
miR-134 Modulates the Proliferation of Human Cardiomyocyte Progenitor Cells by Targeting Meis2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
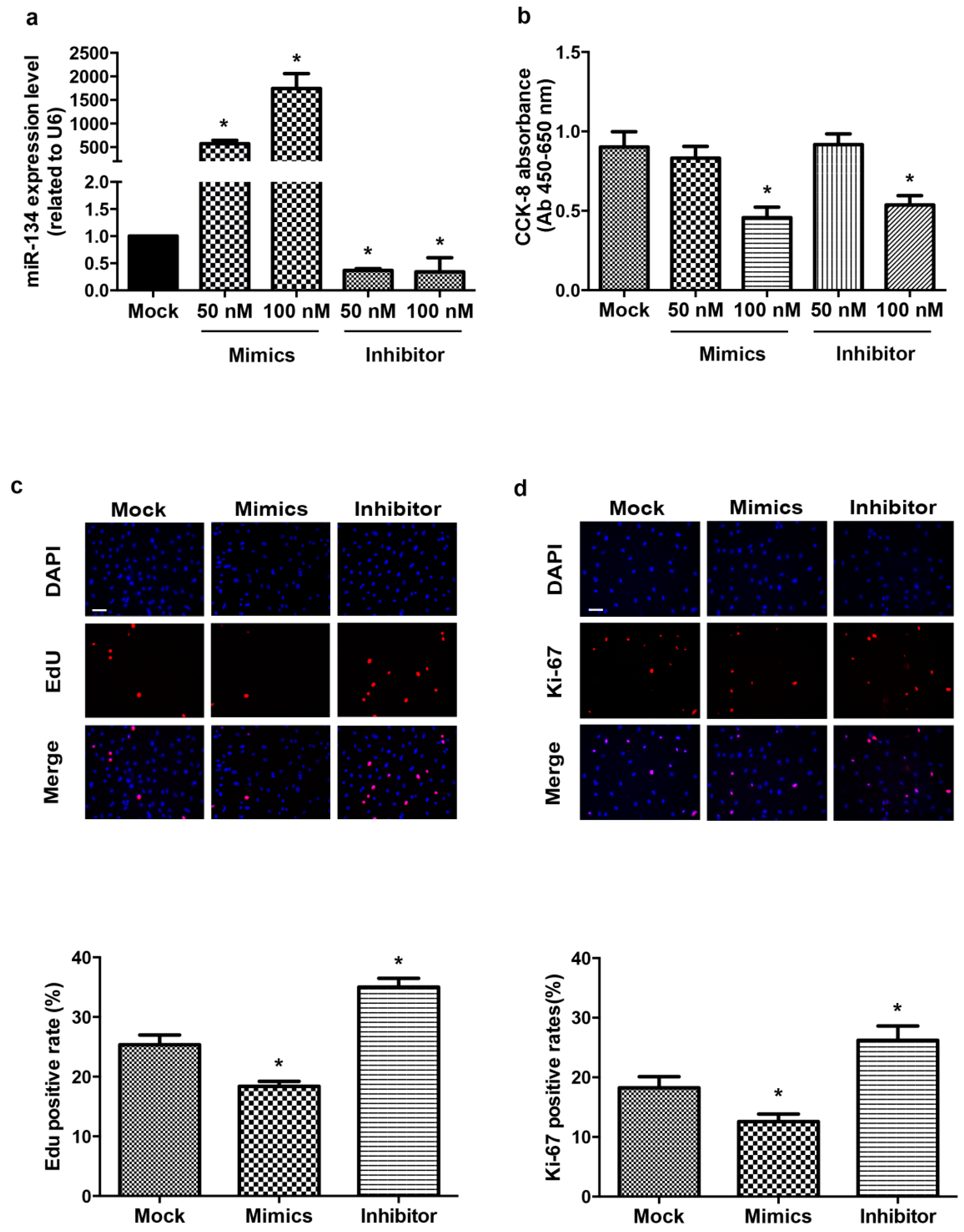
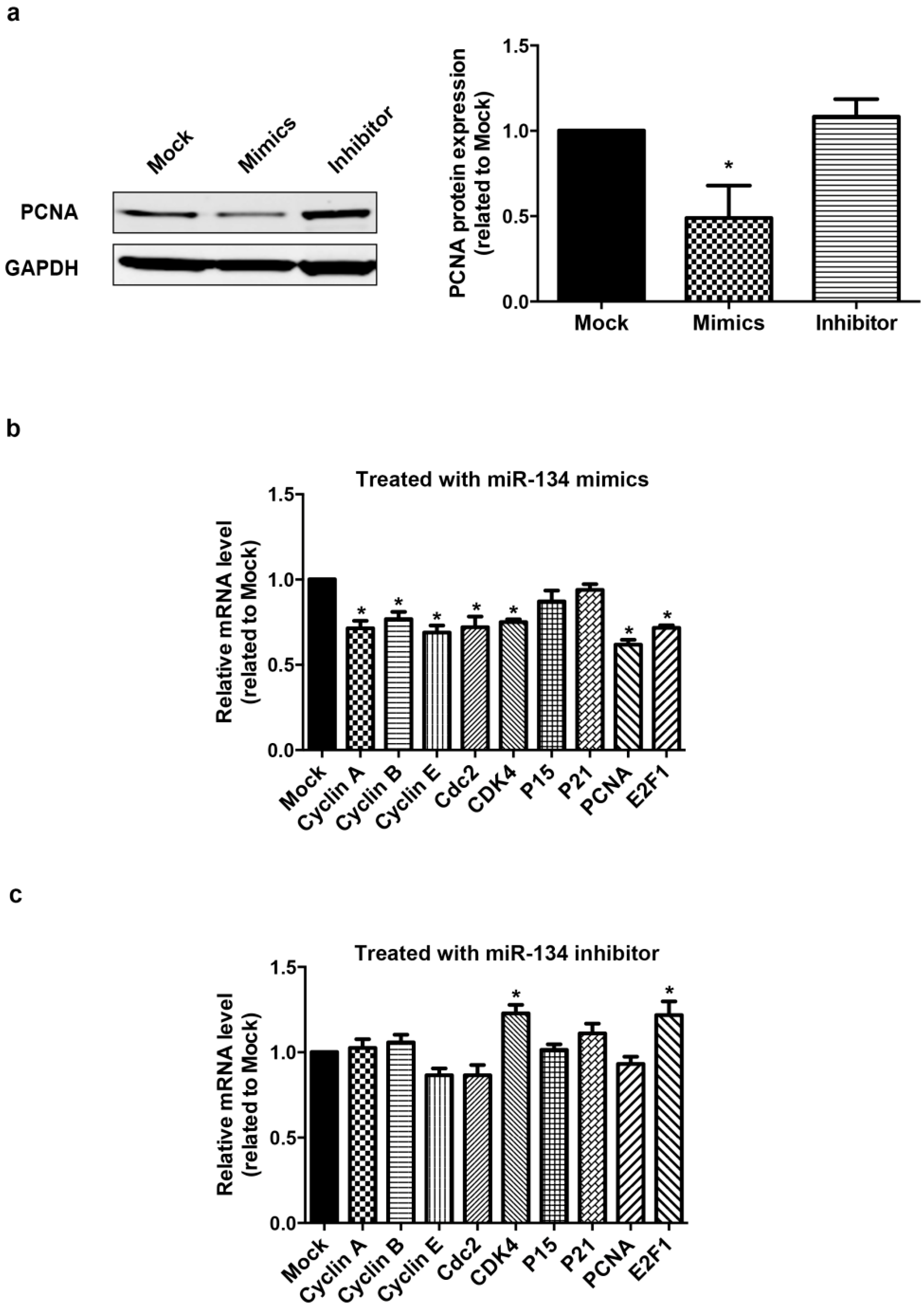
2.1. miR-134 Modulates hCMPCs Proliferation


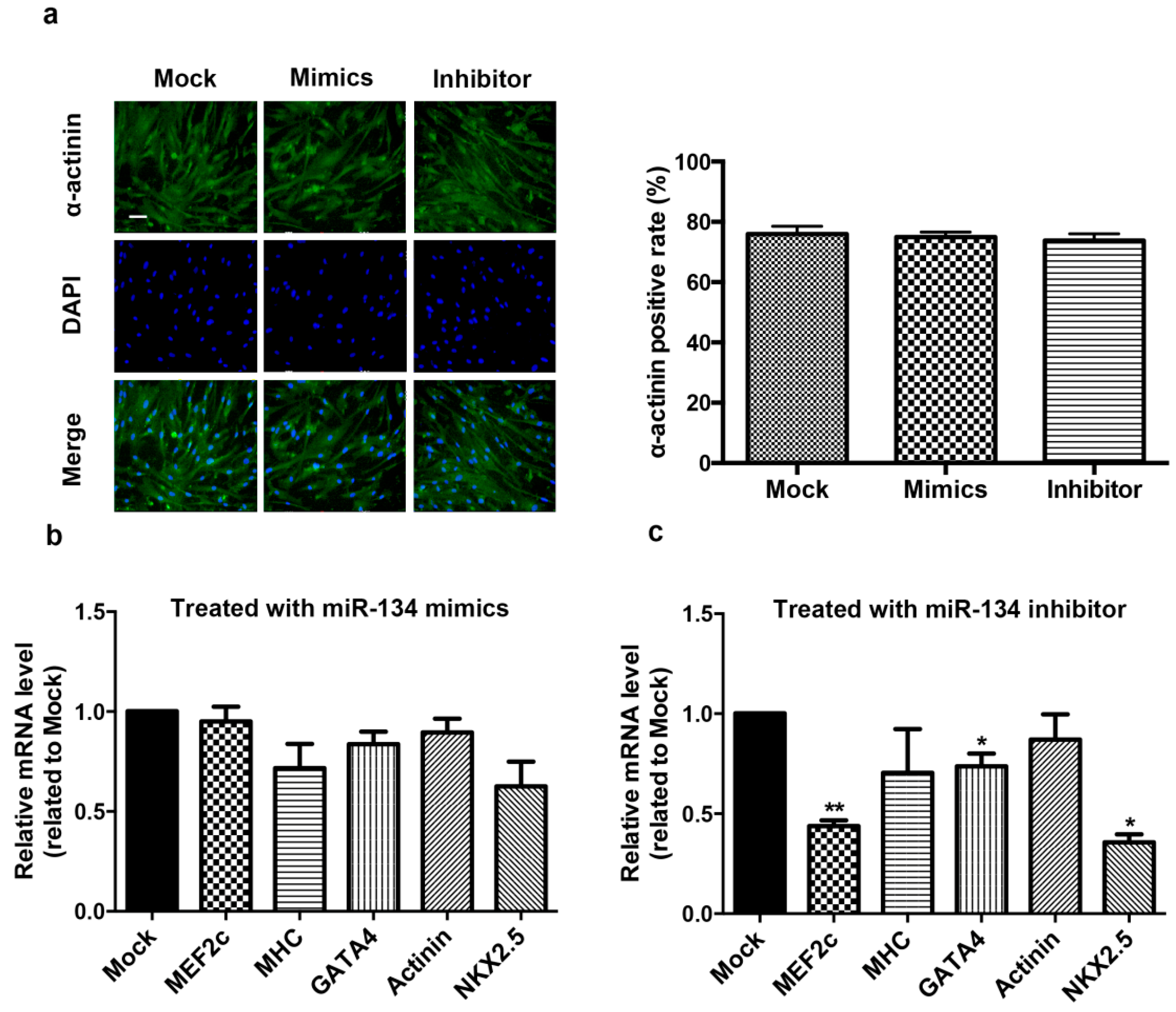
2.2. miR-134 Is Not Required for the Terminal Differentiation of hCMPC into Cardiomyocytes in Our Model
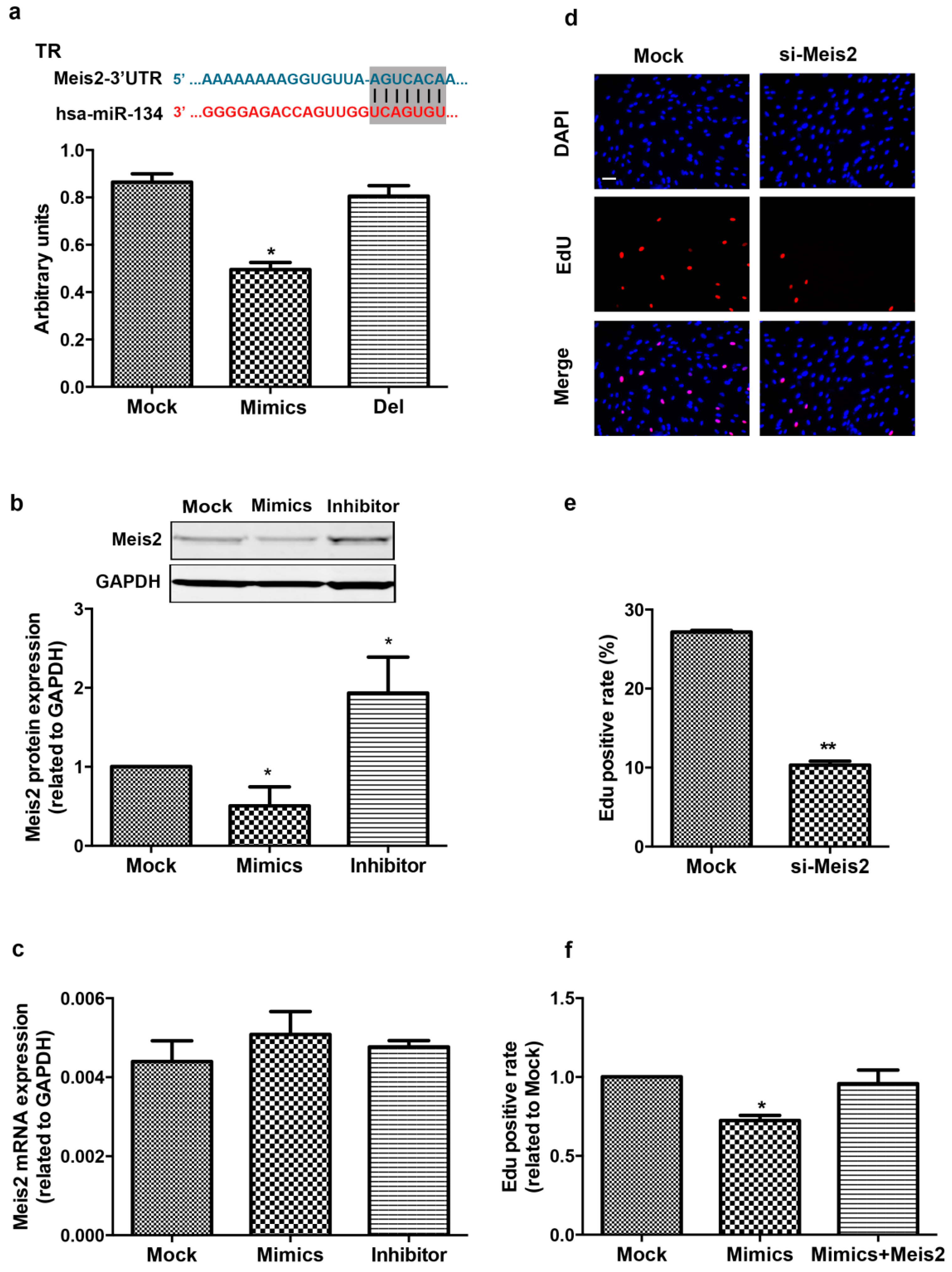
2.3. Meis2 Is Identified as the Target of miR-134

2.4. Meis2 Over-Expression Rescues the Effect of miR-134 on hCMPC Proliferation

3. Discussion
4. Experimental Section
4.1. hCMPC Isolation and Culture
4.2. miRNA Transfection
4.3. hCMPC Proliferation Determination
4.4. Cell Viability and Apoptosis Assay
4.5. Induction and Quantification of hCMPC Differentiation
4.6. Luciferase Experiment
4.7. Modulation of Meis2 in hCMPCs
4.8. RT-PCR
4.9. Western Blot
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rosenthal, N.; Harvey, R.P. Heart Development and Regeneration, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Srivastava, D. Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Hierlihy, A.M.; Seale, P.; Lobe, C.G.; Rudnicki, M.A.; Megeney, L.A. The post-natal heart contains a myocardial stem cell population. FEBS Lett. 2002, 530, 239–243. [Google Scholar] [CrossRef]
