Evaluation of a Method for Nitrotyrosine Site Identification and Relative Quantitation Using a Stable Isotope-Labeled Nitrated Spike-In Standard and High Resolution Fourier Transform MS and MS/MS Analysis

Abstract
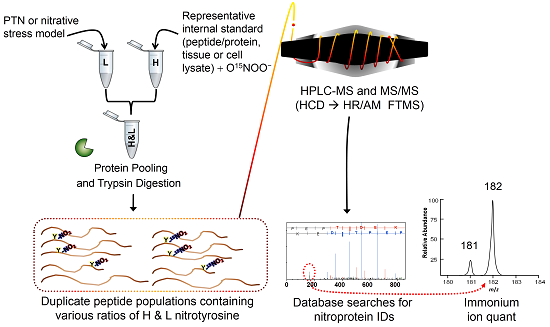
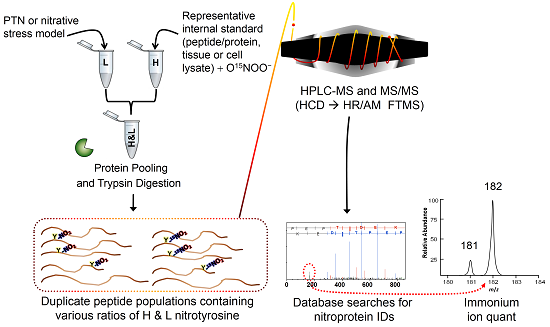
