1. Introduction
Pyruvate dehydrogenase kinases (PDKs) are key enzymes in glucose metabolism since they negatively regulate the activity of pyruvate dehydrogenase complex (PDC) by phosphorylation [
1,
2]. PDC is an important gatekeeper enzyme that catalyzes the oxidative decarboxylation of pyruvate to produce acetyl CoA [
3]. The mammalian PDC is a 9.5 million Dalton multi-enzyme complex consisting of pyruvate dehydrogenase (E1), dihydrolipoyltransacetylase (E2), dihydrolipoamide dehydrogenase (E3) and the E3-bindingprotein (E3BP). PDKs are non-covalently bound to the lipoyl domain (L2) of the E2 subunit of PDC. They phosphorylate the pyruvate dehydrogenase (PDH)-E1α and inactivate PDC [
4,
5,
6,
7]. In mammalians, there are four isoenzymes of PDKs (PDK 1 to 4) with different binding affinity, phosphorylation site specificity and tissue distribution [
8,
9]. The binding affinities of four isoenzymes to L2 are in the following order: PDK3 > PDK1 ≈ PDK2 > PDK4. As for the phosphorylation sites (S264, S271 and S203), only PDK1 is reported to phosphorylate all three sites, while other isoenzymes can only phosphorylate S264 and S271 with different rates. Four isoenzymes have different tissue distribution: PDK1 is present preferentially in the pancreatic islets, heart and skeletal muscles; PDK2 is expressed universally in all tissues; PDK3 is abundant in the kidney, brain and testes; PDK4 is more easily detected in the pancreatic islets, heart, kidney and skeletal muscle.
The PDKs are potential therapeutic targets since the up-regulation of PDKs are associated with many diseases such as cancer, diabetes, obesity and heart failure [
10,
11,
12,
13,
14]. Several recent studies have provided evidence that PDKs play major roles in the metabolism of cancer cells, since the inactivation of PDC is tightly associated with the “Warburg effect” [
15,
16]. It has been reported that, PDKs are universally overexpressed in a variety of cancer cells, such as multiple myeloma and breast cancer. Moreover, the expression and activities of the PDKs are strongly regulated by oncogenes [
17,
18,
19]. Type 2 diabetes and obesity are characterized by dysregulation of glucose and lipid metabolism, in which PDC plays a pivotal role [
20]. Increasing the activity of PDC through inhibition of PDKs could improve glucose oxidation and reduce the blood glucose concentration, thus ultimately promoting insulin activity. Thus, development of potent PDK inhibitors could provide powerful treatment for cancers and metabolic diseases.
In recent years, small molecule inhibitors with different binding sites have been reported to regulate the activities of PDKs [
21]. Dichloroacetate (DCA) is a structural analog of pyruvate, binding to the regulatory domain of PDKs to regulate their activities [
22,
23]. AZD7545 binds to the lipoamide-binding site of PDKs, and disrupts the interaction between PDKs and PDC component, increasing the activity of PDC indirectly [
24,
25]. Radicicol and M77976 bind to the nucleotide-binding pocket and inactivate PDKs by blocking ATP entry to the pocket [
26]. There are also some other small molecule inhibitors with similar mechanisms, such as JX06 [
27] and VER-246608 [
28]. Among all these inhibitors, only DCA has entered clinical trials. Although significant progress has been made in the research of PDK inhibitors with biological activity, design and development of compounds with novel skeleton for therapeutic use are still focus of attention.
The use of natural products has a long history in medicine. Natural products and their derivatives represent a major part of today’s pharmaceutical market [
29,
30]. They also play an important role as tool compounds in molecular biological research, such as pathway screening and validation of target identification [
31,
32,
33]. With the advantage of their innate affinity for biological receptors, natural compounds may provide a valuable source for the development of PDK-based drugs. To discover new PDKs inhibitors, we carried out a virtual screening against the Natural Products database (NP) in the ZINCdatabase [
34]. ADME (absorption, distribution, metabolism, excretion) and toxicity properties of the obtained screened compounds were analyzed to examine drug like properties. Binding modes of the selected candidate compounds obtained from the docking were studied by visual inspection and their binding stabilities were further evaluated by performing 40 ns molecular dynamics simulation via Discovery Studio 3.5.
