subcutaneous small arteries of patients with renovascular hypertension. Hypertension 2000, 35, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J. Small artery remodelling in hypertension. Basic Clin. Pharmacol. Toxicol. 2012, 110, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Heagerty, A.M.; Aalkjaer, C.; Bund, S.J.; Korsgaard, N.; Mulvany, M.J. Small artery structure in hypertension: Dual processes of remodeling and growth. Hypertension 1993, 21, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Heagerty, A.M.; Heerkens, E.H.; Izzard, A.S. Small artery structure and function in hypertension. J. Cell. Mol. Med. 2010, 14, 1037–1043. [Google Scholar] [PubMed]
- Rizzoni, D.; Porteri, E.; Castellano, M.; Bettoni, G.; Muiesan, M.L.; Muiesan, P.; Giulini, S.M.; Agabiti-Rosei, E. Vascular hypertrophy and remodeling in secondary hypertension. Hypertension 1996, 28, 785–690. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.M.; Baumbach, G.L.; Halabi, C.M.; Modrick, M.L.; Lynch, C.M.; Gerhold, T.D.; Ghoneim, S.M.; de Lange, W.J.; Keen, H.L.; Tsai, Y.S.; et al. Interference with PPARγ signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension 2008, 51, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Porteri, E.; Guelfi, D.; Muiesan, M.L.; Valentini, U.; Cimino, A.; Girelli, A.; Rodella, L.; Bianchi, R.; Sleiman, I.; et al. Structural alterations in subcutaneous small arteries of normotensive and hypertensive patients with non-insulin-dependent diabetes mellitus. Circulation 2001, 103, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Agabiti Rosei, E. Small artery remodeling in hypertension and diabetes. Curr. Hypertens. Rep. 2006, 8, 90–5. [Google Scholar] [CrossRef] [PubMed]
- Vessières, E.; Freidja, M.L.; Loufrani, L.; Fassot, C.; Henrion, D. Flow (shear stress)-mediated remodeling of resistance arteries in diabetes. Vascul. Pharmacol. 2012, 57, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, S.L.; Michelini, L.C. Effect of gender on training-induced vascular remodeling in SHR. Braz. J. Med. Biol. Res. 2011, 44, 814–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipolla, M.J.; Sweet, J.G.; Chan, S.L. Cerebral vascular adaptation to pregnancy and its role in the neurological complications of eclampsia. J. Appl. Physiol. 2011, 110, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Hale, S.A.; Weger, L.; Mandala, M.; Osol, G. Reduced NO signaling during pregnancy attenuates outward uterine artery remodeling by altering MMP expression and collagen and elastin deposition. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1266–H1275. [Google Scholar] [CrossRef] [PubMed]
- Tarhouni, K.; Guihot, A.L.; Freidja, M.L.; Toutain, B.; Henrion, B.; Baufreton, C.; Pinaud, F.; Procaccio, V.; Grimaud, L.; Ayer, A.; et al. Key role of estrogens and endothelial estrogen receptor α in blood flow-mediated remodeling of resistance arteries. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Sonoyama, K.; Greenstein, A.; Price, A.; Khavandi, K.; Heagerty, T. Vascular remodeling: Implications for small artery function and target organ damage. Ther. Adv. Cardiovasc. Dis. 2007, 1, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A.; Meininger, G.A.; Davis, M.J.; Laher, I. Therapeutic potential of pharmacologically targeting arteriolar myogenic tone. Trends Pharmacol. Sci. 2009, 30, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Heerkens, E.H. J.; Izzard, A.S.; Heagerty, A.M. Integrins, vascular remodeling, and hypertension. Hypertension 2007, 49, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tuna, B.G.; Schoorl, M.J.; Bakker, E.N.; de Vos, J.; VanBavel, E. Smooth muscle contractile plasticity in rat mesenteric small arteries: Sensitivity to specific vasoconstrictors, distension and inflammatory cytokines. J. Vasc. Res. 2013, 50, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lemus, L.A.; Galinanes, E.L. Matrix metalloproteinases and small artery remodeling. Drug Discov. Today Dis. Models 2011, 8, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Biological roles for the NOX family NADPH oxidases. J. Biol. Chem. 2008, 283, 16961–16965. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Banfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [PubMed]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid. Redox Signal. 2009, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.H. Tissue distribution and putative physiological function of NOX family NADPH oxidases. Jpn. J. Infect. Dis. 2004, 57, S28–S29. [Google Scholar] [PubMed]
- Lassègue, B.; Griendling, K.K. NADPH Oxidases: Functions and Pathologies in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of nox1 and nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Sumimoto, H. Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie 2007, 89, 1133–44. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of NADPH oxidases: The role of Rac proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Janiszewski, M.; Lopes, L.R.; Carmo, A.O.; Pedro, M.A.; Brandes, R.P.; Santos, C.X.; Laurindo, F.R. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 40813–40819. [Google Scholar] [PubMed]
- Miller, F.J., Jr.; Filali, M.; Huss, G.J.; Stanic, B.; Chamseddine, A.; Barna, T.J.; Lamb, F.S. Cytokine activation of nuclear factor kappa B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ. Res. 2007, 101, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Moreland, J.G.; Davis, A.P.; Matsuda, J.J.; Hook, J.S.; Bailey, G.; Nauseef, W.M.; Lamb, F.S. Endotoxin priming of neutrophils requires NADPH oxidase-generated oxidants and is regulated by the anion transporter ClC-3. J. Biol. Chem. 2007, 282, 33958–33967. [Google Scholar] [CrossRef] [PubMed]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassegue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Manickam, N.; Patel, M.; Griendling, K.K.; Gorin, Y.; Barnes, J.L. RhoA/Rho kinase mediates TGF-β-induced kidney myofibroblast activation through Poldip2/Nox4-derived reactive oxygen species. Am. J. Physiol. Renal. Physiol. 2014, 307. [Google Scholar] [CrossRef]
- Martinez-Lemus, L.A.; Zhao, G.; Galiñanes, E.L.; Boone, M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2005–H2015. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Kreuzer, J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc. Res. 2005, 65, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Schroder, K. Differential vascular functions of Nox family NADPH oxidases. Curr. Opin. Lipidol. 2008, 19, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Martinez-Lemus, L.A. Persistent agonist-induced vasoconstriction is not required for angiotensin II to mediate inward remodeling of isolated arterioles with myogenic tone. J. Vasc. Res. 2008, 45, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Tabet, F.; Callera, G.E.; Montezano, A.C.; Yogi, A.; He, Y.; Quinn, M.T.; Salaices, M.; Touyz, R.M. Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J. Am. Soc. Hypertens. 2011, 5, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Beswick, R.A.; Dorrance, A.M.; Leite, R.; Webb, R.C. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 2001, 38, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Nakano, D.; Katsuragi, M.; Ohkita, M.; Takaoka, M.; Ohno, Y.; Matsumura, Y. Role of gp91phox-containing NADPH oxidase in the deoxycorticosterone acetate-salt-induced hypertension. Eur. J. Pharmcol. 2006, 552, 131–134. [Google Scholar] [CrossRef]