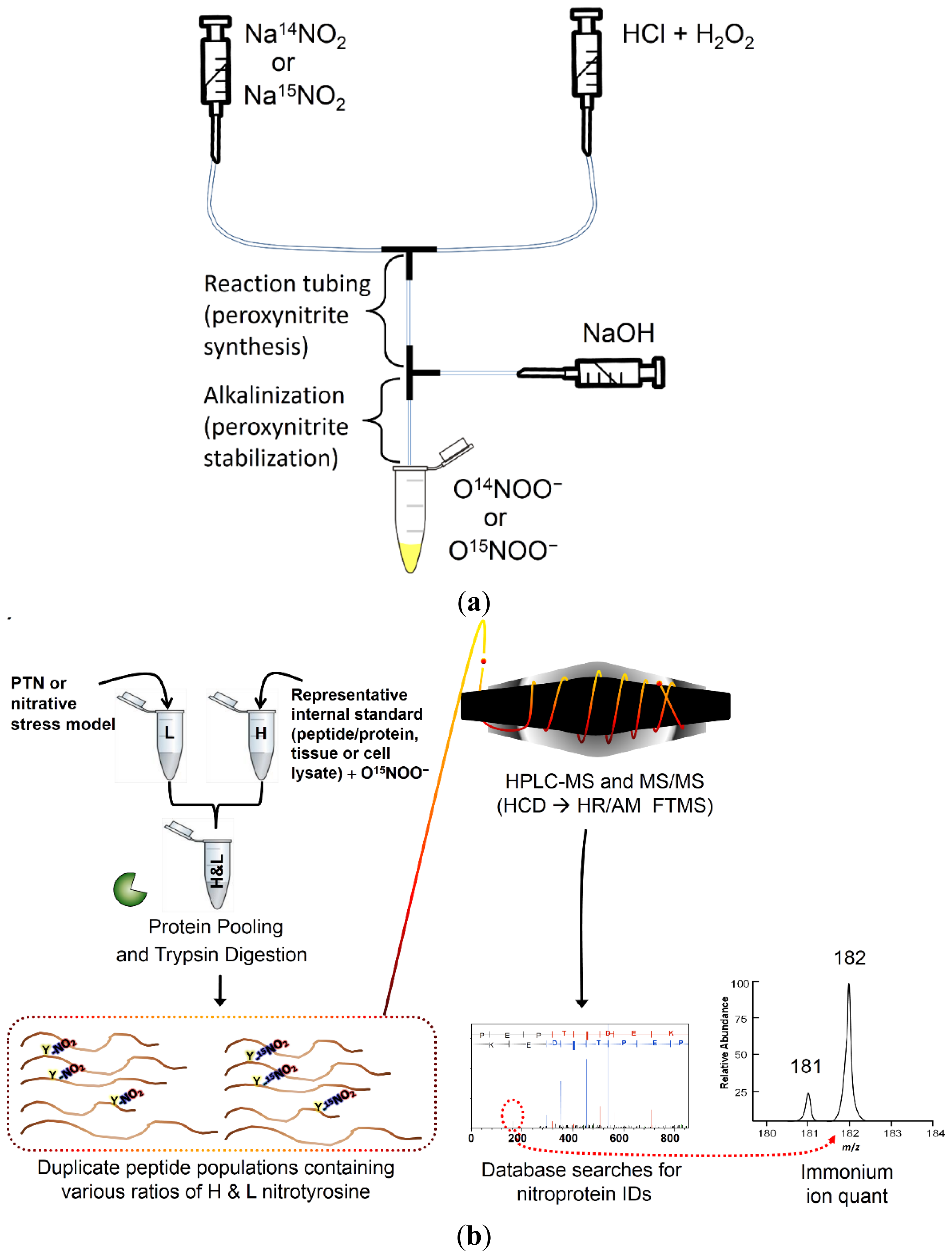
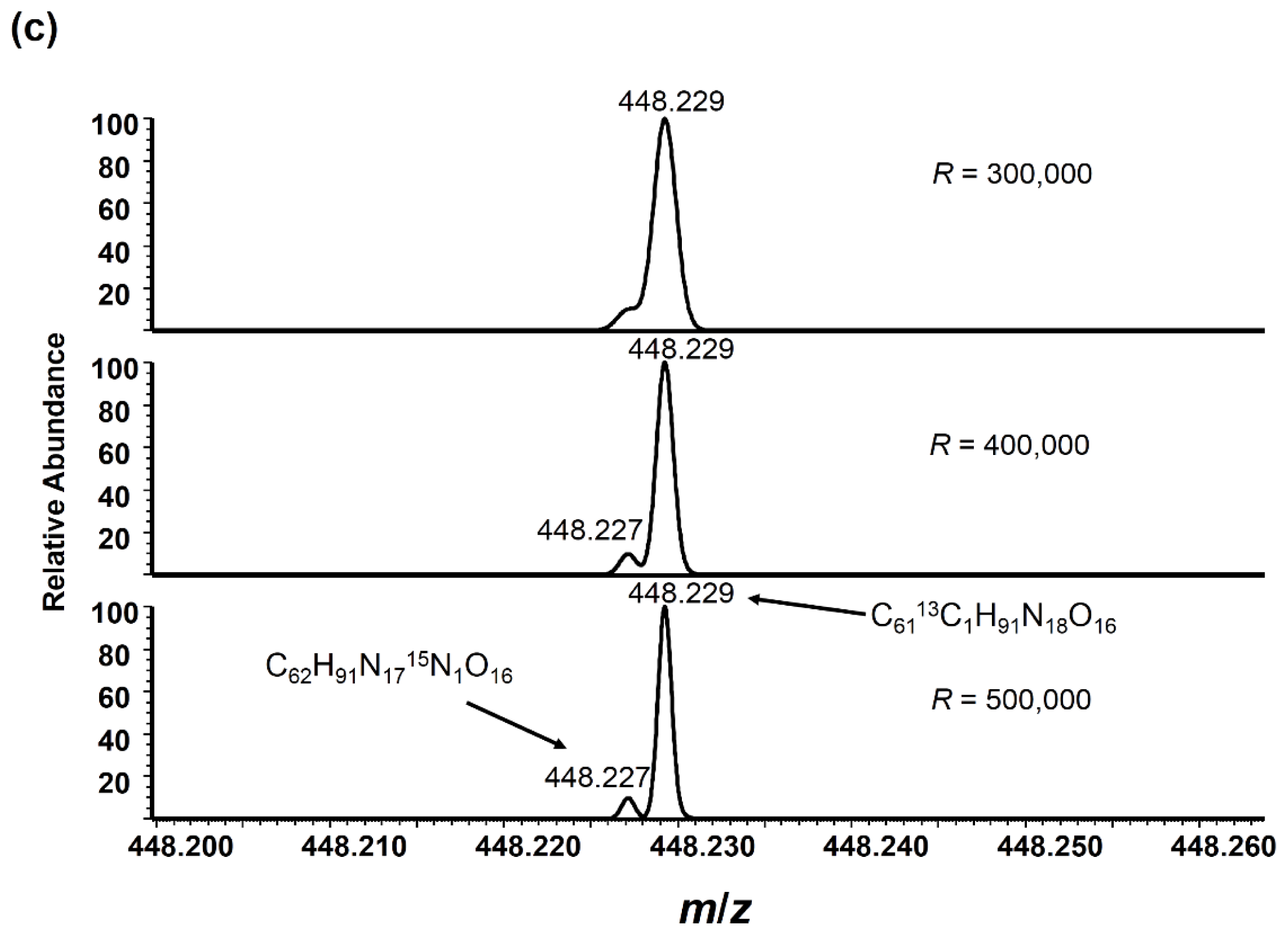
:
1. Introduction
2. Results and Discussion
2.1. Method Overview
2.2. Stable Isotope-Labeled Peroxynitrite Synthesis
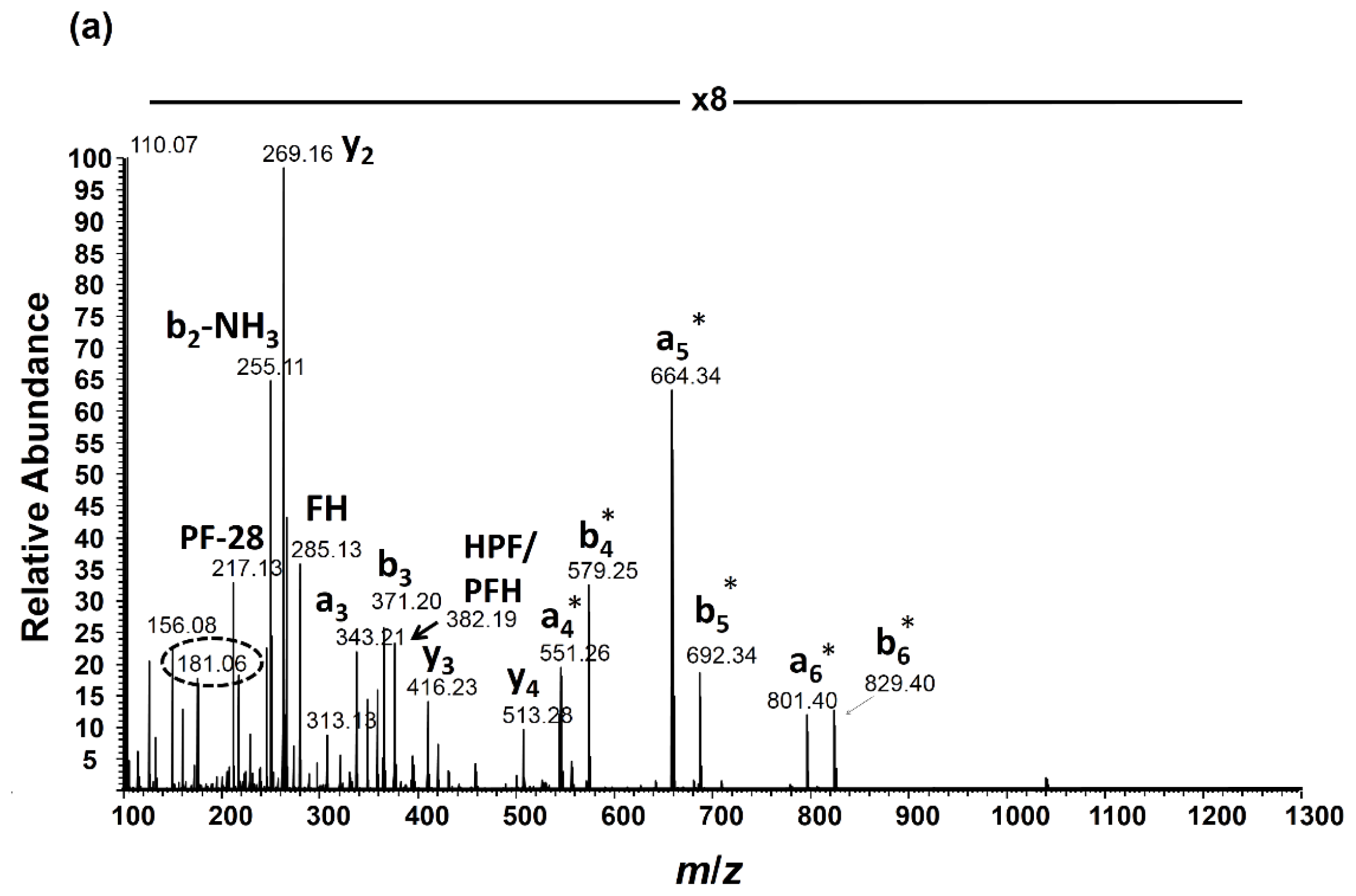
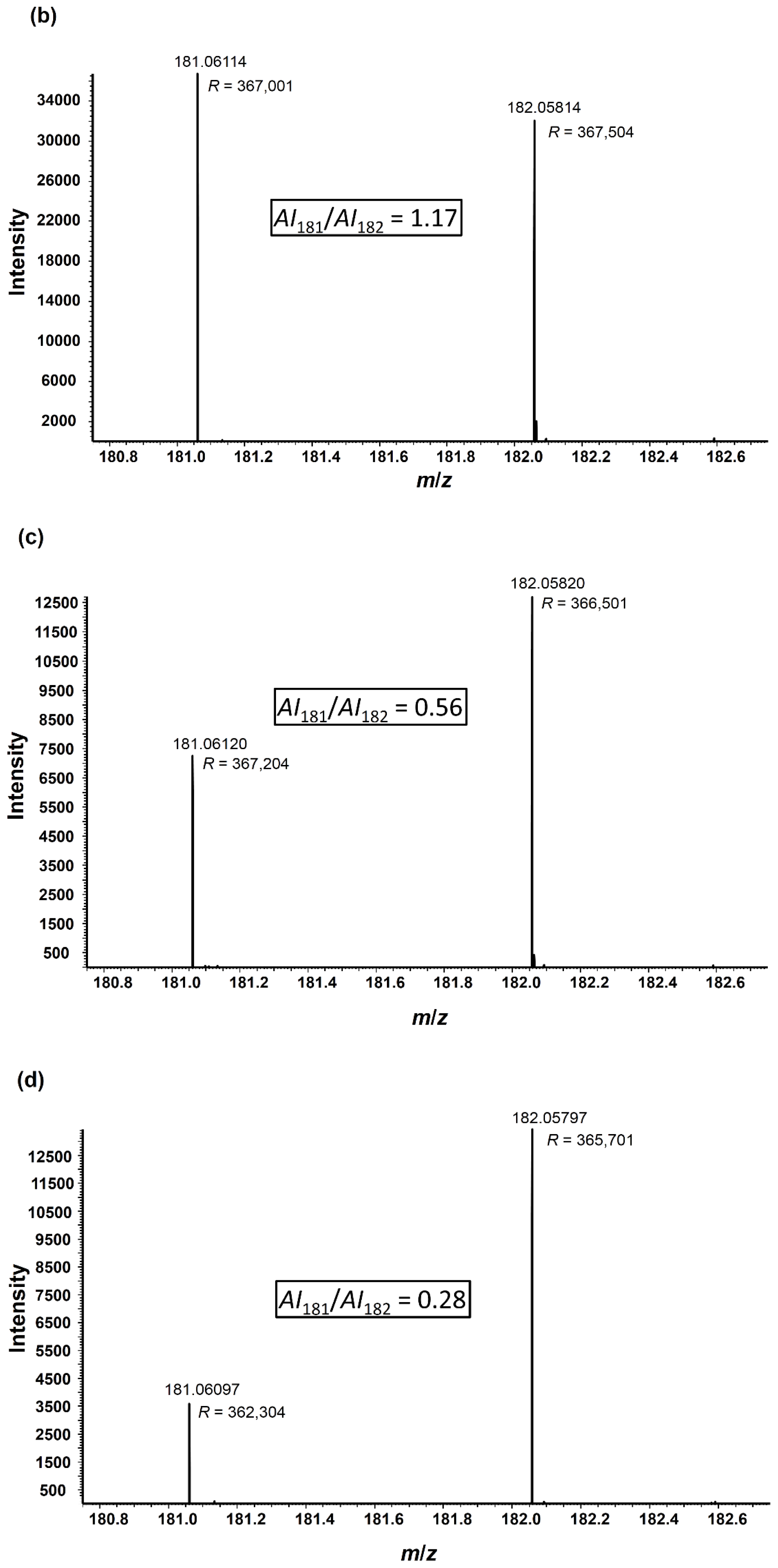
