
Calcium Dyshomeostasis in Tubular Aggregate Myopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
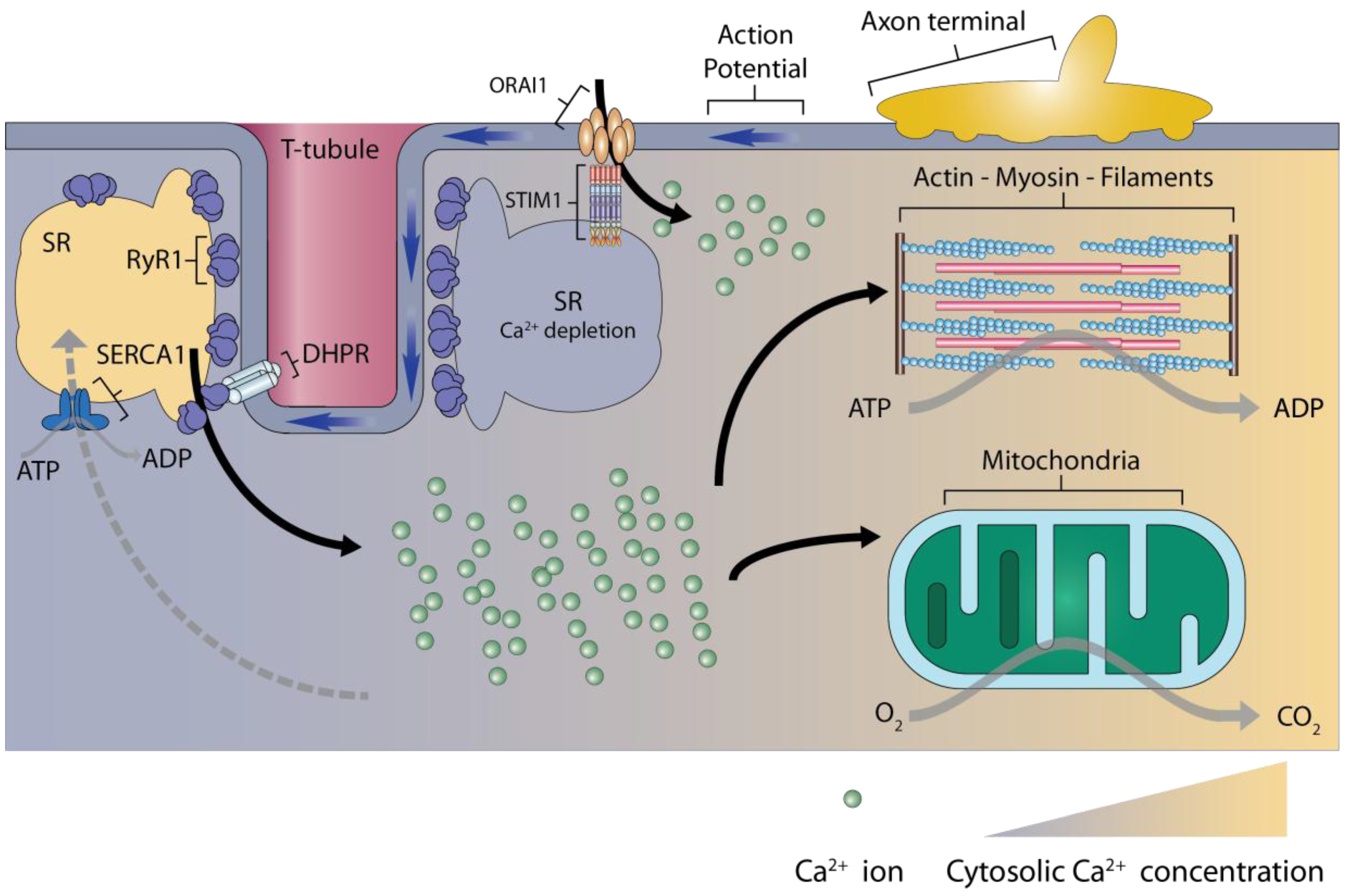
2. Calcium Signaling in Skeletal Muscles
3. Store-Operated Calcium Entry
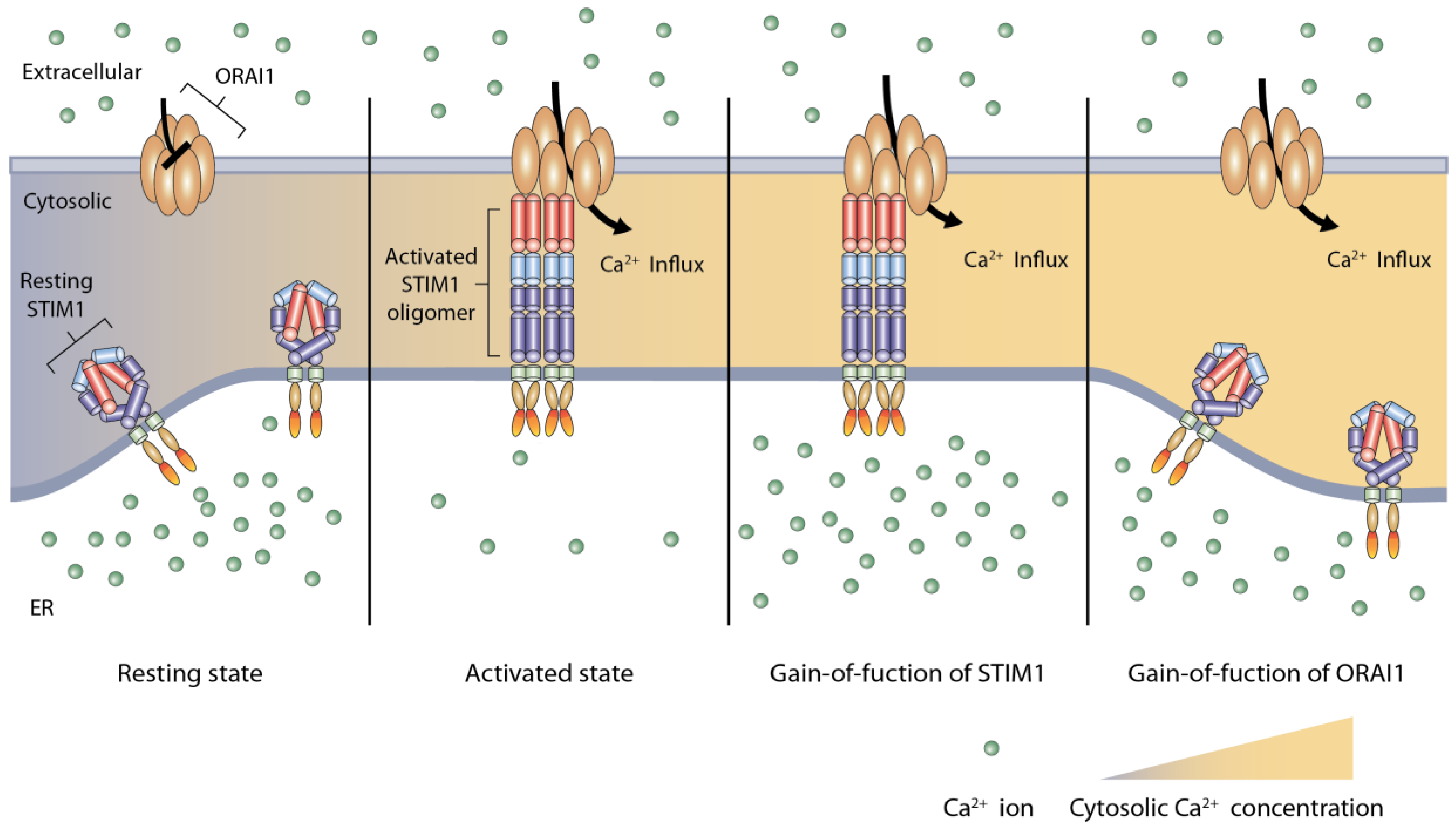
3.1. The Main Components of Store-Operated Calcium Entry
3.2. Activation of Store-Operated Calcium Entry by STIM1 Binding to ORAI1
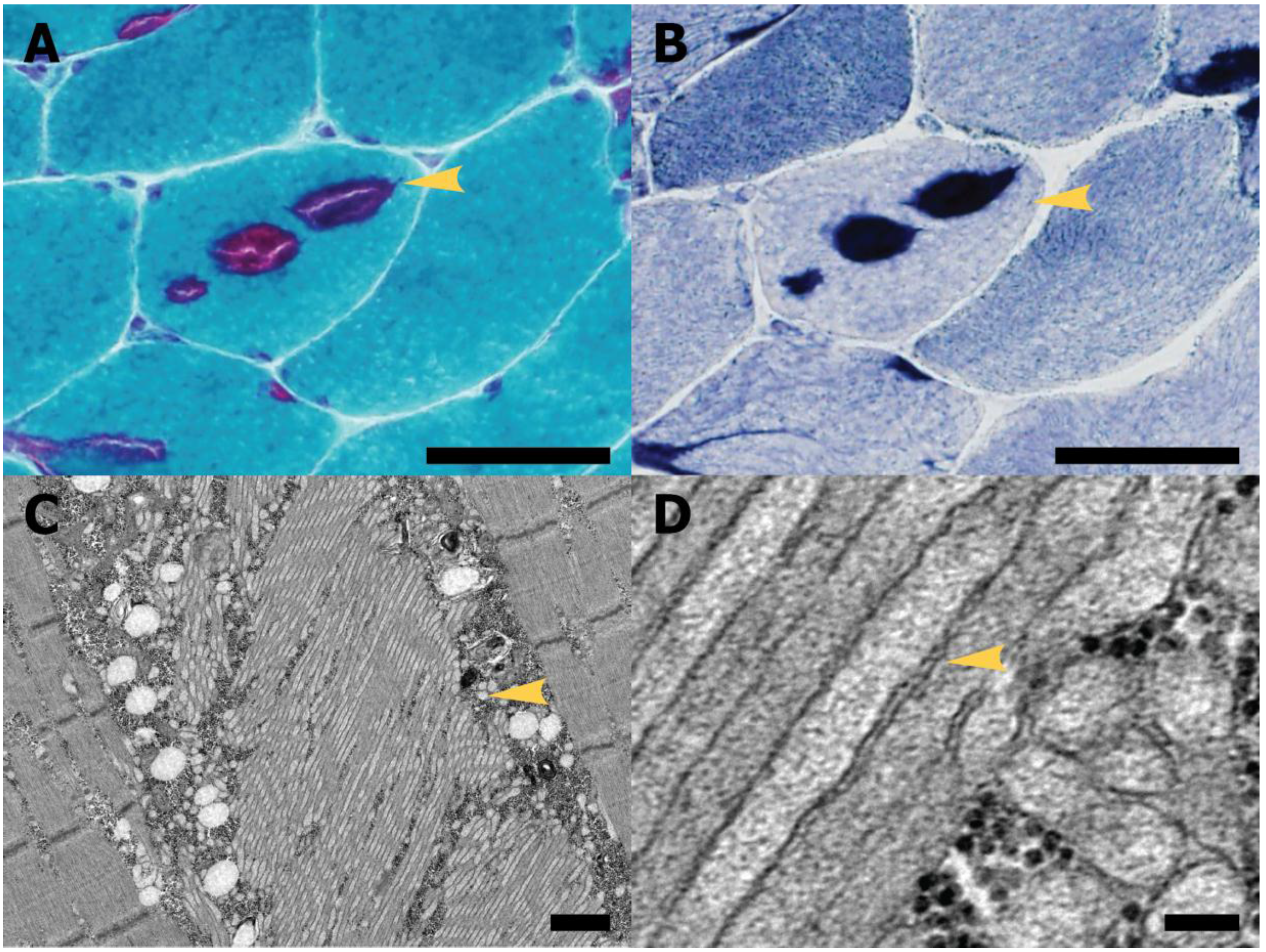
4. Tubular Aggregate Myopathy
4.1. Diagnosis and Pathological Features
4.2. Clinical Manifestations
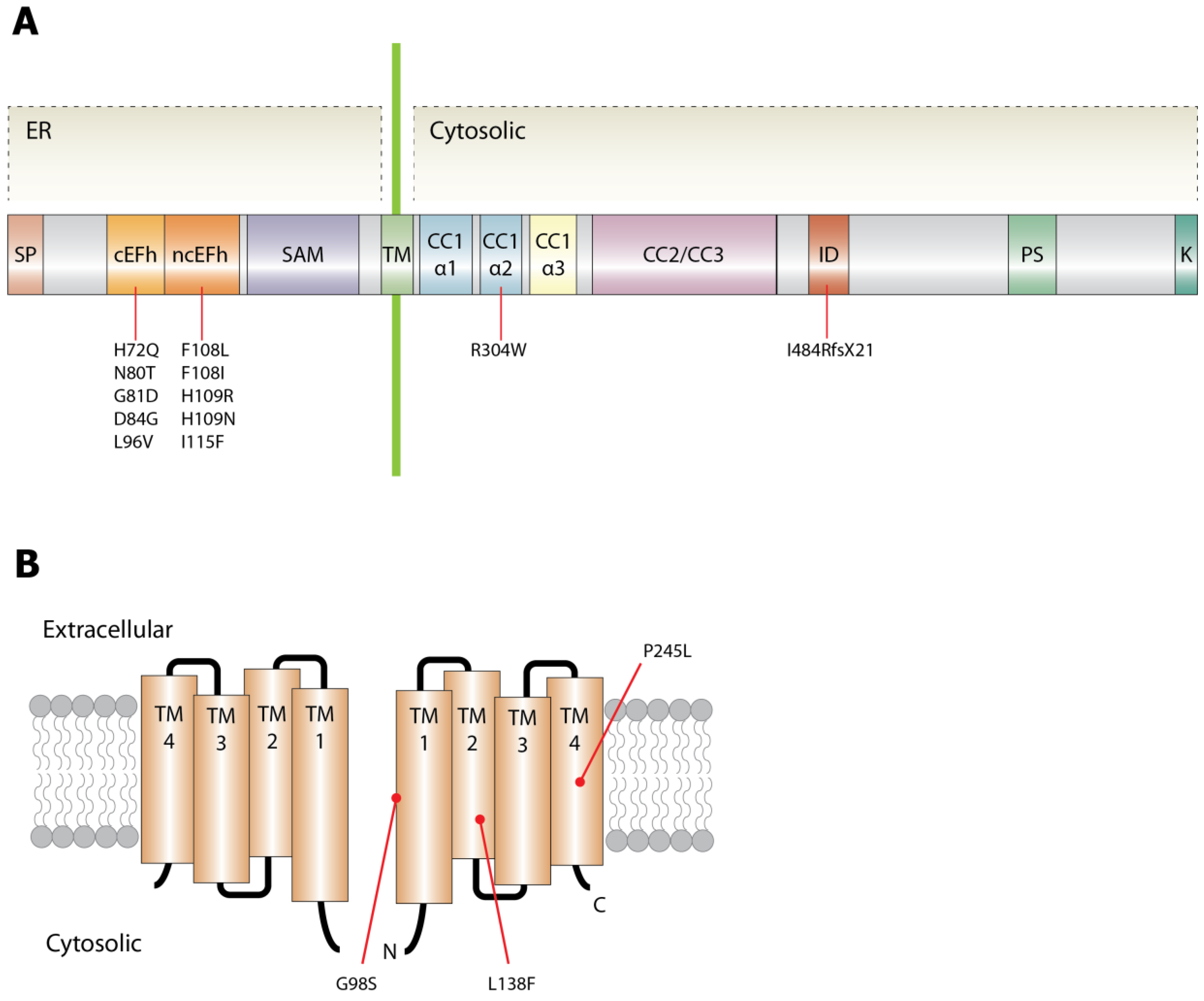
5. Genetic Causes and Possible Molecular Mechanism of Tubular Aggregate Myopathy
5.1. STIM1
5.2. ORAI1
6. Closing Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tanabe, T.; Beam, K.G.; Powell, J.A.; Numa, S. Restoration of excitation–contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature 1988, 336, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Gehlert, S.; Bloch, W.; Suhr, F. Ca2+-dependent regulations and signaling in skeletal muscle: From electro-mechanical coupling to adaptation. Int. J. Mol. Sci. 2015, 16, 1066–1095. [Google Scholar] [CrossRef] [PubMed]
- Franzini-Armstrong, C.; Ferguson, D.G.; Champ, C. Discrimination between fast- and slow-twitch fibres of guinea pig skeletal muscle using the relative surface density of junctional transverse tubule membrane. J. Muscle Res. Cell. Motil. 1988, 9, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Damiani, E.; Margreth, A. Characterization study of the ryanodine receptor and of calsequestrin isoforms of mammalian skeletal muscles in relation to fibre types. J. Muscle Res. Cell. Motil. 1994, 15, 86–101. [Google Scholar] [CrossRef] [PubMed]
- McKillop, D.F.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef]
- MacIntosh, B.R.; Holash, R.J.; Renaud, J.-M. Skeletal muscle fatigue–regulation of excitation–contraction coupling to avoid metabolic catastrophe. J. Cell. Sci. 2012, 125, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.K.; Kerrick, W.G. Characterization of the effects of Mg2+ on Ca2+- and Sr2+-activated tension generation of skinned skeletal muscle fibers. J. Gen. Physiol. 1975, 66, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [PubMed]