- Xiao, Q.; Zeng, L.; Zhang, Z.; Margariti, A.; Ali, Z.A.; Channon, K.M.; Xu, Q.; Hu, Y. Sca-1+ progenitors derived from embryonic stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.M.; van Vliet, P.; Metz, C.H.; Korfage, T.; Sluijter, J.P.; Doevendans, P.A.; Goumans, M.J. Human cardiomyocyte progenitor cells differentiate into functional mature cardiomyocytes: An in vitro model for studying human cardiac physiology and pathophysiology. Nat. Protoc. 2009, 4, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.; van Mil, A.; van Vliet, P.; Metz, C.H.; Liu, J.; Doevendans, P.A.; Goumans, M.J. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Liang, D.; Zhang, H.; Liu, Y.; Zhang, D.; Liu, Y.; Pan, L.; Chen, X.; Doevendans, P.A.; Sun, Y.; et al. MicroRNA-204 is required for differentiation of human-derived cardiomyocyte progenitor cells. J. Mol. Cell. Cardiol. 2012, 3, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Xu, X.; Deng, F.; Feng, J.; Zhang, H.; Liu, Y.; Zhang, Y.; Pan, L.; Liu, Y.; Zhang, D.; et al. miRNA-940 reduction contributes to human Tetralogy of Fallot development. J. Cell. Mol. Med. 2014, 18, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Srivastava, D. MicroRNA Regulation of Cardiac Development and Disease. In Heart Development and Regeneration; Academic Press: Waltham, MA, USA, 2010. [Google Scholar]