- Polichnowski, A.J.; Jin, C.; Yang, C.; Cowley, A.W. Role of renal perfusion pressure versus angiotensin ii on renal oxidative stress in angiotensin II-induced hypertensive rats. Hypertension 2010, 55, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.S.; Jaimes, E.A.; Raij, L. Vascular but not cardiac remodeling is associated with superoxide production in angiotensin II hypertension. J. Hypertens. 2005, 23, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Bonacasa, B.; Sanchez, M.L.; Rodriguez, F.; Lopez, B.; Quesada, T.; Fenoy, F.J.; Hernandez, I. 2-Methoxyestradiol attenuates hypertension and coronary vascular remodeling in spontaneously hypertensive rats. Maturitas 2008, 61, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Chignalia, A.Z.; Schuldt, E.Z.; Camargo, L.L.; Montezano, A.C.; Callera, G.E.; Laurindo, F.R.; Lopes, L.R.; Avellar, M.C.W.; Carvalho, M.H.C.; Fortes, Z.B.; et al. Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c-Src-dependent pathways. Hypertension 2012, 59, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Diep, Q.N.; Amiri, F.; Touyz, R.M.; Cohn, J.S.; Endemann, D.; Neves, M.F.; Schiffrin, E.L. PPARα activator effects on Ang II-induced vascular oxidative stress and inflammation. Hypertension 2002, 40, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, C.; Rehman, A.; Rautureau, Y.; Kasal, D.A.; Briet, M.; Leibowitz, A.; Simeone, S.M.C.; Ebrahimian, T.; Neves, M.F.; Offermanns, S.; et al. Protective role of vascular smooth muscle cell PPARγ in angiotensin II-induced vascular disease. Cardiovasc. Res. 2013, 97, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M.; Lai, E.Y.; Ma, Z.; Steege, A.; Patzak, A.; Eriksson, U.J.; Lundberg, J.O.; Wilcox, C.S.; Persson, A.E.G. Superoxide dismutase 1 limits renal microvascular remodeling and attenuates arteriole and blood pressure responses to angiotensin II via modulation of nitric oxide bioavailability. Hypertension 2010, 56, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Virdis, A.; Neves, M.F.; Amiri, F.; Touyz, R.M.; Schiffrin, E.L. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J. Hypertens. 2004, 22, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Ohishi, M.; Yamamoto, K.; Tatara, Y.; Shiota, A.; Hayashi, N.; Komai, N.; Yanagitani, Y.; Rakugi, H.; Ogihara, T. Renin-angiotensin inhibition reverses advanced cardiac remodeling in aging spontaneously hypertensive rats. Am. J. Hypertens. 2007, 20, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Rodriguez-Criado, N.; Hernanz, R.; Garcia-Redondo, A.B.; Rodrigues-Diez, R.R.; Alonso, M.J.; Egido, J.; Ruiz-Ortega, M.; Salaices, M. Atorvastatin prevents angiotensin II-induced vascular remodeling and oxidative stress. Hypertension 2009, 54, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-X.; Zeng, H.; Tuo, Q.-H.; Yu, H.; Meyrick, B.; Aschner, J.L. NADPH oxidase modulates myocardial Akt, ERK1/2 activation, and angiogenesis after hypoxia-reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1664–H1674. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kim, K.E.; Koh, G.Y.; Ho, Y.-S.; Lee, K.-J. Hydrogen peroxide produced by angiopoietin-1 mediates angiogenesis. Cancer Res. 2006, 66, 6167–6174. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Kamouchi, M.; Sadoshima, J.; Kitazono, T. Pathophysiological roles of NADPH oxidase/Nox family proteins in the vascular system review and perspective. Circ. J. 2011, 75, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Bir, S.C.; Kolluru, G.K.; Fang, K.; Kevil, C.G. Redox balance dynamically regulates vascular growth and remodeling. Semin. Cell Dev. Biol. 2012, 23, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson-Berka, J.L.; Rana, I.; Armani, R.; Agrotis, A. Reactive oxygen species, Nox and angiotensin II in angiogenesis: Implications for retinopathy. Clin. Sci. 2013, 124, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Catani, M.V.; Bernassola, F.; Rossi, A.; Melino, G. Inhibition of clotting factor XIII activity by nitric oxide. Biochem. Biophys. Res. Commun. 1998, 249, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Kanwar, S.; Niu, X.F.; Gaboury, J.P. Nitric oxide synthesis inhibition induces leukocyte adhesion via superoxide and mast cells. FASEB J. 1993, 7, 1293–1299. [Google Scholar] [PubMed]
- Kurose, I.; Wolf, R.; Grisham, M.B.; Tak Yee, A.; Specian, R.D.; Granger, D.N. Microvascular responses to inhibition of nitric oxide production: Role of active oxidants. Circ. Res. 1995, 76, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Cohen, R.A.; Ullrich, V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium 2004, 11, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Tsihlis, N.D.; Oustwani, C.S.; Vavra, A.K.; Jiang, Q.; Keefer, L.K.; Kibbe, M.R. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of ubcH10. Cell. Biochem. Biophys. 2011, 60, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, C.; Ebrahimian, T.; Angulo, O.; Paradis, P.; Schiffrin, E.L. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension 2009, 54, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Romero, M.J.; Shatanawi, A.; Alkilany, A.M.; Caldwell, R.B.; Caldwell, R.W. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br. J. Pharmacol. 2012, 165, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Berkowitz, D.E.; Ryoo, S. Increased arginase II activity contributes to endothelial dysfunction through endothelial nitric oxide synthase uncoupling in aged mice. Exp. Mol. Med. 2012, 44, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Werner-Felmayer, G.; Werner, E.R.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Schmidt, K.; Weiss, G.; Wachter, H. Pteridine biosynthesis in human endothelial cells. Impact on nitric oxide-mediated formation of cyclic GMP. J. Biol. Chem. 1993, 268, 1842–1846. [Google Scholar] [PubMed]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Kashiwagi, A.; Nishio, Y.; Okamura, T.; Yoshida, Y.; Masada, M.; Toda, N.; Kikkawa, R. Abnormal biopterin metabolism is a major cause of impaired endothelium-dependent relaxation through nitric oxide/O2− imbalance in insulin-resistant rat aorta. Diabetes 1999, 48, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.-J.; Hsiao, G.; Cheng, T.-H.; Yen, M.-H. Supplemention with tetrahydrobiopterin suppresses the development of hypertension in spontaneously hypertensive rats. Hypertension 2001, 38, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Krohn, K.; Albers, S.; Meinertz, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 2000, 43, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Fukuda, Y.; Matsuura, H.; Oshima, T.; Chayama, K. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am. J. Hypertens. 2002, 15, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Milstien, S.; Katusic, Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Somers, M.; Kurz, S.; McCann, L.; Warnholtz, A.; Freeman, B.A.; Tarpey, M.; Fukai, T.; Harrison, D.G. Endothelial regulation of vasomotion in apoE-deficient mice: Implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 2001, 103, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.H.; Guan, Y.Y.; Alp, N.J.; Channon, K.M.; Chen, A.F. Endothelium-specific GTP cyclohydrolase i overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation 2008, 117, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Tatham, A.L.; Hale, A.B.; Alp, N.J.; Channon, K.M. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: Relative importance of the de novo biopterin synthesis versus salvage pathways. J. Biol. Chem. 2009, 284, 28128–28136. [Google Scholar] [CrossRef] [PubMed]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Gao, L.; Cai, H. The p47 phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 2012, 55, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Stipek, S.; Novak, L.; Crkovska, J.; Zima, T.; Platenik, J. Xanthine oxidoreductase. Biochemical, biological and pathogenic functions. Sb. Lek. 1994, 95, 289–295. [Google Scholar] [PubMed]