- Sacchetto, R.; Margreth, A.; Pelosi, M.; Carafoli, E. Colocalization of the dihydropyridine receptor, the plasma-membrane calcium ATPase isoform 1 and the sodium/calcium exchanger to the junctional-membrane domain of transverse tubules of rabbit skeletal muscle. Eur. J. Biochem. 1996, 237, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fujii, J.; Phillips, M.S.; Chen, H.S.; Karpati, G.; Yee, W.C.; Schrank, B.; Cornblath, D.R.; Boylan, K.B.; MacLennan, D.H. Characterization of cDNA and genomic DNA encoding SERCA1, the Ca2+-ATPase of human fast-twitch skeletal muscle sarcoplasmic reticulum, and its elimination as a candidate gene for Brody disease. Genomics 1995, 30, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Oyamada, H.; Demaurex, N.; Grinstein, S.; McCarthy, T.V.; MacLennan, D.H. Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J. Biol. Chem. 1997, 272, 26332–26339. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Donoso, P. Luminal calcium regulation of calcium release from sarcoplasmic reticulum. Biosci. Rep. 1995, 15, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Mickelson, J.R.; Louis, C.F. Malignant hyperthermia: excitation–contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol. Rev. 1996, 76, 537–592. [Google Scholar] [PubMed]
- Berchtold, M.W.; Brinkmeier, H.; Müntener, M. Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 2000, 80, 1215–1265. [Google Scholar] [PubMed]
- Jurkat-Rott, K.; Lehmann-Horn, F. Muscle channelopathies and critical points in functional and genetic studies. J. Clin. Investig. 2005, 115, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.; Carpenter, D.; Shaw, M.-A.; Halsall, J.; Hopkins, P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum. Mutat. 2006, 27, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, M.; van Engelen, B.G.M.; Küsters, B.; Lammens, M.; Meijer, R.; Molenaar, J.P.F.; Raaphorst, J.; Verschuuren-Bemelmans, C.C.; Straathof, C.S.M.; Sie, L.T.L.; et al. RYR1-related myopathies: A wide spectrum of phenotypes throughout life. Eur. J. Neurol. 2015, 22, 1094–1112. [Google Scholar] [CrossRef] [PubMed]
- Lynch, P.J.; Tong, J.; Lehane, M.; Mallet, A.; Giblin, L.; Heffron, J.J.; Vaughan, P.; Zafra, G.; MacLennan, D.H.; McCarthy, T.V. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc. Natl. Acad. Sci. USA. 1999, 96, 4164–4169. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, H.; Muller, C.R.; Halliger-Keller, B.; Brockington, M.; Brown, S.C.; Feng, L.; Chattopadhyay, A.; Mercuri, E.; Manzur, A.Y.; Ferreiro, A.; et al. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology 2002, 59, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, H.; Sewry, C.A.; Muntoni, F. Core myopathies. Semin. Pediatr. Neurol. 2011, 18, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Avila, G.; Dirksen, R.T. Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. J. Gen. Physiol. 2001, 118, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Avila, G.; O’Brien, J.J.; Dirksen, R.T. Excitation--contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 4215–4220. [Google Scholar] [CrossRef] [PubMed]