3. Discussion
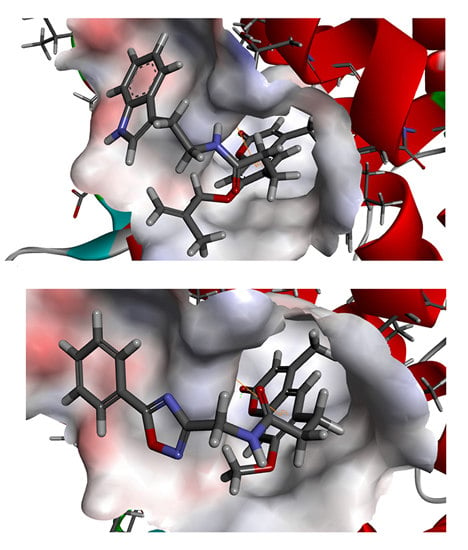
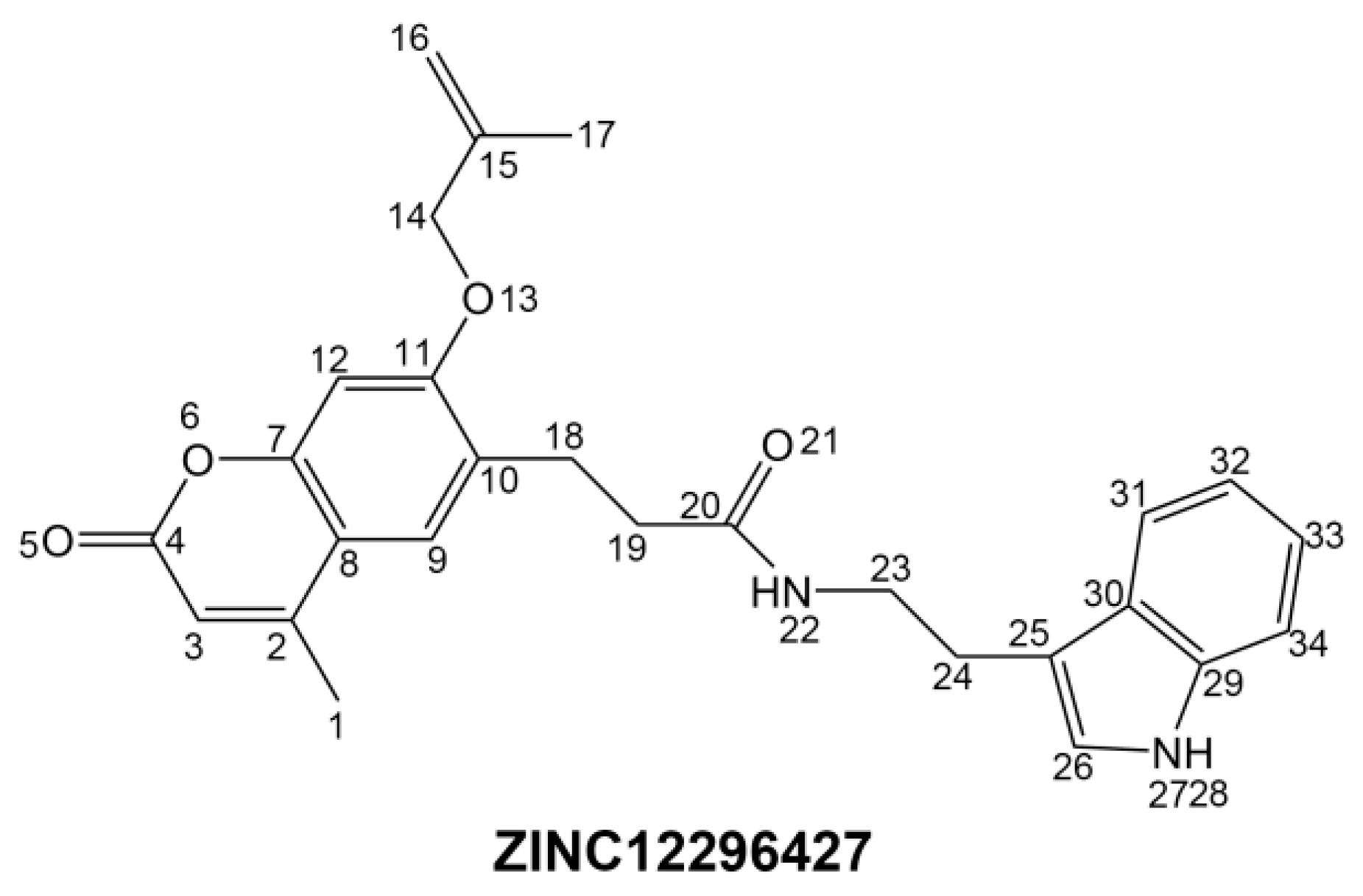
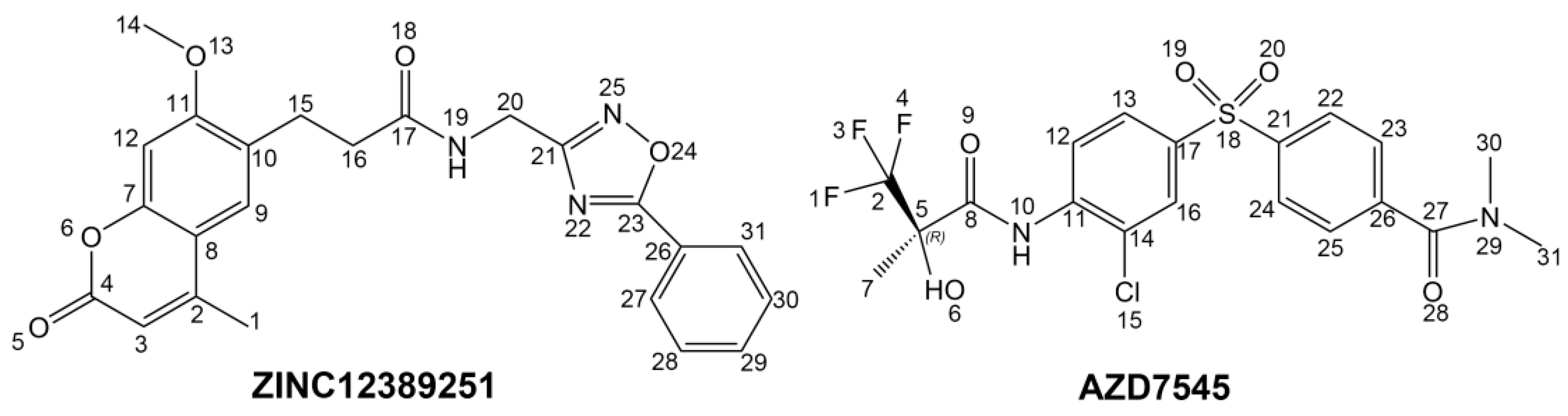
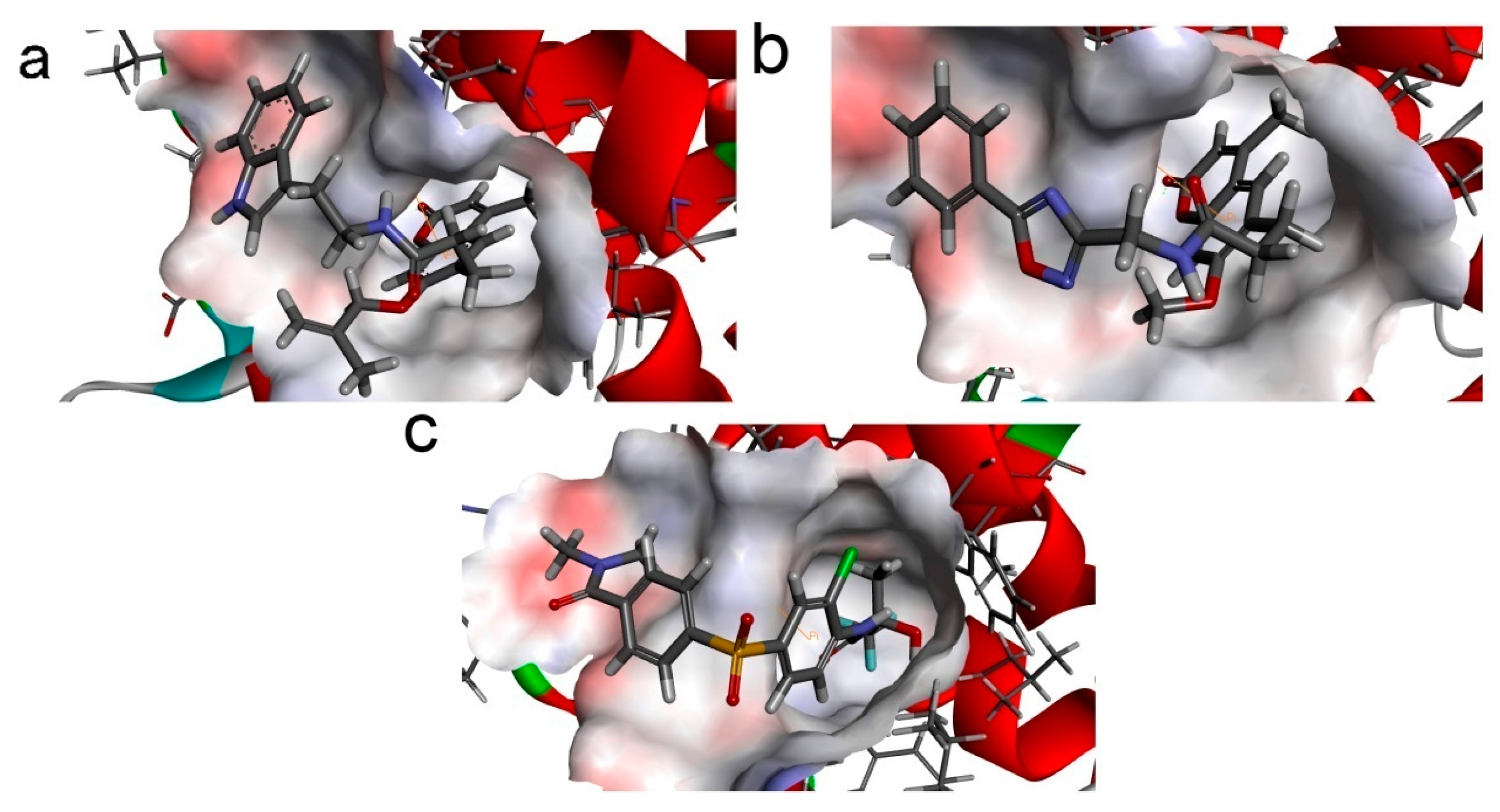
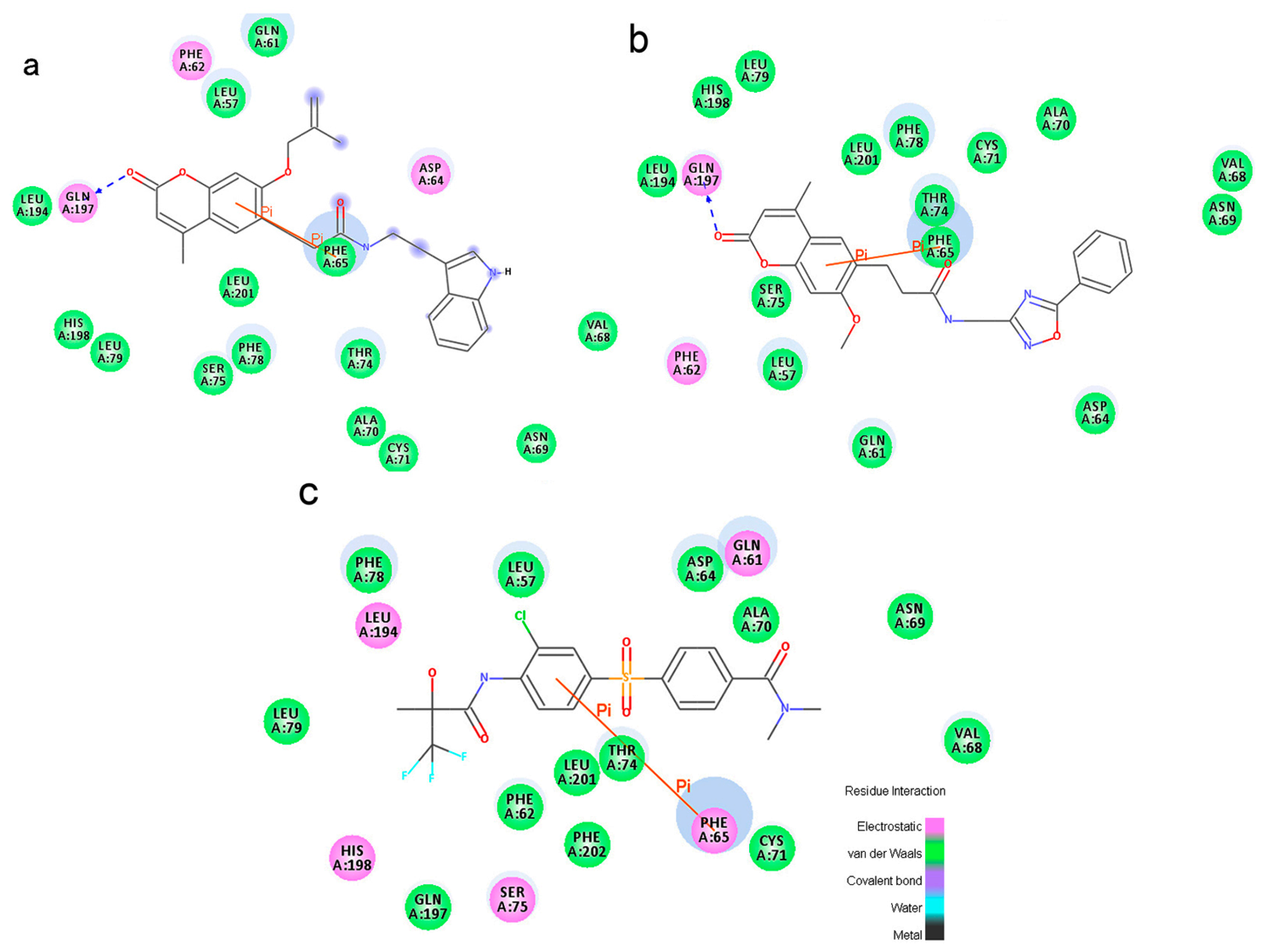
To phosphorylate the pyruvate dehydrogenase (E1 of PDC), PDKs need to interact with the L2 domain of dihydrolipoyltransacetylase (E2 of PDC) through their lipoamide binding pockets. Inhibitors binding to the lipoamide binding pockets can uncouple the link between PDKs and PDC, and then upregulate PDC activity indirectly. In this study, we identified two novel natural compounds, ZINC12296427 and ZINC12389251, from ZINC database to effectively inhibit PDKs through virtual screening technique. Molecular docking study showed that the two novel compounds could bind to the lipoamide-binding pocket of PDKs through forming of hydrogen bonds and Pi-Pi interactions with receptors. Moreover, these two compounds were predicted with no hepatotoxicity, Ames mutagenicity, rodent carcinogenicity and developmental toxicity. Thus, our findings provided potential mechanism of two novel inhibitors of PDKs for clinical drug design. Since the compounds were virtually screened and their inhibitory activities have not been reported, experiments such as IC50 and EC50 measurements will be carried out in our further study to examine their bioactivities.