- Garattini, E.; Mendel, R.; Romão, M.J.; Wright, R.; Terao, M. Mammalian molybdo-flavoenzymes, an expanding family of proteins: Structure, genetics, regulation, function and pathophysiology. Biochem. J. 2003, 372, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, S.; Fujimoto, Y.; Sakamoto, Y.; Uchiyama, T.; Yoshioka, K.; Nishida, H.; Fujita, T. Peroxynitrite induces the conversion of xanthine dehydrogenase to oxidase in rabbit liver. Biochem. Biophys. Res. Commun. 1997, 230, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, Y.; Nishino, T.; Okamoto, K.; Matsumura, T.; Eger, B.T.; Pai, E.F. Unique amino acids cluster for switching from the dehydrogenase to oxidase form of xanthine oxidoreductase. Proc. Natl. Acad. Sci. USA 2003, 100, 8170–8175. [Google Scholar] [CrossRef] [PubMed]
- Kelley, E.E.; Khoo, N.K. H.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [PubMed]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [PubMed]
- Kou, B.; Ni, J.; Vatish, M.; Singer, D.R. Xanthine oxidase interaction with vascular endothelial growth factor in human endothelial cell angiogenesis. Microcirculation 2008, 15, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Jankov, R.P.; Kantores, C.; Pan, J.; Belik, J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L233–L245. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Delano, F.A.; Parks, D.A.; Jamshidi, N.; Granger, D.N.; Ishii, H.; Suematsu, M.; Zweifach, B.W.; Schmid-Schönbein, G.W. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc. Natl. Acad. Sci. USA 1998, 95, 4754–4759. [Google Scholar] [CrossRef] [PubMed]
- Swei, A.; Lacy, F.; Delano, F.A.; Parks, D.A.; Schmid-Schönbein, G.W. A mechanism of oxygen free radical production in the Dahl hypertensive rat. Microcirculation 1999, 6, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.L. H.; Vickers, J.J.; Zhang, Y.; McKenzie, K.U. S.; Walsh, C.E.; Whitworth, J.A. Role of xanthine oxidase in dexamethasone-induced hypertension in rats. Clin. Exp. Pharmacol. Physiol. 2007, 34, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, Y.; Wu, C.; Gunst, S.J. Integrin-linked kinase regulates N-WASp-mediated actin polymerization and tension development in tracheal smooth muscle. J. Biol. Chem. 2007, 282, 34568–34580. [Google Scholar] [CrossRef] [PubMed]
- Lustgarten, M.S.; Bhattacharya, A.; Muller, F.L.; Jang, Y.C.; Shimizu, T.; Shirasawa, T.; Richardson, A.; van Remmen, H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 2012, 422, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Percolation and criticality in a mitochondrial network. Proc. Natl. Acad. Sci. USA 2004, 101, 4447–4452. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; Zheng, Y.-M.; Niu, C.-F.; Liu, Q.-H.; Korde, A.; Ho, Y.-S.; Wang, Y.-X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCɛ signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Viel, E.C.; Benkirane, K.; Javeshghani, D.; Touyz, R.M.; Schiffrin, E.L. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H281–H288. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Tostes, R.C.; Webb, R.C. Mitochondrial aldehyde dehydrogenase prevents ROS-induced vascular contraction in angiotensin-II hypertensive mice. J. Am. Soc. Hypertens. 2011, 5, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.N.; Shi, N.; Chen, S.Y. Manganese superoxide dismutase inhibits neointima formation through attenuation of migration and proliferation of vascular smooth muscle cells. Free Radic. Biol. Med. 2012, 52, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Intengan, H.D.; Schiffrin, E.L. Vascular remodeling in hypertension: Roles of apoptosis, inflammation, and fibrosis. Hypertension 2001, 38, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Kumai, Y.; Ooboshi, H.; Ago, T.; Ishikawa, E.; Takada, J.; Kamouchi, M.; Kitazono, T.; Ibayashi, S.; Iida, M. Protective effects of angiotensin II Type 1 receptor blocker on cerebral circulation independent of blood pressure. Exp. Neurol. 2008, 210, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Bonacasa, B.; Hernández, I.; Fenoy, F.J.; Quesada, T.; López, B. Effect of tempol on myocardial vascular remodeling in female spontaneously hypertensive rats. Histol. Histopathol. 2012, 27, 1047–1054. [Google Scholar] [PubMed]
- Pires, P.W.; Deutsch, C.; McClain, J.L.; Rogers, C.T.; Dorrance, A.M. Tempol, a superoxide dismutase mimetic, prevents cerebral vessel remodeling in hypertensive rats. Microvasc. Res. 2010, 80, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: Role of phospholipase d-dependent NAD(P)H oxidase-sensitive pathways. J. Hypertens. 2001, 19, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Belin De Chantemèle, E.J.; Vessières, E.; Guihot, A.L.; Toutain, B.; Maquignau, M.; Loufrani, L.; Henrion, D. Type 2 diabetes severely impairs structural and functional adaptation of rat resistance arteries to chronic changes in blood flow. Cardiovasc. Res. 2009, 81, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Baron-Menguy, C.; Toutain, B.; Cousin, M.; Dumont, O.; Guihot, A.L.; Vessières, E.; Subra, J.F.; Custaud, M.A.; Loufrani, L.; Henrion, D. Involvement of angiotensin II in the remodeling induced by a chronic decrease in blood flow in rat mesenteric resistance arteries. Hypertens. Res. 2010, 33, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freidja, M.L.; Vessieres, E.; Clere, N.; Desquiret, V.; Guihot, A.L.; Toutain, B.; Loufrani, L.; Jardel, A.; Procaccio, V.; Faure, S.; et al. Heme oxygenase-1 induction restores high-blood-flow-dependent remodeling and endothelial function in mesenteric arteries of old rats. J. Hypertens. 2011, 29, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freidja, M.L.; Toutain, B.; Caillon, A.; Desquiret, V.; Lambert, D.; Loufrani, L.; Procaccio, V.; Henrion, D. Heme oxygenase 1 is differentially involved in blood flow-dependent arterial remodeling: Role of inflammation, oxidative stress, and nitric oxide. Hypertension 2011, 58, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Cousin, M.; Custaud, M.A.; Baron-Menguy, C.; Toutain, B.; Dumont, O.; Guihot, A.L.; Vessieres, E.; Subra, J.F.; Henrion, D.; Loufrani, L. Role of angiotensin II in the remodeling induced by a chronic increase in flow in rat mesenteric resistance arteries. Hypertension 2010, 55, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, C.; de Chantemèle, E.B.; Guihot, A.L.; Vessières, E.; Bocquet, A.; Dumont, O.; Jardel, A.; Loufrani, L.; Moreau, P.; Henrion, D. Flow-induced remodeling in resistance arteries from obese Zucker rats is associated with endothelial dysfunction. Hypertension 2007, 50, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Belin De Chanteméle, E.; Vessiéres, E.; Dumont, O.; Guihot, A.L.; Toutain, B.; Loufrani, L.; Henrion, D. Reactive oxygen species are necessary for high flow (shear Stress)-induced diameter enlargement of rat resistance arteries. Microcirculation 2009, 16, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.; Miao, S.B.; Shu, Y.N.; Dong, L.H.; Liu, G.; Xie, X.L.; Gao, M.; Wang, Y.C.; Yin, Y.J.; Wang, X.J.; et al. Phosphorylation of smooth muscle 22α facilitates angiotensin II-induced ROS production via activation of the PKCδ-P47phox axis through release of PKCδ and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circ. Res. 2012, 111, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Alom-Ruiz, S.P.; Wang, M.; Zhang, M.; Walker, S.; Yu, B.; Brewer, A.; Shah, A.M. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res. Cardiol. 2011, 106, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Sakurada, T.; Ishizawa, K.; Imanishi, M.; Izawa-Ishizawa, Y.; Fujii, S.; Tominaga, E.; Tsuneishi, T.; Horinouchi, Y.; Kihira, Y.; Ikeda, Y.; et al. Nitrosonifedipine ameliorates angiotensin II-induced vascular remodeling via antioxidative effects. Naunyn. Schmiedebergs Arch. Pharmacol. 2013, 386, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.N.T.P.; Matlung, H.L.; Bonta, P.; de Vries, C.J.; van Rooijen, N.; Vanbavel, E. Blood flow-dependent arterial remodelling is facilitated by inflammation but directed by vascular tone. Cardiovasc. Res. 2008, 78, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pakeerappa, P.; Lee, H.J.; Fisher, S.A. Induction of PDE5 and de-sensitization to endogenous NO signaling in a systemic resistance artery under altered blood flow. J. Mol. Cell. Cardiol. 2009, 47, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Doyle, J.L.; Distasi, M.R.; Norton, L.E.; Sheridan, K.M.; Unthank, J.L. Involvement of MMPs in the outward remodeling of collateral mesenteric arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2429–H2437. [Google Scholar] [CrossRef] [PubMed]