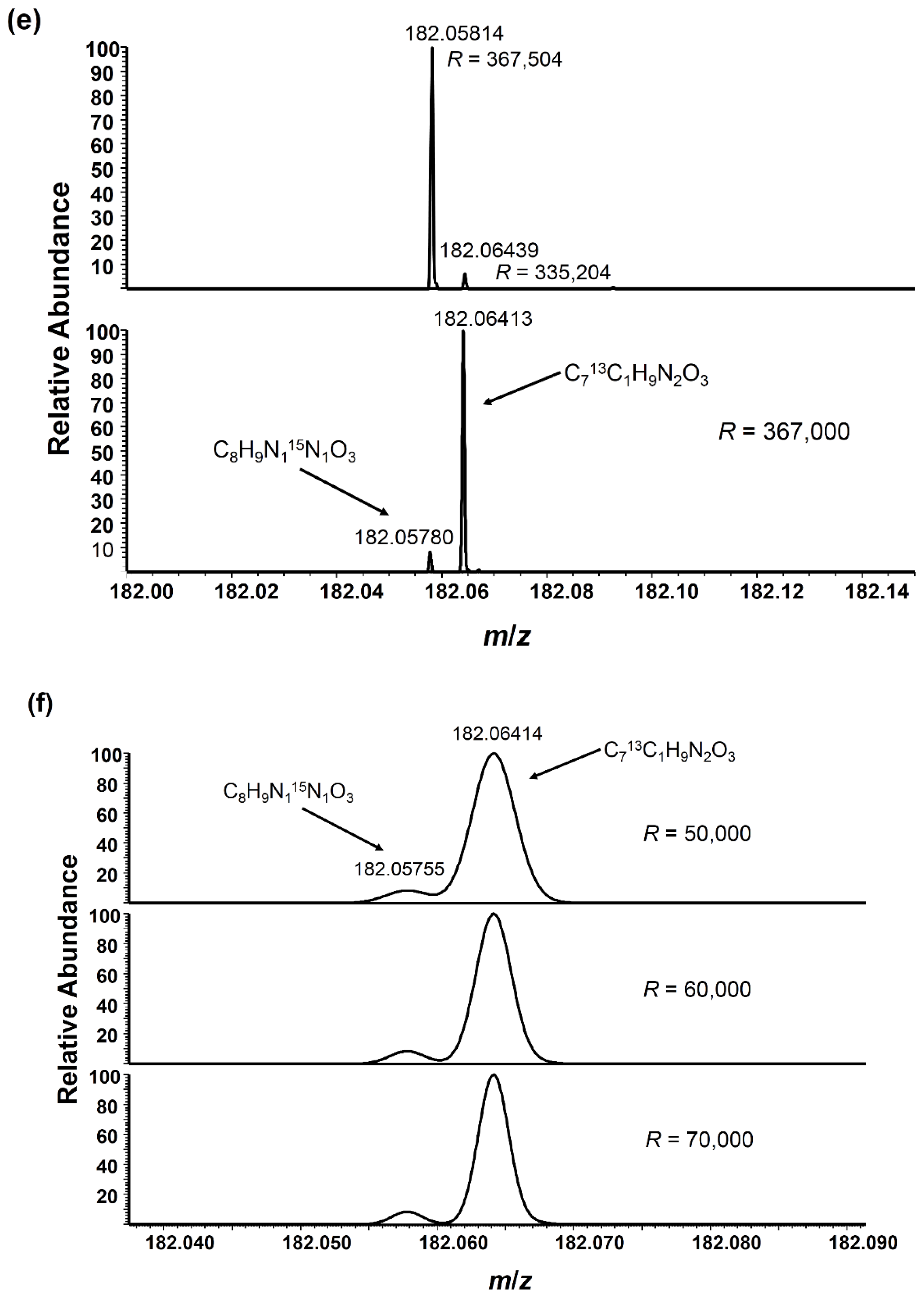
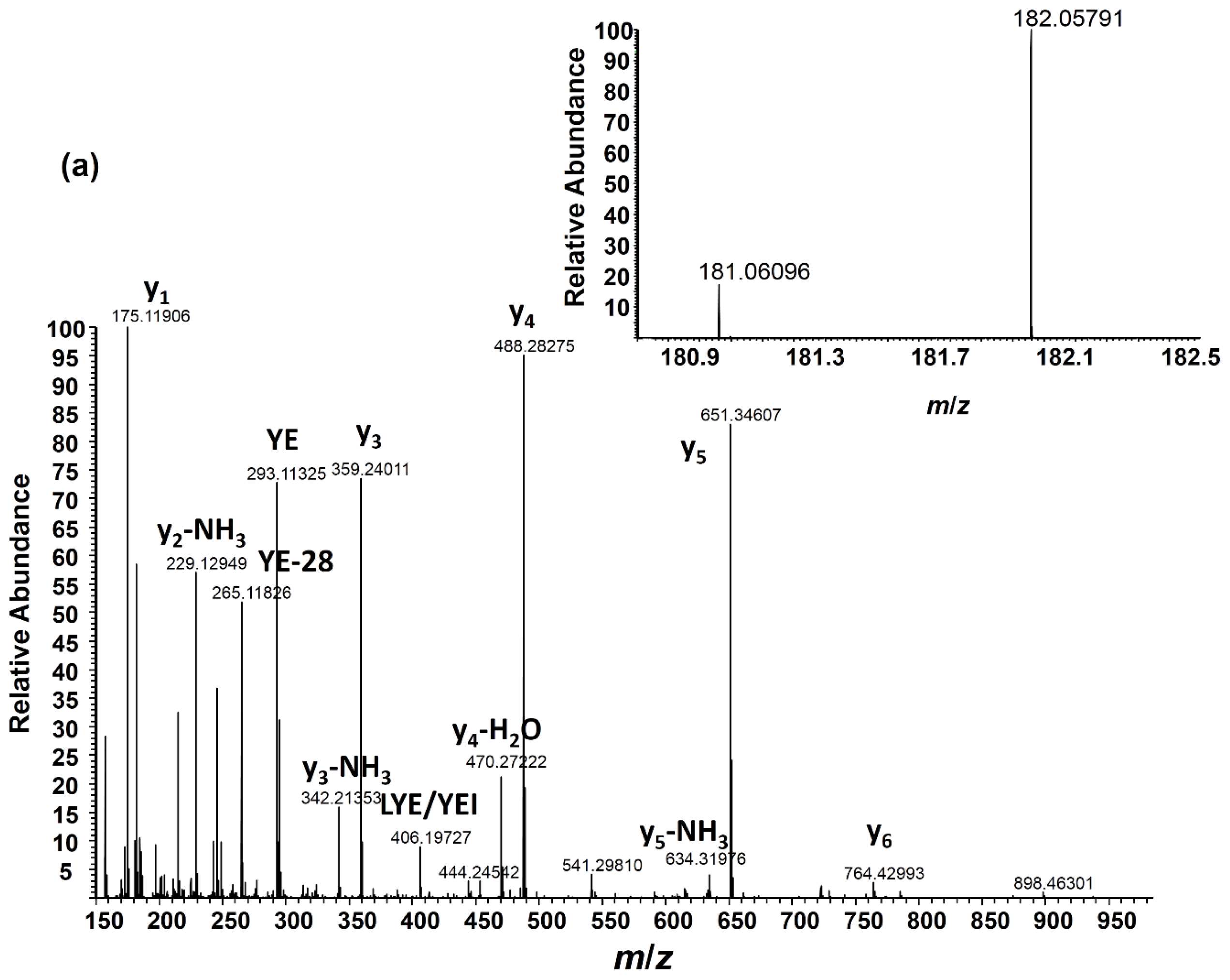
2.3. Angiotensin I Peptide Testing
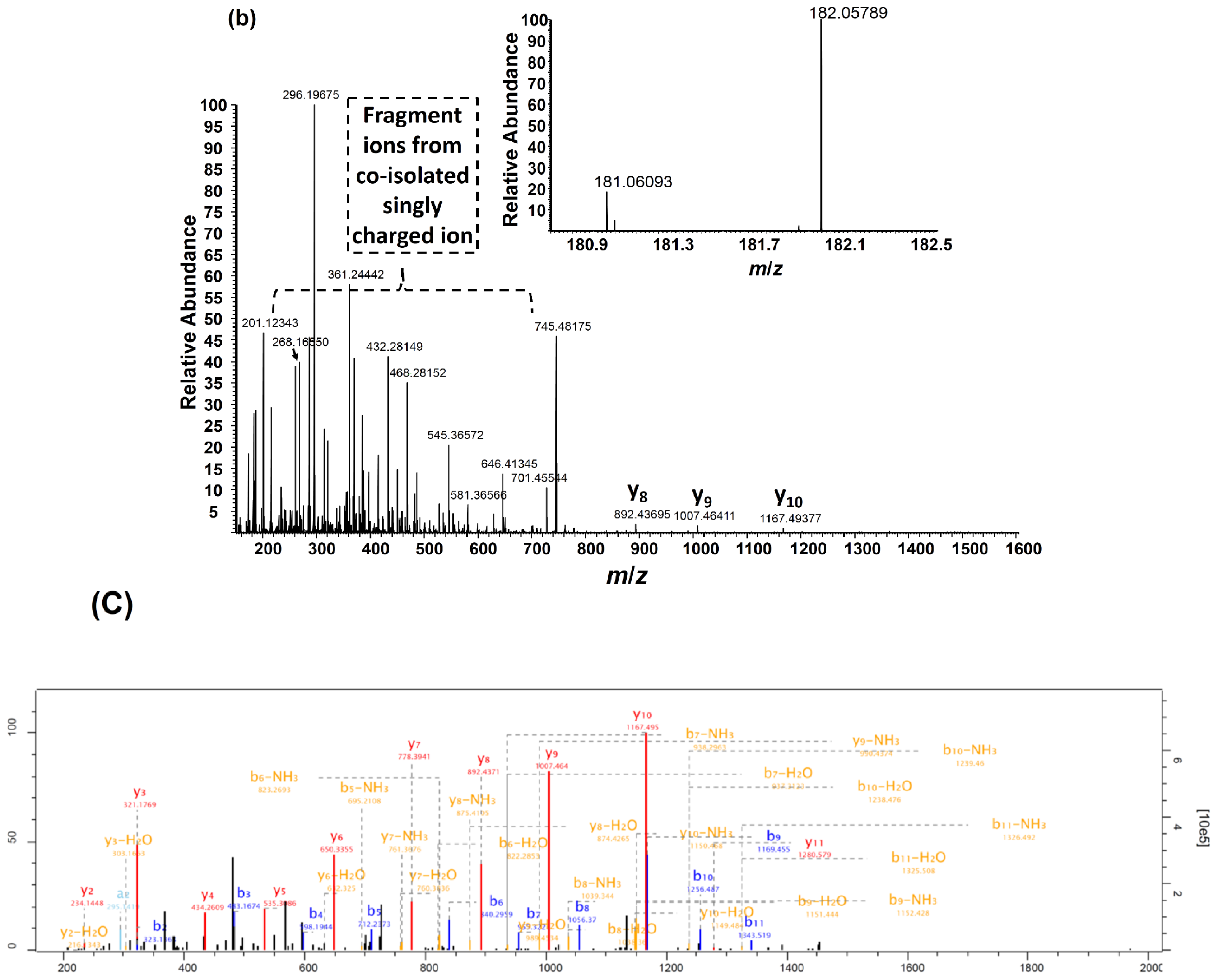
2.4. Protein Standard Testing
2.5. Method Evaluation Summary: Implications in Global-Scale Nitration Analysis