- Brody, I.A. Muscle contracture induced by exercise. A syndrome attributable to decreased relaxing factor. N. Engl. J. Med. 1969, 281, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, A.; Taschner, P.E.; Khanna, V.K.; Busch, H.F.; Karpati, G.; Jablecki, C.K.; Breuning, M.H.; MacLennan, D.H. Mutations in the gene-encoding SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+ ATPase, are associated with Brody disease. Nat. Genet. 1996, 14, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.E.; Dirksen, R.T. Sarcoplasmic reticulum: The dynamic calcium governor of muscle. Muscle Nerve 2006, 33, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, B. Regulation of oxidative phosphorylation through parallel activation. Biophys. Chem. 2007, 129, 93–110. [Google Scholar] [CrossRef] [PubMed]
- McMillin, J.B.; Madden, M.C. The role of calcium in the control of respiration by muscle mitochondria. Med. Sci. Sports Exerc. 1989, 21, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Robert, V.; Massimino, M.L.; Tosello, V.; Marsault, R.; Cantini, M.; Sorrentino, V.; Pozzan, T. Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 2001, 276, 4647–4651. [Google Scholar] [CrossRef] [PubMed]
- Territo, P.R.; French, S.A.; Dunleavy, M.C.; Evans, F.J.; Balaban, R.S. Calcium activation of heart mitochondrial oxidative phosphorylation: Rapid kinetics of mVO2, NADH, AND light scattering. J. Biol. Chem. 2001, 276, 2586–2599. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Gómez, L.J.; Salido, G.M.; Rosado, J.A. Dynamic interaction of hTRPC6 with the Orai1–STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca(2+) entry pathways. Biochem. J. 2009, 420, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.T.; Hwang, S.-Y.; Tomita, T.; DeHaven, W.I.; Mercer, J.C.; Putney, J.W. Activation and regulation of store-operated calcium entry. J. Cell. Mol. Med. 2010, 14, 2337–2349. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.-H.; Bonilla-Abadía, F.; Cañas, C.A.; Tobón, G.J. Calcium, channels, intracellular signaling and autoimmunity. Reumatol. Clin. 2014, 10, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D. Calcium signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011171. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Erxleben, C.; Yildirim, E.; Abramowitz, J.; Armstrong, D.L.; Birnbaumer, L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 4682–4687. [Google Scholar] [CrossRef] [PubMed]