- Castier, Y.; Brandes, R.P.; Leseche, G.; Tedgui, A.; Lehoux, S. p47phox-dependent NADPH oxidase regulates flow-induced vascular remodeling. Circ. Res. 2005, 97, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Freidja, M.L.; Tarhouni, K.; Toutain, B.; Fassot, C.; Loufrani, L.; Henrion, D. The AGE-breaker ALT-711 restores high blood flow-dependent remodeling in mesenteric resistance arteries in a rat model of type 2 diabetes. Diabetes 2012, 61, 1562–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Mann, G.E. Vascular NAD(P)H oxidase activation in diabetes: A double-edged sword in redox signalling. Cardiovasc. Res. 2009, 82, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Rensen, S.S.M.; Doevendans, P.A.F.M.; van Eys, G.J.J.M. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Heart J. 2007, 15, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.Y.; Fukuda, N.; Kanmatsuse, K. Growth characteristics, angiotensin II generation, and microarray-determined gene expression in vascular smooth muscle cells from young spontaneously hypertensive rats. J. Hypertens. 2002, 20, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.H.; Fukuda, N.; Jin, X.Q.; Yao, E.H.; Ueno, T.; Endo, M.; Saito, S.; Matsumoto, K.; Mugishima, H. Complement 3 is involved in the synthetic phenotype and exaggerated growth of vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension 2004, 44, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Lin, J.X.; Takahashi, R.; Tomimoto, H. Cilostazol alleviates cerebral small-vessel pathology and white-matter lesions in stroke-prone spontaneously hypertensive rats. Brain Res. 2008, 1203, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Saito, Y.; Hamada, K.; Hara, N.; Nakanishi, K.; Masaki, K.; Tanaka, M.; Ger, Y.C.; Nakamura, K. Renal vascular walls in patients with preeclampsia superimposed on essential hypertension. Am. J. Kidney Dis. 2001, 37, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Quarck, R.; Wynants, M.; Ronisz, A.; Sepulveda, M.R.; Wuytack, F.; van Raemdonck, D.; Meyns, B.; Delcroix, M. Characterization of proximal pulmonary arterial cells from chronic thromboembolic pulmonary hypertension patients. Respir. Res. 2012, 13. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lemus, L.A.; Hill, M.A.; Bolz, S.S.; Pohl, U.; Meininger, G.A. Acute mechanoadaptation of vascular smooth muscle cells in response to continuous arteriolar vasoconstriction: Implications for functional remodeling. FASEB J. 2004, 18, 708–710. [Google Scholar] [PubMed]
- De la Cuesta, F.; Zubiri, I.; Maroto, A.S.; Posada, M.; Padial, L.R.; Vivanco, F.; Alvarez-Llamas, G.; Barderas, M.G. Deregulation of smooth muscle cell cytoskeleton within the human atherosclerotic coronary media layer. J. Proteomics 2013, 82, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Umemoto, S.; Hiromoto, M.; Toma, Y.; Tomochika, Y.; Aoyagi, S.; Tanaka, M.; Fujii, T.; Matsuzaki, M. Importance of NAD(P)H oxidase-mediated oxidative stress and contractile type smooth muscle myosin heavy chain SM2 at the early stage of atherosclerosis. Circulation 2002, 105, 2288–2295. [Google Scholar] [CrossRef] [PubMed]
- Absood, A.; Furutani, A.; Kawamura, T.; Graham, L.M. Differential PDGF secretion by graft and aortic SMC in response to oxidized LDL. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H725–H732. [Google Scholar] [PubMed]
- Ashino, T.; Yamamoto, M.; Yoshida, T.; Numazawa, S. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Cardneau, J.D.; Colles, S.M.; Graham, L.M. Synthetic smooth muscle cell phenotype is associated with increased nicotinamide adenine dinucleotide phosphate oxidase activity: Effect on collagen secretion. J. Vasc. Surg. 2006, 43, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Touré, F.; Fritz, G.; Li, Q.; Rai, V.; Daffu, G.; Zou, Y.S.; Rosario, R.; Ramasamy, R.; Alberts, A.S.; Yan, S.F.; et al. Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ. Res. 2012, 110, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.J.; Eskin, S.G.; Sakurai, Y.; Yee, A.; Kataoka, N.; McIntire, L.V. Oxidative stress produced with cell migration increases synthetic phenotype of vascular smooth muscle cells. Ann. Biomed. Eng. 2005, 33, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Mitra, S.; Gregg, H.; Flavahan, S.; Chotani, M.A.; Clark, K.R.; Goldschmidt-Clermont, P.J.; Flavahan, N.A. Redox regulation of vascular smooth muscle cell differentiation. Circ. Res. 2001, 89, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Griendling, K.K. Redox signaling, vascular function, and hypertension. Antioxid. Redox Signal. 2008, 10, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.H.W.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Clempus, R.E.; Sorescu, D.; Dikalova, A.E.; Pounkova, L.; Jo, P.; Sorescu, G.P.; Lassègue, B.; Griendling, K.K. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Zuckerbraun, B.S.; Stoyanovsky, D.A.; Sengupta, R.; Shapiro, R.A.; Ozanich, B.A.; Rao, J.; Barbato, J.E.; Tzeng, E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am. J. Physiol.Cell. Physiol. 2007, 292, C824–C831. [Google Scholar] [CrossRef] [PubMed]
- Majesky, M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Facemire, C.S.; Banes, A.J.; Faber, J.E. Different alpha-adrenoceptors mediate migration of vascular smooth muscle cells and adventitial fibroblasts in vitro. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2364–H2370. [Google Scholar] [PubMed]
- Nishio, E.; Watanabe, Y. The involvement of reactive oxygen species and arachidonic acid in alpha 1-adrenoceptor-induced smooth muscle cell proliferation and migration. Br. J. Pharmacol 1997, 121, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.P.; Graf, K.; Goetze, S.; Fleck, E.; Hsueh, W.A.; Law, R.E. Central role of the MAPK pathway in ang II-mediated DNA synthesis and migration in rat vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Hwangbo, C.; Lee, S.; Lee, J.H. Eupatolide inhibits PDGF-induced proliferation and migration of aortic smooth muscle cells through ROS-dependent heme oxygenase-1 induction. Phytother. Res. 2013, 27, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Castresana, M.R.; Newman, W.H. Reactive oxygen and NF-κB in VEGF-induced migration of human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2001, 285, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Castresana, M.R.; Newman, W.H. Reactive oxygen species-sensitive p38 MAPK controls thrombin-induced migration of vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2004, 36, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Shriver, A.S.; Jagadeesha, D.K.; Chamseddine, A.H.; Szócs, K.; Weintraub, N.L.; Griendling, K.K.; Bhalla, R.C.; Miller, F.J., Jr. Increased expression of Nox1 in neointimal smooth muscle cells promotes activation of matrix metalloproteinase-9. J. Vasc. Res. 2012, 49, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Lau, Y.T. Migration of vascular smooth muscle cells is enhanced in cultures derived from spontaneously hypertensive rat. Pflugers Arch. 1998, 435, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Kim, H.J.; Won, K.J.; Choi, W.S.; Lee, K.Y.; Bae, Y.M.; Park, P.J.; Park, T.K.; Lee, Y.L.; Lee, C.K.; et al. Contribution of soluble intercellular adhesion molecule-1 to the migration of vascular smooth muscle cells. Eur. J. Pharmcol. 2008, 579, 260–268. [Google Scholar] [CrossRef]