3. Experimental Section
3.1. Peroxynitrite Synthesis
3.2. Stable Isotope Labeling Using Synthetic Peroxynitrite
3.2.1. Peptide Standard Reactions
3.2.2. Protein Standard Reactions
3.3. Liquid Chromatography and Fourier Transform Mass Spectrometry (FTMS) Analyses
3.3.1. Direct Infusion Electrospray Ionization (ESI)-FTMS
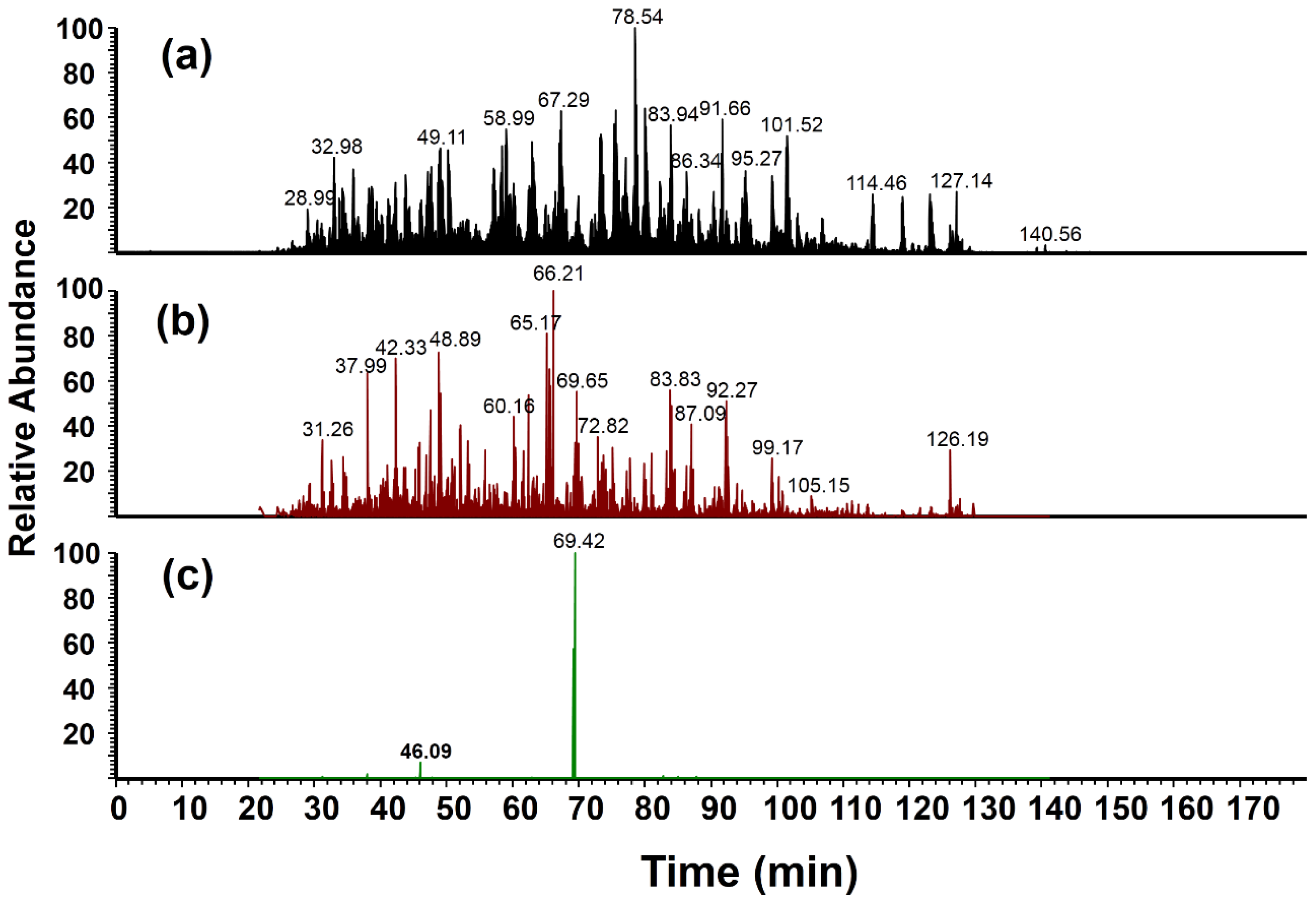
3.3.2. Liquid Chromatography (LC)-MS/MS Analysis of a Microglial Cell Lysate with Nitrated Bovine Serum Albumin (BSA) Spike-In
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsS.M.S. conceived of and designed the experiments. K.W.S. carried out the peroxynitrite synthesis and initial mass spectrometric analyses. A.R.F. and J.P.C.P. performed the labeling experiments. C.P.D. performed the FTMS analyses. S.M.S. and K.W.S. analyzed the data and wrote the manuscript.