- Stiber, J.; Hawkins, A.; Zhang, Z.-S.; Wang, S.; Burch, J.; Graham, V.; Ward, C.C.; Seth, M.; Finch, E.; Malouf, N.; et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Numaga, T.; Nishida, M.; Kiyonaka, S.; Kato, K.; Katano, M.; Mori, E.; Kurosaki, T.; Inoue, R.; Hikida, M.; Putney, J.W.; et al. Ca2+ influx and protein scaffolding via TRPC3 sustain PKCβ and ERK activation in B cells. J. Cell. Sci. 2010, 123, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Kiviluoto, S.; Decuypere, J.-P.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet. Muscle 2011, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Xu, P.; Li, Z.; Lu, J.; Liu, L.; Zhan, Y.; Chen, Y.; Hille, B.; Xu, T.; Chen, L. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc. Natl. Acad. Sci. USA 2008, 105, 13668–13673. [Google Scholar] [CrossRef] [PubMed]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J. Physiol. 2008, 586, 419–425. [Google Scholar] [CrossRef] [PubMed]
- McCarl, C.-A.; Picard, C.; Khalil, S.; Kawasaki, T.; Röther, J.; Papolos, A.; Kutok, J.; Hivroz, C.; LeDeist, F.; Plogmann, K.; et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 2009, 124, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; McCarl, C.-A.; Papolos, A.; Khalil, S.; Lüthy, K.; Hivroz, C.; LeDeist, F.; Rieux-Laucat, F.; Rechavi, G.; Rao, A.; et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N. Engl. J. Med. 2009, 360, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.; Abhyankar, A.; Lelarge, V.; Plancoulaine, S.; Palanduz, A.; Telhan, L.; Boisson, B.; Picard, C.; Dewell, S.; Zhao, C.; et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010, 207, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. Immunodeficiency due to defects in store-operated calcium entry. Ann. N. Y. Acad. Sci. 2011, 1238, 74–90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ramachandran, S.; Oh-Hora, M.; Rao, A.; Hogan, P.G. Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 4896–4901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yeromin, A.V.; Hu, J.; Amcheslavsky, A.; Zheng, H.; Cahalan, M.D. Mutations in Orai1 transmembrane segment 1 cause STIM1-independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc. Natl. Acad. Sci. USA 2011, 108, 17838–17843. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhou, M.