- Zhang, L.; Xie, P.; Wang, J.; Yang, Q.; Fang, C.; Zhou, S.; Li, J. Impaired peroxisome proliferator-activated receptor-γ contributes to phenotypic modulation of vascular smooth muscle cells during hypertension. J. Biol. Chem. 2010, 285, 13666–13677. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kyotani, Y.; Ito, S.; Nagayama, K.; Ozawa, K.; Yoshizumi, M. Comparison of the effects of Src inhibition and mitogen-activated protein kinase inhibition on the migration of vascular smooth muscle cells stimulated by angiotensin II. J. Nara Med. Assoc. 2012, 63, 25–35. [Google Scholar]
- Panchatcharam, M.; Miriyala, S.; Yang, F.; Leitges, M.; Chrzanowska-Wodnicka, M.; Quilliam, L.A.; Anaya, P.; Morris, A.J.; Smyth, S.S. Enhanced proliferation and migration of vascular smooth muscle cells in response to vascular injury under hyperglycemic conditions is controlled by β3 integrin signaling. Int. J. Biochem. Cell. Biol. 2010, 42, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Nesterova, A.; Burant, C.F.; Klemm, D.J.; Reusch, J.E.B. Diabetes-related changes in cAMP response element-binding protein content enhance smooth muscle cell proliferation and migration. J. Biol. Chem. 2001, 276, 46142–46150. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Igarashi, M.; Hirata, A.; Sugae, N.; Tsuchiya, H.; Jimbu, Y.; Tominaga, M.; Kato, T. Altered PDGF-BB-induced p38 MAP kinase activation in diabetic vascular smooth muscle cells: Roles of protein kinase C-δ. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2095–2101. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Abhijit, S.; Bhaskaran, R.; Narayanasamy, A.; Chakroborty, A.; Manickam, N.; Dixit, M.; Mohan, V.; Balasubramanyam, M. Hyperinsulinemia-induced vascular smooth muscle cell (VSMC) migration and proliferation is mediated by converging mechanisms of mitochondrial dysfunction and oxidative stress. Mol. Cell. Biochem. 2013, 373, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesha, D.K.; Takapoo, M.; Banfi, B.; Bhalla, R.C.; Miller, F.J., Jr. Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovasc. Res. 2012, 93, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Mac Gabhann, F.; Peirce, S.M. Collateral capillary arterialization following arteriolar ligation in murine skeletal muscle. Microcirculation 2010, 17, 333–347. [Google Scholar] [PubMed]
- Unthank, J.L.; Fath, S.W.; Burkhart, H.M.; Miller, S.C.; Dalsing, M.C. Wall remodeling during luminal expansion of mesenteric arterial collaterals in the rat. Circ. Res. 1996, 79, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Cai, W.J.; Vosschulte, R.; Koltai, S.; Mousavipour, D.; Scholz, D.; Afsah-Hedjri, A.; Schaper, W.; Schaper, J. Vascular remodeling and altered protein expression during growth of coronary collateral arteries. J. Mol. Cell. Cardiol. 1998, 30, 2291–305. [Google Scholar] [CrossRef] [PubMed]
- Gerthoffer, W.T. Mechanisms of vascular smooth muscle cell migration. Circ. Res. 2007, 100, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Sun, Z.; Li, M.; Li, Z.; Bunyak, F.; Ersoy, I.; Trzeciakowski, J.P.; Staiculescu, M.C.; Jin, M.; Martinez-Lemus, L.; et al. Vasoactive agonists exert dynamic and coordinated effects on vascular smooth muscle cell elasticity, cytoskeletal remodelling and adhesion. J. Physiol. 2014, 592, 1249–1266. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Sun, Z.; Li, Z.; Mesquitta, W.T.; Trzeciakowski, J.P.; Meininger, G.A. Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovasc. Res. 2012, 96, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Yamin, R.; Morgan, K.G. Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J. Physiol. 2012, 590, 4145–4154. [Google Scholar] [CrossRef] [PubMed]
- Lehman, W.; Morgan, K.G. Structure and dynamics of the actin-based smooth muscle contractile and cytoskeletal apparatus. J. Muscle Res. Cell. Motil. 2012, 33, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Castorena-Gonzalez, J.A.; Staiculescu, M.C.; Foote, C.A.; Polo-Parada, L.; Martinez-Lemus, L.A. The obligatory role of the actin cytoskeleton on inward remodeling induced by dithiothreitol activation of endogenous transglutaminase in isolated arterioles. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H485–H495. [Google Scholar] [CrossRef] [PubMed]
- Gunst, S.J.; Zhang, W. Actin cytoskeletal dynamics in smooth muscle: A new paradigm for the regulation of smooth muscle contraction. Am. J. Physiol. Cell. Physiol. 2008, 295, C576–C587. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.E.; Milliken, E.E.; Irani, K.; Zweier, J.L.; Becker, L.C.; Johnson, T.M.; Eissa, N.T.; Crystal, R.G.; Finkel, T.; Goldschmidt-Clermont, P.J. Superoxide-mediated actin response in post-hypoxic endothelial cells. J. Biol. Chem. 1996, 271, 26863–26867. [Google Scholar] [CrossRef] [PubMed]
- Zweier, J.L.; Broderick, R.; Kuppusamy, P.; Thompson-Gorman, S.; Lutty, G.A. Determination of the mechanism of free radical generation in human aortic endothelial cells exposed to anoxia and reoxygenation. J. Biol. Chem. 1994, 269, 24156–24162. [Google Scholar] [PubMed]
- Moldovan, L.; Moldovan, N.I.; Sohn, R.H.; Parikh, S.A.; Goldschmidt-Clermont, P.J. Redox changes of cultured endothelial cells and actin dynamics. Circ. Res. 2000, 86, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Firat-Karalar, E.N.; Welch, M.D. New mechanisms and functions of actin nucleation. Curr. Opin. Cell. Biol. 2011, 23, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, Y.; Du, L.; Tang, D.D.; Gunst, S.J. Activation of the Arp2/3 complex by N-WASp is required for actin polymerization and contraction in smooth muscle. Am. J. Physiol. Cell. Physiol. 2005, 288, C1145–C1460. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.D.; Zhang, W.; Gunst, S.J. The adapter protein CrkII regulates neuronal Wiskott-Aldrich syndrome protein, actin polymerization, and tension development during contractile stimulation of smooth muscle. J. Biol. Chem. 2005, 280, 23380–23389. [Google Scholar] [CrossRef] [PubMed]
- Anfinogenova, Y.; Wang, R.; Li, Q.F.; Spinelli, A.M.; Tang, D.D. Abl silencing inhibits CAS-mediated process and constriction in resistance arteries. Circ. Res. 2007, 101, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Al Ghouleh, I.; Rodriguez, A.; Pagano, P.J.; Csanyi, G. Proteomic analysis identifies an NADPH oxidase 1 (Nox1)-mediated role for actin-related protein 2/3 complex subunit 2 (ARPC2) in promoting smooth muscle cell migration. Int. J. Mol. Sci. 2013, 14, 20220–20235. [Google Scholar] [CrossRef] [PubMed]
- Kaverina, I.; Stradal, T.E.; Gimona, M. Podosome formation in cultured A7r5 vascular smooth muscle cells requires Arp2/3-dependent de-novo actin polymerization at discrete microdomains. J. Cell Sci. 2003, 116, 4915–4924. [Google Scholar] [CrossRef] [PubMed]