Our results of the binding mechanism study also showed that the functional groups of these two compounds are their coumarins group. Coincidentally, recent studies have reported the antitumor activities of coumarins and their derivatives [
41]. Our results suggest that upregulation of mitochondria aerobic metabolism of cancer cells through inhibition of PDKs might contribute to the anticancer activities of this kind of compound. This is also probably one possible mechanism for the anticancer activity of coumarones, which have similar structure to coumarins. Further studies are needed to determine the exact mechanism. Coumarins have broad pharmacological activities, such as anti-coagulant, anti-viral, anti-inflammatory and anti-microbial effects [
42]. Therefore, development of novel bioactivities of natural and synthetic coumarins based on their PDKs inhibition activities is of great significance for the treatment of metabolic diseases.
4. Materials and Methods
4.1. Docking Software and Ligand Library
Libdock, and ADMET modules of Discovery Studio 3.5 software (DS3.5, Accelrys, Inc.) were used for virtual screening and ADMET analyzing of inhibitors. CDOCKER and AutoDock were used for docking study. A Natural Products database (NP) in the ZINC database was employed to screen PDKs inhibitors.
4.2. Structure-Based Virtual Screening Using Libdock
Libdock is a rigid-based docking program. It calculates hotspots for the protein using a grid placed into the binding site and using polar and apolar probes (San Diego, CA, USA). Then the hot spots are further used to align the ligands to form favourable interaction. The Smart Minimiser algorithm and CHARMm force field (Cambridge, MA, USA) were used for ligands minimization. After minimized, all the ligand poses are ranked based on the ligands score. The 1.9 Å crystal structure of human pyruvate dehydrogenase kinase 1 (PDK1) in complex with AZD7545 (PDB ID: 2Q8G) were downloaded from protein data bank (PDB) and imported to the working environment of Libdock. The protein was prepared by removing crystal water and other hetero atoms (except AZD7545), followed by addition of hydrogen, protonation, ionization and energy minimization. The CHARMm [
43] force field and the Smart Minimiser algorithm were applied for energy minimization. The minimization performed 2000 steps with an RMS gradient tolerance of 0.1, and the final RMS gradient was 0.09463. The prepared protein was used to define the binding site from the “Edit binding site” option on the receptor-ligand interaction tool bar. Using the bound ligands (AZD7545) binding positions, the active sites for docking were generated. Virtual screening was carried out by docking all the prepared ligands at the defined active site using Libdock. Based on the Libdock score, all the docked poses were ranked and grouped by name. All compounds were ranked according to their Libdock score.