- Giraldez, A.J.; Cinalli, R.M.; Glasner, M.E.; Enright, A.J.; Thomson, J.M.; Baskerville, S.; Hammond, S.M.; Bartel, D.P.; Schier, A.F. MicroRNAs regulate brain morphogenesis in zebrafish. Science 2005, 308, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA thattargets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; de Boer, T.P.; Smits, A.M.; van Laake, L.W.; van Vliet, P.; Metz, C.H.; Korfage, T.H.; Kats, K.P.; Hochstenbach, R.; Pasterkamp, G.; et al. TGF-β1 induces efficient differentiation of human cardiomyocyte progenitor cells into functional cardiomyocytes in vitro. Stem Cell Res. 2007, 1, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Zhen, L.; Yuan, T.; Huang, J.; Deng, F.; Wuyahan; Zhang, H.; Pan, L.; Liu, Y.; The, E.; et al. miR-10a regulates proliferation of human cardiomyocyte progenitor cells by targeting GATA6. PLoS ONE 2014, 9, e103097. [Google Scholar] [CrossRef] [PubMed]
- Reinecke, H.; Minami, E.; Zhu, W.Z.; Laflamme, M.A. Cardiogenic differentiation and transdifferentiation of progenitor cells. Circ. Res. 2008, 103, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; van Mil, A.; Vrijsen, K.; Zhao, J.; Gao, L.; Metz, C.H.; Goumans, M.J.; Doevendans, P.A.; Sluijter, J.P. MicroRNA-155 