- DalleDonne, I.; Milzani, A.; Colombo, R. H2O2-treated actin: Assembly and polymer interactions with cross-linking proteins. Biophys. J. 1995, 69, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Milzani, A.; DalleDonne, I.; Colombo, R. Prolonged oxidative stress on actin. Arch. Biochem. Biophys. 1997, 339, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.; Houle, F.; Marceau, F.; Landry, J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ. Res. 1997, 80, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, A.; Wittchen, E.S.; Campbell, S.L.; Burridge, K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One 2009, 4, e8045. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell. Dev. Biol. 2005, 21, 247–69. [Google Scholar] [CrossRef] [PubMed]
- Ten Klooster, J.P.; Evers, E.E.; Janssen, L.; Machesky, L.M.; Michiels, F.; Hordijk, P.; Collard, J.G. Interaction between Tiam1 and the Arp2/3 complex links activation of Rac to actin polymerization. Biochem. J. 2006, 397, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Millius, A.; Watanabe, N.; Weiner, O.D. Diffusion, capture and recycling of SCAR/WAVE and Arp2/3 complexes observed in cells by single-molecule imaging. J. Cell. Sci 2012, 125, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, A.; Goodwin, M.; Verma, S.; Yap, A.S.; Ali, R.G. Rac is a dominant regulator of cadherin-directed actin assembly that is activated by adhesive ligation independently of Tiam1. Am. J. Physiol. Cell. Physiol. 2007, 292, C1061–C1069. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Schlondorff, J.S.; Brown, E.J.; Higgs, H.N.; Pollak, M.R. Rho activation of mDia formins is modulated by an interaction with inverted formin 2 (INF2). Proc. Natl. Acad. Sci. USA 2011, 108, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Otomo, C.; Tomchick, D.R.; Machius, M.; Rosen, M.K. Structural basis of Rho GTPase-mediated activation of the formin mDia1. Mol. Cell. 2005, 18, 273–2781. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.D.; Gunst, S.J. The small GTPase Cdc42 regulates actin polymerization and tension development during contractile stimulation of smooth muscle. J. Biol. Chem. 2004, 279, 51722–51728. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Ying, Z.; Webb, R.C. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am. J. Physiol Heart Circ. Physiol. 2004, 287, H1495–H1500. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Walker, B.R.; Resta, T.C. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L515–L529. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Nugent, W.H.; Mahavadi, S.; Walsh, S.W. Mechanisms of enhanced vascular reactivity in preeclampsia. Hypertension 2011, 58, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.F.; Tang, D.D. Role of p47(phox) in regulating Cdc42GAP, vimentin, and contraction in smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2009, 297, C1424–C1433. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.F.; Spinelli, A.M.; Tang, D.D. Cdc42GAP, reactive oxygen species, and the vimentin network. Am. J. Physiol. Cell. Physiol. 2009, 297, C299–C309. [Google Scholar] [CrossRef] [PubMed]
- Li, P.F.; Dietz, R.; von Harsdorf, R. Differential effect of hydrogen peroxide and superoxide anion on apoptosis and proliferation of vascular smooth muscle cells. Circulation 1997, 96, 3602–3609. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.N.; Berk, B.C. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ. Res. 1992, 70, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, N.N.; Sorescu, D.; Seshiah, P.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Griendling, K.K. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid. Redox Signal. 2002, 4, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, W.; Su, J.; Liu, W.; Altura, B.T.; Altura, B.M. Hydrogen peroxide induces apoptosis in cerebral vascular smooth muscle cells: Possible relation to neurodegenerative diseases and strokes. Brain Res. Bull. 2003, 62, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K.K. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension 1998, 32, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Al Ghouleh, I.; Frazziano, G.; Rodriguez, A.I.; Csányi, G.; Maniar, S.; St Croix, C.M.; Kelley, E.E.; Egaña, L.A.; Song, G.J.; Bisello, A.; et al. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc. Res. 2013, 97, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Okada, M.; Hara, Y.; Yamawaki, H. Death-associated protein kinase 3 mediates vascular inflammation and development of hypertension in spontaneously hypertensive rats. Hypertension 2012, 60, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.D.; Johns, D.G.; Xu, S.; Cohen, R.A. Role of superoxide anion in regulating pressor and vascular hypertrophic response to angiotensin II. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1697–H1702. [Google Scholar] [PubMed]
- Zhang, Y.; Griendling, K.K.; Dikalova, A.; Owens, G.K.; Taylor, W.R. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension 2005, 46, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Matesanz, N.; Lafuente, N.; Azcutia, V.; Martín, D.; Cuadrado, A.; Nevado, J.; Rodríguez-Mañas, L.; Sánchez-Ferrer, C.F.; Peiró, C. Xanthine oxidase-derived extracellular superoxide anions stimulate activator protein 1 activity and hypertrophy in human vascular smooth muscle via c-Jun N-terminal kinase and p38 mitogen-activated protein kinases. J. Hypertens. 2007, 25, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Ushio-Fukai, M.; Hitomi, H.; Rocic, P.; Kingsley, M.J.; Pfahnl, C.; Weber, D.S.; Alexander, R.W.; Griendling, K.K. Role of p38 MAPK and MAPKAPK-2 in angiotensin II-induced Akt activation in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2004, 287, C494–C499. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Griendling, K.K. p38 mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J. Biol. Chem. 1998, 273, 15022–15029. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Yang, H.; Motley, E.D.; Guo, Z. Overexpression of Cu/Zn-superoxide dismutase and/or catalase in mice inhibits aorta smooth muscle cell proliferation. Am. J. Hypertens. 2004, 17, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Whaley-Connell, A.T.; Chen, K.; Habibi, J.; Uptergrove, G.M.E.; Clark, S.E.; Stump, C.S.; Ferrario, C.M.; Sowers, J.R. NADPH oxidase contributes to vascular inflammation, insulin resistance, and remodeling in the transgenic (mRen2) rat. Hypertension 2007, 50, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Mactaggart, J.; Knispel, R.; Worth, J.; Zhu, Z.; Li, Y.; Sun, Y.; Baxter, B.T.; Johanning, J. Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model. Atherosclerosis 2009, 202, 128–34. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Rodella, L.; Porteri, E.; Rezzani, R.; Guelfi, D.; Piccoli, A.; Castellano, M.; Muiesan, M.L.; Bianchi, R.; Rosei, E.A. Time course of apoptosis in small resistance arteries of spontaneously hypertensive rats. J. Hypertens. 2000, 18, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Diep, Q.N.; Li, J.S.; Schiffrin, E.L. In vivo study of AT1 and AT2 angiotensin receptors in apoptosis in rat blood vessels. Hypertension 1999, 34, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Irani, K. Oxidant signaling in vascular cell growth, death, and survival: A review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ. Res. 2000, 87, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ. Res. 1998, 83, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhang, Y.; Tuck, W.S.; Li, S. Urocortin II inhibits the apoptosis of mesenteric arterial smooth muscle cells via L-type calcium channels in spontaneously hypertensive rats. Cell. Physiol. Biochem. 2006, 17, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.F.; Tran, E.D.; Fortes, Z.B.; Schmid-Schönbein, G.W. Matrix metalloproteinases cleave the β2-adrenergic receptor in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H25–H35. [Google Scholar] [CrossRef] [PubMed]