References
- Abello, N.; Kerstjens, H.A.M.; Postma, D.S.; Bischoff, R. Protein tyrosine nitration: Selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. J. Proteome Res 2009, 8, 3222–3238. [Google Scholar]
- Anantharaman, M.; Tangpong, J.; Keller, J.N.; Murphy, M.P.; Markesbery, W.R.; Kiningham, K.K.; St. Clair, D.K. B-amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLh/NLh X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol 2006, 168, 1608–1618. [Google Scholar]
- Aoyama, K.; Matsubara, K.; Fujikawa, Y.; Nagahiro, Y.; Shimizu, K.; Umegae, N.; Hayase, N.; Shiono, H.; Kobayashi, S. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann. Neurol 2000, 47, 524–527. [Google Scholar]
- Ckless, K.; Lampert, A.; Reiss, J.; Kasahara, D.; Poynter, M.E.; Irvin, C.G.; Lundblad, L.K.A.; Norton, R.; van der Vliet, A.; Janssen-Heininger, Y.M.W. Inhibition of arginase activity enhances inflammation in mice with allergic airway disease, in association with increases in protein S-nitrosylation and tyrosine nitration. J. Immunol 2008, 181, 4255–4264. [Google Scholar]
- Ito, K.; Hanazawa, T.; Tomita, K.; Barnes, P.J.; Adcock, I.M. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: Role of tyrosine nitration. Biochem. Biophys. Res. Commun 2004, 315, 240–245. [Google Scholar]
- Neumann, H.; Hazen, J.L.; Weinstein, J.; Mehl, R.A.; Chin, J.W. Genetically encoding protein oxidative damage. J. Am. Chem. Soc 2008, 130, 4028–4033. [Google Scholar]
- Ohmori, H.; Kanayama, N. Immunogenicity of an inflammation-associated product, tyrosine nitrated self-proteins. Autoimmun. Rev 2005, 4, 224–229. [Google Scholar]
- Reynolds, M.R.; Reyes, J.F.; Fu, Y.; Bigio, E.H.; Guillozet-Bongaarts, A.L.; Berry, R.W.; Binder, L.I. Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer’s disease and other tauopathies. J. Neurosci 2006, 26, 10636–10645. [Google Scholar]
- Turko, I.V.; Li, L.; Aulak, K.S.; Stuehr, D.J.; Chang, J.-Y.; Murad, F. Protein tyrosine nitration in the mitochondria from diabetic mouse heart: Implications to dysfunctional mitochondria in diabetes. J. Biol. Chem 2003, 278, 33972–33977. [Google Scholar]
- Abriata, L.A.; Cassina, A.; Tórtora, V.; Marín, M.; Souza, J.M.; Castro, L.; Vila, A.J.; Radi, R. Nitration of solvent-exposed tyrosine 74 on cytochrome c triggers heme iron-methionine 80 bond disruption: Nuclear magnetic resonance and optical spectroscopy studies. J. Biol. Chem 2009, 284, 17–26. [Google Scholar]
- Aulak, K.S.; Koeck, T.; Crabb, J.W.; Stuehr, D.J. Dynamics of protein nitration in cells and mitochondria. Am. J. Physiol 2003, 286, H30–H38. [Google Scholar]
- Koeck, T.; Fu, X.; Hazen, S.L.; Crabb, J.W.; Stuehr, D.J.; Aulak, K.S. Rapid and selective oxygen-regulated protein tyrosine denitration and nitration in mitochondria. J. Biol. Chem 2004, 279, 27257–27262. [Google Scholar]
- Khan, J.; Brennan, D.; Bradley, N.; Gao, B.; Bruckdorfer, R.; Jacobs, M. 3-Nitrotyrosine in the proteins of human plasma determined by an ELISA method. Biochem. J 1998, 330, 795–801. [Google Scholar]
- Shigenaga, M.K.; Lee, H.H.; Blount, B.C.; Christen, S.; Shigeno, E.T.; Yip, H.; Ames, B.N. Inflammation and nox-induced nitration: Assay for 3-nitrotyrosine by HPLC with electrochemical detection. Proc. Natl. Acad. Sci. USA 1997, 94, 3211–3216. [Google Scholar]
- Nikov, G.; Bhat, V.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrated proteins by nitrotyrosine-specific affinity probes and mass spectrometry. Anal. Biochem 2003, 320, 214–222. [Google Scholar]
- Prokai, L.; Guo, J.; Prokai-Tatrai, K. Selective chemoprecipitation to enrich nitropeptides from complex proteomes for mass-spectrometric analysis. Nat. Protoc 2014, 9, 882–895. [Google Scholar]
- Prokai-Tatrai, K.; Guo, J.; Prokai, L. Selective chemoprecipitation and subsequent release of tagged species for the analysis of nitropeptides by liquid chromatography—Tandem mass spectrometry. Mol. Cell. Proteomics 2011. [Google Scholar] [CrossRef]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol 2003, 21, 255–261. [Google Scholar]
- Nørregaard Jensen, O. Modifi qacation-specific proteomics: Characterization of post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol 2004, 8, 33–41. [Google Scholar]
- Petersson, A.S.; Steen, H.; Kalume, D.E.; Caidahl, K.; Roepstorff, P. Investigation of tyrosine nitration in proteins by mass spectrometry. J. Mass Spectrom 2001, 36, 616–625. [Google Scholar]
- Falick, A.; Hines, W.; Medzihradszky, K.; Baldwin, M.; Gibson, B. Low-mass ions produced from peptides by high-energy collision-induced dissociation in tandem mass spectrometry. J. Am. Soc. Mass Spectrom 1993, 4, 882–893. [Google Scholar]
- Papayannopoulos, I.A. The interpretation of collision-induced dissociation tandem mass spectra of peptides. Mass Spectrom. Rev 1995, 14, 49–73. [Google Scholar]
- Hohmann, L.J.; Eng, J.K.; Gemmill, A.; Klimek, J.; Vitek, O.; Reid, G.E.; Martin, D.B. Quantification of the compositional information provided by immonium ions on a quadrupole-time-of-flight mass spectrometer. Anal. Chem 2008, 80, 5596–5606. [Google Scholar]
- Steen, H.; Küster, B.; Fernandez, M.; Pandey, A.; Mann, M. Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole tof mass spectrometry in positive ion mode. Anal. Chem 2001, 73, 1440–1448. [Google Scholar]
- Bateman, R.; Carruthers, R.; Hoyes, J.; Jones, C.; Langridge, J.; Millar, A.; Vissers, J. A novel precursor ion discovery method on a hybrid quadrupole orthogonal acceleration time-of-flight (Q-TOF) mass spectrometer for studying protein phosphorylation. J. Am. Soc. Mass Spectrom 2002, 13, 792–803. [Google Scholar]