-H.; Hu, C.; Kuo, E.; Peng, X.; Hu, J.; Kuo, L.; Zhang, S.L. Differential roles of the C and N termini of Orai1 protein in interacting with stromal interaction molecule 1 (STIM1) for Ca2+ release-activated Ca2+ (CRAC) channel activation. J. Biol. Chem. 2013, 288, 11263–11272. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Rensing-Ehl, A.; Speckmann, C.; Bengsch, B.; Schmitt-Graeff, A.; Bondzio, I.; Maul-Pavicic, A.; Bass, T.; Vraetz, T.; Strahm, B.; et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J. Immunol. 2012, 188, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Pan, Z. Retrograde activation of store-operated calcium channel. Cell Calcium. 2003, 33, 375–384. [Google Scholar] [CrossRef]
- Kundu, M.; Thompson, C.B. Autophagy: basic principles and relevance to disease. Annu. Rev. Pathol. 2008, 3, 427–455. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Yang, D.; Nagaraj, R.Y.; Nosek, T.A.; Nishi, M.; Takeshima, H.; Cheng, H.; Ma, J. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat. Cell Biol. 2002, 4, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Schaballie, H.; Rodriguez, R.; Martin, E.; Moens, L.; Frans, G.; Lenoir, C.; Dutré, J.; Canioni, D.; Bossuyt, X.; Fischer, A.; et al. A novel hypomorphic mutation in STIM1 results in a late-onset immunodeficiency. J. Allergy Clin. Immunol. 2015, 136, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Partiseti, M.; Le Deist, F.; Hivroz, C.; Fischer, A.; Korn, H.; Choquet, D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J. Biol. Chem. 1994, 269, 32327–32335. [Google Scholar] [PubMed]
- Wei-LaPierre, L.; Carrell, E.M.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 2013, 4, 2805. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Stathopulos, P.B.; Schindl, R.; Li, G.-Y.; Romanin, C.; Ikura, M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc. Natl. Acad. Sci. USA 2011, 108, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Srinivasan, P.; Razavi, S.; Seymour, S.; Meraner, P.; Gudlur, A.; Stathopulos, P.B.; Ikura, M.; Rao, A.; Hogan, P.G. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat. Struct. Mol. Biol. 2013, 20, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Noguchi, S.; Hara, Y.; Hayashi, Y.K.; Motomura, K.; Miyatake, S.; Murakami, N.; Tanaka, S.; Yamashita, S.; Kizu, R.; et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca2+ channels. Hum. Mol. Genet. 2015, 24, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Covington, E.D.; Wu, M.M.; Lewis, R.S. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol. Biol. Cell 2010, 21, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Yee, M.