- Abdalvand, A.; Morton, J.S.; Bourque, S.L.; Quon, A.L.; Davidge, S.T. Matrix metalloproteinase enhances big-endothelin-1 constriction in mesenteric vessels of pregnant rats with reduced uterine blood flow. Hypertension 2013, 61, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, P.; Libert, C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Martín, A.; Jiménez-Altayó, F.; Dantas, A.P.; Caracuel, L.; Planas, A.M.; Vila, E. Middle cerebral artery alterations in a rat chronic hypoperfusion model. J. Appl. Physiol. 2012, 112, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Spiers, J.P.; Kelso, E.J.; Siah, W.F.; Edge, G.; Song, G.; McDermott, B.J.; Hennessy, M. Alterations in vascular matrix metalloproteinase due to ageing and chronic hypertension: Effects of endothelin receptor blockade. J. Hypertens. 2005, 23, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, C.; Dentali, F.; Nicolini, E.; Maresca, A.M.; Tayebjee, M.H.; Franz, M.; Guasti, L.; Venco, A.; Schiffrin, E.L.; Lip, G.Y.H.; et al. Plasma levels of matrix metalloproteinases and their inhibitors in hypertension: A systematic review and meta-analysis. J. Hypertens. 2012, 30, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.E.; Vrabas, I.S.; Sailer, N.; Kapelouzou, A.; Fotiadis, G.; Noussios, G.; Karayannacos, P.E.; Angelopoulou, N. Exercise ameliorates serum MMP-9 and TIMP-2 levels in patients with type 2 diabetes. Diabetes Metab. 2010, 36, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Tayebjee, M.H.; Karalis, I.; Nadar, S.K.; Beevers, D.G.; MacFadyen, R.J.; Lip, G.Y.H. Circulating matrix metalloproteinase-9 and tissue inhibitors of metalloproteinases-1 and -2 levels in gestational hypertension. Am. J. Hypertens. 2005, 18, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Fontana, V.; Silva, P.S.; Belo, V.A.; Antonio, R.C.; Ceron, C.S.; Biagi, C.; Gerlach, R.F.; Tanus-Santos, J.E. Consistent alterations of circulating matrix metalloproteinases levels in untreated hypertensives and in spontaneously hypertensive rats: A relevant pharmacological target. Basic Clin. Pharmacol. Toxicol. 2011, 109, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Gutierrez, G.; Cappello, R.E.; Mishra, N.; Romero, R.; Strauss, J.F., III; Walsh, S.W. Increased expression of matrix metalloproteinase-1 in systemic vessels of preeclamptic women: A critical mediator of vascular dysfunction. Am. J. Pathol. 2011, 178, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Luan, R.; Zhang, H.; Lau, W.B.; Wang, Q.; Zhang, Y.; Wang, H.C.; Tao, L. Hydrogen peroxide enhances osteopontin expression and matrix metalloproteinase activity in aortic vascular smooth muscle cells. Clin. Exp. Pharmacol. Physiol. 2009, 36, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Chow, F.L.; Hao, L.; Wang, X.; Nishimura, T.; MacLeod, K.M.; McNeill, J.H.; Fernandez-Patron, C. Maintenance of adrenergic vascular tone by MMP transactivation of the EGFR requires PI3K and mitochondrial ATP synthesis. Cardiovasc. Res. 2009, 84, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Kamioka, M.; Ishibashi, T.; Ohkawara, H.; Nagai, R.; Sugimoto, K.; Uekita, H.; Matsui, T.; Yamagishi, S.I.; Ando, K.; Sakamoto, T; et al. Involvement of membrane type 1-matrix metalloproteinase (MT1-MMP) in RAGE activation signaling pathways. J. Cell. Physiol. 2011, 226, 1554–1563. [Google Scholar] [CrossRef] [PubMed]
- Valentin, F.; Bueb, J.L.; Kieffer, P.; Tschirhart, E.; Atkinson, J. Oxidative stress activates MMP-2 in cultured human coronary smooth muscle cells. Fundam. Clin. Pharmacol. 2005, 19, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Browatzki, M.; Larsen, D.; Pfeiffer, C.A. M.; Gehrke, S.G.; Schmidt, J.; Kranzhöfer, A.; Katus, H.A.; Kranzhöfer, R. Angiotensin II stimulates matrix metalloproteinase secretion in human vascular smooth muscle cells via nuclear factor-κB and activator protein 1 in a redox-sensitive manner. J. Vasc. Res. 2005, 42, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Luchtefeld, M.; Grote, K.; Grothusen, C.; Bley, S.; Bandlow, N.; Selle, T.; Struber, M.; Haverich, A.; Bavendiek, U.; Drexler, H.; et al. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochem. Biophys. Res. Commun. 2005, 328, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.S.; Wang, S.Q. Salvianolic acid B from Salvia miltiorrhiza inhibits tumor necrosis factor-α (TNF-α)-induced MMP-2 upregulation in human aortic smooth muscle cells via suppression of NAD(P)H oxidase-derived reactive oxygen species. J. Mol. Cell. Cardiol. 2006, 41, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.M.; Rizzi, E.; Ceron, C.S.; Guimaraes, D.A.; Rodrigues, G.J.; Bendhack, L.M.; Gerlach, R.F.; Tanus-Santos, J.E. Doxycycline ameliorates 2K-1C hypertension-induced vascular dysfunction in rats by attenuating oxidative stress and improving nitric oxide bioavailability. Nitric Oxide 2012, 26, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Cau, S.B. A.; Guimaraes, D.A.; Rizzi, E.; Ceron, C.S.; Souza, L.L.; Tirapelli, C.R.; Gerlach, R.F.; Tanus-Santos, J.E. Pyrrolidine dithiocarbamate down-regulates vascular matrix metalloproteinases and ameliorates vascular dysfunction and remodelling in renovascular hypertension. Br. J. Pharmacol. 2011, 164, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.M.; Rizzi, E.; Rodrigues, G.J.; Ceron, C.S.; Bendhack, L.M.; Gerlach, R.F; Tanus-Santos, J.E. Antioxidant treatment reduces matrix metalloproteinase-2-induced vascular changes in renovascular hypertension. Free Radic. Biol. Med. 2009, 46, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Feihl, F.; Liaudet, L.; Waeber, B.; Levy, B.I. Hypertension: A disease of the microcirculation? Hypertension 2006, 48, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Vogt, C.J.; Schmid-Schönbein, G.W. Microvascular endothelial cell death and rarefaction in the glucocorticoid-induced hypertensive rat. Microcirculation 2001, 8, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Gobé, G.; Browning, J.; Howard, T.; Hogg, N.; Winterford, C.; Cross, R. Apoptosis occurs in endothelial cells during hypertension-induced microvascular rarefaction. J. Struct. Biol. 1997, 118, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Dikalov, S.I. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid. Redox Signal. 2007, 9, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.N.; Geng, Y.J.; Li, F.; Yang, T.; Su, D.F.; Duan, J.L.; Li, Y. Insulin-like growth factor-1 receptor activation prevents hydrogen peroxide-induced oxidative stress, mitochondrial dysfunction and apoptosis. Apoptosis 2011, 16, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Nako, H.; Kataoka, K.; Koibuchi, N.; Dong, Y.F.; Toyama, K.; Yamamoto, E.; Yasuda, O.; Ichijo, H.; Ogawa, H.; Kim-Mitsuyama, S. Novel mechanism of angiotensin II-induced cardiac injury in hypertensive rats: the critical role of ASK1 and VEGF. Hypertens. Res. 2012, 35, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; DeLano, F.A.; Schmid-Schönbein, G.W. Oxidative stress promotes endothelial cell apoptosis and loss of microvessels in the spontaneously hypertensive rats. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.J.; Norton, L.E.; Murphy, M.P.; Dalsing, M.C.; Unthank, J.L. The role of the renin-angiotensin system and oxidative stress in spontaneously hypertensive rat mesenteric collateral growth impairment. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2523–H2531. [Google Scholar] [CrossRef] [PubMed]