prevents ne-crotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J. Cell. Mol. Med. 2011, 15, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, K.A.; Coxon, C.H.; Brooks, G. Can the cardiomyocyte cell cycle be reprogrammed? J. Mol. Cell. Cardiol. 2007, 42, 706–702. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, P.; Sdek, P.; MacLellan, W.R. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 2007, 87, 521–544. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.V.; Brooks, G. The mammalian cell cycle: An overview. Methods Mol. Biol. 2005, 296, 113–153. [Google Scholar] [PubMed]
- Leone, G.; DeGregori, J.; Jakoi, L.; Cook, J.G.; Nevins, J.R. Collaborative role of E2F transcriptional activity and G1 cyclin dependent kinase activity in the induction of S phase. Proc. Natl. Acad. Sci. USA 1999, 96, 6626–6631. [Google Scholar] [CrossRef] [PubMed]
- Ebelt, H.; Hufnagel, N.; Neuhaus, P.; Neuhaus, H.; Gajawada, P.; Simm, A.; Müller-Werdan, U.; Werdan, K.; Braun, T. Divergent siblings: E2F2 and E2F4 but not E2F1 and E2F3 induce DNA synthesis in cardiomyocytes without activation of apoptosis. Circ. Res. 2005, 96, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.D.; Lee, M.R.; Broxmeyer, H.E. 5-Aminoimidazole-4-carboxyamide