-K.W.; Gwack, Y.; Ribalet, B. The third transmembrane segment of orai1 protein modulates Ca2+ release-activated Ca2+ (CRAC) channel gating and permeation properties. J. Biol. Chem. 2011, 286, 35318–35328. [Google Scholar] [CrossRef] [PubMed]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.-C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Lange, I.; Feske, S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 2009, 385, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Stanley, C.; Isacoff, E.Y. Critical role for Orai1 C-terminal domain and TM4 in CRAC channel gating. Cell Res. 2015, 25, 963–980. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, J.; Xu, P.; Xie, X.; Chen, L.; Xu, T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J. Biol. Chem. 2007, 282, 29448–29456. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and regulation of TRPC channels in store-operated Ca2+ entry. Curr. Top. Membr. 2013, 71, 149–179. [Google Scholar] [PubMed]
- Sours-Brothers, S.; Ding, M.; Graham, S.; Ma, R. Interaction between TRPC1/TRPC4 assembly and STIM1 contributes to store-operated Ca2+ entry in mesangial cells. Exp. Biol. Med. (Maywood) 2009, 234, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Narayanappa, G.; Taly, A.B.; Chickbasavaiya, Y.T.; Mahadevan, A.; Vani, S.; Atchayaram, N.; Mohapatra, I.; Susarala, K.S. Tubular aggregate myopathy: A phenotypic spectrum and morphological study. Neurol. India 2010, 58, 747–751. [Google Scholar] [PubMed]
- Morgan-Hughes, J.A. Tubular aggregates in skeletal muscle: Their functional significance and mechanisms of pathogenesis. Curr. Opin. Neurol. 1998, 11, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Sharma, M.C.; Sarkar, C.; Suri, V.; Sharma, S.K.; Singh, S.; Das, T.K. Tubular aggregate myopathy: A rare form of myopathy. J. Clin. Neurosci. 2008, 15, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Chevessier, F.; Bauché-Godard, S.; Leroy, J.-P.; Koenig, J.; Paturneau-Jouas, M.; Eymard, B.; Hantaï, D.; Verdière-Sahuqué, M. The origin of tubular aggregates in human myopathies. J. Pathol. 2005, 207, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S. Tubular aggregates in skeletal muscle: just a special type of protein aggregates? Neuromuscul. Disord. 2012, 22, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Böhm, J.; Chevessier, F.; Maues De Paula, A.; Koch, C.; Attarian, S.; Feger, C.; Hantaï, D.; Laforet, P.; Ghorab, K.; Vallat, J.-M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar]