- Chiappetta, G.; Corbo, C.; Palmese, A.; Marino, G.; Amoresano, A. Quantitative identification of protein nitration sites. Proteomics 2009, 9, 1524–1537. [Google Scholar]
- Guo, J.; Prokai-Tatrai, K.; Prokai, L. Relative quantitation of protein nitration by liquid chromatography-mass spectrometry using isotope-coded dimethyl labeling and chemoprecipitation. J. Chromatogr 2012, 1232, 266–275. [Google Scholar]
- Colzani, M.; Schütz, F.; Potts, A.; Waridel, P.; Quadroni, M. Relative protein quantification by isobaric silac with immonium ion splitting (ISIS). Mol. Cell. Proteomics 2008, 7, 927–937. [Google Scholar]
- Zhang, G.; Neubert, T.A. Automated comparative proteomics based on multiplex tandem mass spectrometry and stable isotope labeling. Mol. Cell. Proteomics 2006, 5, 401–411. [Google Scholar]
- Robinson, K.M.; Beckman, J.S. Synthesis of peroxynitrite from nitrite and hydrogen peroxide. Methods Enzymol 2005, 396, 207–214. [Google Scholar]
- Hebert, A.S.; Merrill, A.E.; Bailey, D.J.; Still, A.J.; Westphall, M.S.; Strieter, E.R.; Pagliarini, D.J.; Coon, J.J. Neutron-encoded mass signatures for multiplexed proteome quantification. Nat. Methods 2013, 10, 332–334. [Google Scholar]
- Bell-Temin, H.; Barber, D.S.; Zhang, P.; Liu, B.; Stevens, S.M. Proteomic analysis of rat microglia establishes a high - confidence reference data set of over 3000 proteins. Proteomics 2012, 12, 246–250. [Google Scholar]
- Bell-Temin, H.; Zhang, P.; Chaput, D.; King, M.A.; You, M.; Liu, B.; Stevens, S.M. Quantitative proteomic characterization of ethanol-responsive pathways in rat microglial cells. J. Proteome Res 2013, 12, 2067–2077. [Google Scholar]
- Cox, J.; Mann, M. Maxquant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–1372. [Google Scholar]
- Seeley, K.W.; Stevens, S.M., Jr. Investigation of local primary structure effects on peroxynitrite-mediated tyrosine nitration using targeted mass spectrometry. J. Proteomics 2012, 75, 1691–1700. [Google Scholar]
- Stevens, S.M.; Prokai-Tatrai, K.; Prokai, L. Factors that contribute to the misidentification of tyrosine nitration by shotgun proteomics. Mol. Cell. Proteomics 2008, 7, 2442–2451. [Google Scholar]
- Rose, C.M.; Merrill, A.E.; Bailey, D.J.; Hebert, A.S.; Westphall, M.S.; Coon, J.J. Neutron encoded labeling for peptide identification. Anal. Chem 2013, 85, 5129–5137. [Google Scholar]
- Cook, S.; Jackson, G.P. Characterization of tyrosine nitration and cysteine nitrosylation modifications by metastable atom-activation dissociation mass spectrometry. J. Am. Soc. Mass Spectrom 2011, 22, 221–232. [Google Scholar]
- Jones, A.; Mikhailov, V.; Iniesta, J.; Cooper, H. Electron capture dissociation mass spectrometry of tyrosine nitrated peptides. J. Am. Soc. Mass Spectrom 2010, 21, 268–277. [Google Scholar] [Green Version]
- Mikhailov, V.A.; Iniesta, J.; Cooper, H.J. Top-down mass analysis of protein tyrosine nitration: Comparison of electron capture dissociation with “slow-heating” tandem mass spectrometry methods. Anal. Chem 2010, 82, 7283–7292. [Google Scholar]
- Hughes, M.N.; Nicklin, H.G. The chemistry of pernitrites. Part I. Kinetics of decomposition of pernitrous acid. J. Chem. Soc 1968, 450–452. [Google Scholar]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar]
- UniProt Knowledgebase. Available online: http://www.uniprot.org/uniprot/?query=organism:10116+keyword:1185 (accessed on 10 September 2013).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dilution | 181/182 | Normalized ratio |
|---|---|---|
| 1:1 | 0.69 ± 0.02 | 1 ± 0.04 |
| 0.25:1 (a) | 0.20 ± 0.04 | 0.28 ± 0.06 |
| 0.25:1 (b) | 0.16 ± 0.01 | 0.23 ± 0.02 |
| 0.25:1 (c) | 0.17± 0.01 | 0.24 ± 0.02 |
| 0.10:1 | 0.066 ± 0.004 | 0.094 ± 0.007 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Seeley, K.W.; Fertig, A.R.; Dufresne, C.P.; Pinho, J.P.C.; Jr., S.M.S. Evaluation of a Method for Nitrotyrosine Site Identification and Relative Quantitation Using a Stable Isotope-Labeled Nitrated Spike-In Standard and High Resolution Fourier Transform MS and MS/MS Analysis. Int. J. Mol. Sci. 2014, 15, 6265-6285. https://doi.org/10.3390/ijms15046265
Seeley KW, Fertig AR, Dufresne CP, Pinho JPC, Jr. SMS. Evaluation of a Method for Nitrotyrosine Site Identification and Relative Quantitation Using a Stable Isotope-Labeled Nitrated Spike-In Standard and High Resolution Fourier Transform MS and MS/MS Analysis. International Journal of Molecular Sciences. 2014; 15(4):6265-6285. https://doi.org/10.3390/ijms15046265
Chicago/Turabian StyleSeeley, Kent W., Alison R. Fertig, Craig P. Dufresne, Joao P. C. Pinho, and Stanley M. Stevens Jr. 2014. "Evaluation of a Method for Nitrotyrosine Site Identification and Relative Quantitation Using a Stable Isotope-Labeled Nitrated Spike-In Standard and High Resolution Fourier Transform MS and MS/MS Analysis" International Journal of Molecular Sciences 15, no. 4: 6265-6285. https://doi.org/10.3390/ijms15046265
APA StyleSeeley, K. W., Fertig, A. R., Dufresne, C. P., Pinho, J. P. C., & Jr., S. M. S. (2014). Evaluation of a Method for Nitrotyrosine Site Identification and Relative Quantitation Using a Stable Isotope-Labeled Nitrated Spike-In Standard and High Resolution Fourier Transform MS and MS/MS Analysis. International Journal of Molecular Sciences, 15(4), 6265-6285. https://doi.org/10.3390/ijms15046265