ribonucleoside induces G1/S arrest and Nanog downregulation via p53 and enhances erythroid differentiation. Stem Cells 2012, 30, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.M.; Tam, W.L.; Ang, Y.S.; Gaughwin, P.M.; Yang, H.; Wang, W.; Liu, R.; George, J.; Ng, H.H.; Perera, R.J.; et al. MicroRNA-134 modulates the differentiation of mouse embryonic stem cells, where it causes post-transcriptional attenuation of Nanog and LRH1. Stem Cells 2008, 26, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Heine, P.; Dohle, E.; Bumsted-O’Brien, K.; Engelkamp, D.; Schulte, D. Evidence for an evolutionary conserved role of homothorax/Meis1/2 during vertebrate retina development. Development 2008, 135, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Louw, J.J.; Corveleyn, A.; Jia, Y.; Hens, G.; Gewillig, M.; Devriendt, K. MEIS2 involvement in cardiac development, cleft palate, and intellectual disability. Am. J. Med. Genet. 2015, 167, 1142–1146. [Google Scholar]
- Zha, Y.; Xia, Y.; Ding, J.; Choi, J.H.; Yang, L.; Dong, Z.; Yan, C.; Huang, S.; Ding, H.F. MEIS2 is essential for neuroblastoma cell survival and proliferation by transcriptional control of M-phase progression. Cell Death Dis. 2014, 5, e1417. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-H.; Zhao, H.; Zhou, L.