- Khodo, S.N.; Dizin, E.; Sossauer, G.; Szanto, I.; Martin, P.Y.; Feraille, E.; Krause, K.H.; de Seigneux, S. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J. Am. Soc. Nephrol. 2012, 23, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Antonios, T.F.T.; Rattray, F.M.; Singer, D.R.J.; Markandu, N.D.; Mortimer, P.S.; MacGregor, G.A. Rarefaction of skin capillaries in normotensive offspring of individuals with essential hypertension. Heart 2003, 89, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Antonios, T.F.T.; Raghuraman, R.P.; D’Souza, R.; Nathan, P.; Wang, D.; Manyonda, I.T. Capillary remodeling in infants born to hypertensive pregnancy: Pilot study. Am. J. Hypertens. 2012, 25, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Valcarcel-Ares, M.N.; Gautam, T.; Warrington, J.P.; Bailey-Downs, L.; Sosnowska, D.; de Cabo, R.; Losonczy, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: Implications for microvascular aging. J. Gerontol. 2012, 67, 821–829. [Google Scholar] [CrossRef]
- Wagatsuma, A. Effect of aging on expression of angiogenesis-related factors in mouse skeletal muscle. Exp. Gerontol. 2006, 41, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Li, T.S.; Kurazumi, H.; Takemoto, Y.; Ohshima, M.; Murata, T.; Katsura, S.; Morikage, N.; Furutani, A.; Hamano, K. Hypoxic preconditioning enhances angiogenic potential of bone marrow cells with aging-related functional impairment. Circ. J. 2012, 76, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.E.; Kleinman, H.K.; DiPietro, L.A. Impaired wound repair and delayed angiogenesis in aged mice. Lab. Investig. 1999, 79, 1479–1487. [Google Scholar] [PubMed]
- Choudhery, M.S.; Khan, M.; Mahmood, R.; Mehmood, A.; Khan, S.N.; Riazuddin, S. Bone marrow derived mesenchymal stem cells from aged mice have reduced wound healing, angiogenesis, proliferation and anti-apoptosis capabilities. Cell. Biol. Int. 2012, 36, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Jesmin, S.; Hattori, Y.; Togashi, H.; Ueno, K.I.; Yoshioka, M.; Sakuma, I. Age-related changes in cardiac expression of VEGF and its angiogenic receptor KDR in stroke-prone spontaneously hypertensive rats. Mol. Cell. Biochem. 2005, 272, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Bravard, A.; Bonnard, C.; Durand, A.; Chauvin, M.A.; Favier, R.; Vidal, H.; Rieusset, J. Inhibition of xanthine oxidase reduces hyperglycemia-induced oxidative stress and improves mitochondrial alterations in skeletal muscle of diabetic mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E581–E591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rask-Madsen, C.; King, G.L. Mechanisms of disease: Endothelial dysfunction in insulin resistance and diabetes. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002, 106, 2781–2786. [Google Scholar] [CrossRef] [PubMed]
- Aicher, A.; Heeschen, C.; Mildner-Rihm, C.; Urbich, C.; Ihling, C.; Technau-Ihling, K.; Zeiher, A.M.; Dimmeler, S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 2003, 9, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Chan, P.-S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007. [Google Scholar] [CrossRef]
- Lu, M.; Kuroki, M.; Amano, S.; Tolentino, M.; Keough, K.; Kim, I.; Bucala, R.; Adamis, A.P. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J. Clin. Investig. 1998, 101, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Heerkens, E.H.; Shaw, L.; Ryding, A.; Brooker, G.; Mullins, J.J.; Austin, C.; Ohanian, V.; Heagerty, A.M. alphaV integrins are necessary for eutrophic inward remodeling of small arteries in hypertension. Hypertension 2006, 47, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, K.; Oemar, B.S.; Yang, Z.; Luscher, T.F. Pulsatile stretch stimulates superoxide production and activates nuclear factor-kappa B in human coronary smooth muscle. Circ. Res. 1997, 81, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Grote, K.; Flach, I.; Luchtefeld, M.; Akin, E.; Holland, S.M.; Drexler, H.; Schieffer, B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ. Res. 2003, 92, e80–e86. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Loscalzo, J. Cyclic strain modulates resistance to oxidant stress by increasing G6PDH expression in smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2477–H2485. [Google Scholar] [PubMed]
- Lehoux, S. Redox signalling in vascular responses to shear and stretch. Cardiovasc. Res. 2006, 71, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, S.; Esposito, B.; Merval, R.; Loufrani, L.; Tedgui, A. Pulsatile stretch-induced extracellular signal-regulated kinase 1/2 activation in organ culture of rabbit aorta involves reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Edwards, J.G.; Kaminski, P.M.; Wolin, M.S.; Kaley, G.; Koller, A. Increased superoxide production in coronary arteries in hyperhomocysteinemia: Role of tumor necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Huang, A.; Kaminski, P.M.; Wolin, M.S.; Koller, A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation 2003, 108, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Oeckler, R.A.; Kaminski, P.M.; Wolin, M.S. Stretch enhances contraction of bovine coronary arteries via an NAD(P)H oxidase-mediated activation of the extracellular signal-regulated kinase mitogen-activated protein kinase cascade. Circ. Res. 2003, 92, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Kaminski, P.M.; Wolin, M.S.; Koller, A. Chronic high pressure-induced arterial oxidative stress: Involvement of protein kinase C-dependent NAD(P)H oxidase and local renin-angiotensin system. Am. J. Pathol. 2004, 165, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Lemarie, C.A.; Tharaux, P.L.; Esposito, B.; Tedgui, A.; Lehoux, S. Transforming growth factor-α mediates nuclear factor kappaB activation in strained arteries. Circ. Res. 2006, 99, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Gebremedhin, D.; Terashvili, M.; Wickramasekera, N.; Zhang, D.X.; Rau, N.; Miura, H.; Harder, D.R. Redox signaling via oxidative inactivation of PTEN modulates pressure-dependent myogenic tone in rat middle cerebral arteries. PLoS One 2013, 8, e68498. [Google Scholar] [CrossRef] [PubMed]
- Schleifenbaum, J.; Kassmann, M.; Szijarto, I.A.; Hercule, H.C.; Tano, J.Y.; Weinert, S.; Heidenreich, M.; Pathan, A.R.; Anistan, Y.M.; Alenina, N.; et al. Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ. Res. 2014, 115, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Staiculescu, M.C.; Ramirez-Perez, F.I.; Castorena-Gonzalez, J.A.; Hong, Z.; Sun, Z.; Meininger, G.A.; Martinez-Lemus, L.A. Lysophosphatidic acid induces integrin activation in vascular smooth muscle and alters arteriolar myogenic vasoconstriction. Front. Physiol. 2014, 5, 413. [Google Scholar] [CrossRef]
- Martinez-Lemus, L.A.; Crow, T.; Davis, M.J.; Meininger, G.A. αvβ3- and α5β1-integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H322–H329. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staiculescu, M.C.; Foote, C.; Meininger, G.A.; Martinez-Lemus, L.A. The Role of Reactive Oxygen Species in Microvascular Remodeling. Int. J. Mol. Sci. 2014, 15, 23792-23835. https://doi.org/10.3390/ijms151223792
Staiculescu MC, Foote C, Meininger GA, Martinez-Lemus LA. The Role of Reactive Oxygen Species in Microvascular Remodeling. International Journal of Molecular Sciences. 2014; 15(12):23792-23835. https://doi.org/10.3390/ijms151223792
Chicago/Turabian StyleStaiculescu, Marius C., Christopher Foote, Gerald A. Meininger, and Luis A. Martinez-Lemus. 2014. "The Role of Reactive Oxygen Species in Microvascular Remodeling" International Journal of Molecular Sciences 15, no. 12: 23792-23835. https://doi.org/10.3390/ijms151223792
APA StyleStaiculescu, M. C., Foote, C., Meininger, G. A., & Martinez-Lemus, L. A. (2014). The Role of Reactive Oxygen Species in Microvascular Remodeling. International Journal of Molecular Sciences, 15(12), 23792-23835. https://doi.org/10.3390/ijms151223792