- Böhm, J.; Chevessier, F.; Koch, C.; Peche, G.A.; Mora, M.; Morandi, L.; Pasanisi, B.; Moroni, I.; Tasca, G.; Fattori, F.; et al. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J. Med. Genet. 2014, 51, 824–833. [Google Scholar]
- Hedberg, C.; Niceta, M.; Fattori, F.; Lindvall, B.; Ciolfi, A.; D'Amico, A.; Tasca, G.; Petrini, S.; Tulinius, M.; Tartaglia, M.; et al. Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J. Neurol. 2014, 261, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Okuma, H.; Saito, F.; Mitsui, J.; Hara, Y.; Hatanaka, Y.; Ikeda, M.; Shimizu, T.; Matsumura, K.; Shimizu, J.; Tsuji, S.; et al. Tubular aggregate myopathy caused by a novel mutation in the cytoplasmic domain of STIM1. Neurol. Genet. 2016, 2, e50. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Rossius, M.; Zitzelsberger, M.; Vorgerd, M.; Müller-Felber, W.; Ertl-Wagner, B.; Zhang, Y.; Brinkmeier, H.; Senderek, J.; Schoser, B. 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul. Disord. 2015, 25, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Böhm, J.; Vasli, N.; Malfatti, E.; Le Gras, S.; Feger, C.; Jost, B.; Monnier, N.; Brocard, J.; Karasoy, H.; Gérard, M.; et al. An integrated diagnosis strategy for congenital myopathies. PLoS ONE 2013, 8, e67527. [Google Scholar]
- Tasca, G.; D’Amico, A.; Monforte, M.; Nadaj-Pakleza, A.; Vialle, M.; Fattori, F.; Vissing, J.; Ricci, E.; Bertini, E. Muscle imaging in patients with tubular aggregate myopathy caused by mutations in STIM1. Neuromuscul. Disord. 2015, 25, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Stormorken, H.; Sjaastad, O.; Langslet, A.; Sulg, I.; Egge, K.; Diderichsen, J. A new syndrome: thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis. Clin. Genet. 1985, 28, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Misceo, D.; Holmgren, A.; Louch, W.E.; Holme, P.A.; Mizobuchi, M.; Morales, R.J.; De Paula, A.M.; Stray-Pedersen, A.; Lyle, R.; Dalhus, B.; et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum. Mutat. 2014, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Morin, G.; Bruechle, N.O.; Singh, A.R.; Knopp, C.; Jedraszak, G.; Elbracht, M.; Brémond-Gignac, D.; Hartmann, K.; Sevestre, H.; Deutz, P.; et al. Gain-of-function mutation in STIM1 (P.R304W) is associated with Stormorken syndrome. Hum. Mutat. 