-P.; Zhao, C.-X.; Wu, Y.-F.; Zhen, L.-X.; Li, J.; Ge, D.-X.; Xu, L.; Lin, L.; et al. miR-134 Modulates the Proliferation of Human Cardiomyocyte Progenitor Cells by Targeting Meis2. Int. J. Mol. Sci. 2015, 16, 25199-25213. https://doi.org/10.3390/ijms161025199
Wu Y-H, Zhao H, Zhou L-P, Zhao C-X, Wu Y-F, Zhen L-X, Li J, Ge D-X, Xu L, Lin L, et al. miR-134 Modulates the Proliferation of Human Cardiomyocyte Progenitor Cells by Targeting Meis2. International Journal of Molecular Sciences. 2015; 16(10):25199-25213. https://doi.org/10.3390/ijms161025199
Chicago/Turabian StyleWu, Ya-Han, Hong Zhao, Li-Ping Zhou, Chun-Xia Zhao, Yu-Fei Wu, Li-Xiao Zhen, Jun Li, Dong-Xia Ge, Liang Xu, Li Lin, and et al. 2015. "miR-134 Modulates the Proliferation of Human Cardiomyocyte Progenitor Cells by Targeting Meis2" International Journal of Molecular Sciences 16, no. 10: 25199-25213. https://doi.org/10.3390/ijms161025199
APA StyleWu, Y. -H., Zhao, H., Zhou, L. -P., Zhao, C. -X., Wu, Y. -F., Zhen, L. -X., Li, J., Ge, D. -X., Xu, L., Lin, L., Liu, Y., Liang, D. -D., & Chen, Y. -H. (2015). miR-134 Modulates the Proliferation of Human Cardiomyocyte Progenitor Cells by Targeting Meis2. International Journal of Molecular Sciences, 16(10), 25199-25213. https://doi.org/10.3390/ijms161025199