2014, 35, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Shahrizaila, N.; Lowe, J.; Wills, A. Familial myopathy with tubular aggregates associated with abnormal pupils. Neurology 2004, 63, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Grosse, J.; Braun, A.; Varga-Szabo, D.; Beyersdorf, N.; Schneider, B.; Zeitlmann, L.; Hanke, P.; Schropp, P.; Mühlstedt, S.; Zorn, C.; et al. An EF hand mutation in Stim1 causes premature platelet activation and bleeding in mice. J. Clin. Investig. 2007, 117, 3540–3550. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 2016, 310, C643–C662. [Google Scholar] [PubMed]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.-J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Lhermusier, T.; Chap, H.; Payrastre, B. Platelet membrane phospholipid asymmetry: From the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. J. Thromb. Haemost. 2011, 9, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Markello, T.; Chen, D.; Kwan, J.Y.; Horkayne-Szakaly, I.; Morrison, A.; Simakova, O.; Maric, I.; Lozier, J.; Cullinane, A.R.; Kilo, T.; et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol. Genet. Metab. 2015, 114, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Wu, M.J.; Ju, Y.K.; Marciniec, T.; Yeoh, T.; Allen, D.G.; Harvey, R.P.; Graham, R.M. Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signalling pathway. Nature 1999, 400, 576–581. [Google Scholar] [PubMed]
- Sharma, S.; Quintana, A.; Findlay, G.M.; Mettlen, M.; Baust, B.; Jain, M.; Nilsson, R.; Rao, A.; Hogan, P.G. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 2013, 499, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Jung, H.-J.; Kim, K.-D.; Souda, P.; Whitelegge, J.; Gwack, Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat. Cell Biol. 2010, 12, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER-PM junctions identifies STIMATE as a regulator of Ca²⁺ influx. Nat. Cell Biol. 2015, 17, 1339–1347. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-M.; Noguchi, S. Calcium Dyshomeostasis in Tubular Aggregate Myopathy. Int. J. Mol. Sci. 2016, 17, 1952. https://doi.org/10.3390/ijms17111952
Lee J-M, Noguchi S. Calcium Dyshomeostasis in Tubular Aggregate Myopathy. International Journal of Molecular Sciences. 2016; 17(11):1952. https://doi.org/10.3390/ijms17111952
Chicago/Turabian StyleLee, Jong-Mok, and Satoru Noguchi. 2016. "Calcium Dyshomeostasis in Tubular Aggregate Myopathy" International Journal of Molecular Sciences 17, no. 11: 1952. https://doi.org/10.3390/ijms17111952
APA StyleLee, J. -M., & Noguchi, S. (2016). Calcium Dyshomeostasis in Tubular Aggregate Myopathy. International Journal of Molecular Sciences, 17(11), 1952. https://doi.org/10.3390/ijms17111952