4.3. Molecular Docking
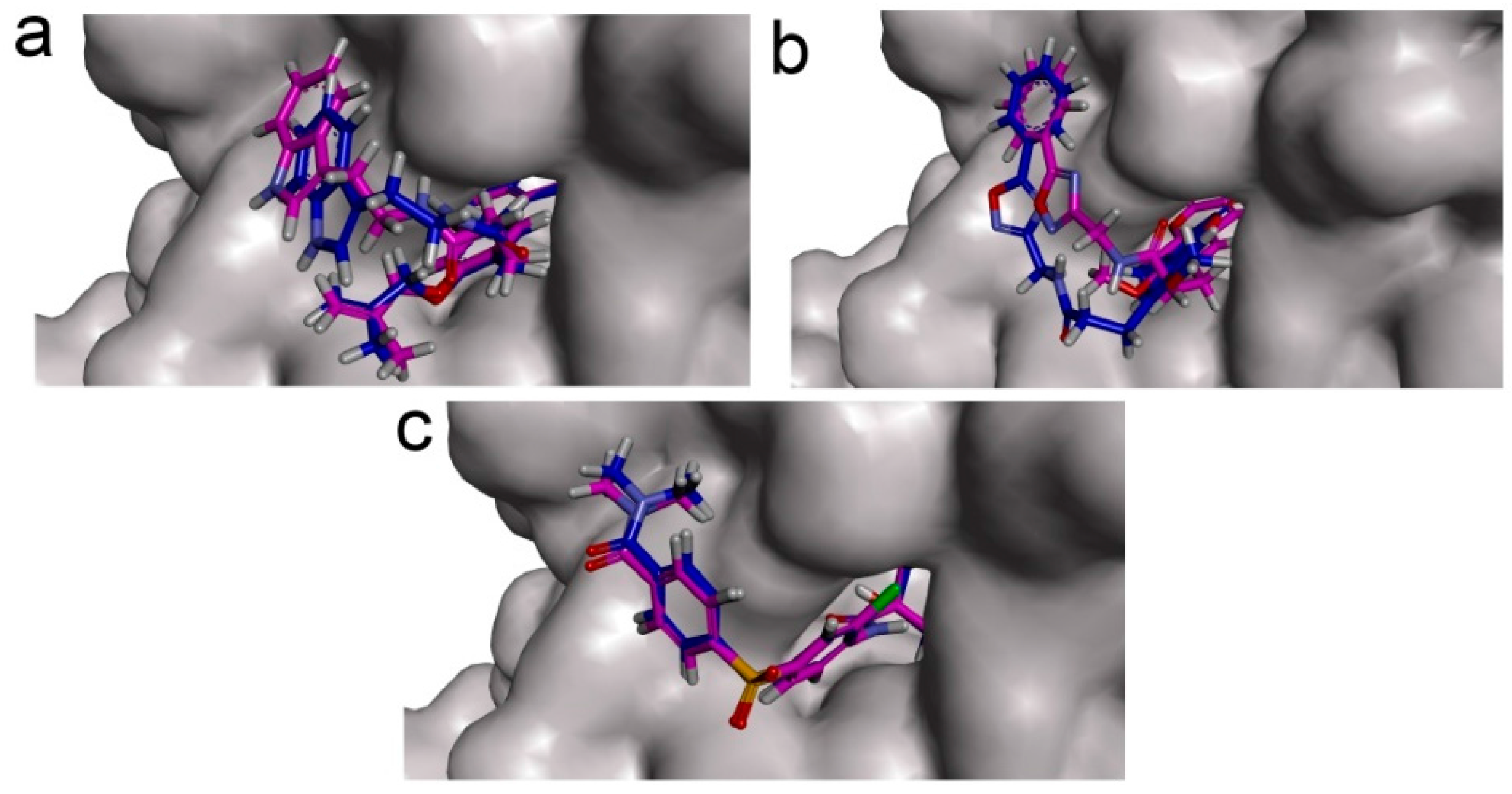
CDOCKER module of Discovery Studio was used for molecular docking study. CDOCKER is an implementation of a CHARMm based docking tool. The receptor is held rigid while the ligands are allowed to flex during the docking process. For each complex pose, the CHARMm energy (interaction energy plus ligand strain) and the interaction energy, which indicate ligand binding affinity, are calculated. Crystal structure of PDK1 (PDB ID: 2Q8G, 1.9 Å), PDK2 (PDB ID: 2BU6, 2.4 Å) [
44], PDK3 (PDB ID: 2Q8I, 2.6 Å) [
25] and PDK4 (PDB ID: 2ZDX, 2.5 Å) [
26] were obtained from the protein data bank. The crystal water molecules are generally removed in rigid and semi-flexible docking process [
45,
46], since the fixed water molecules might affect the formation of receptor-ligand complex. The water molecules were removed and hydrogen atoms were added to the protein. The 3D structures of ZINC12296427 and ZINC12389251 were obtained from ZINC database (no experimental crystal structures available in crystallographic databases). The CHARMm forcefield was used for receptors and ligands. The binding site spheres of PDK1 and PDK2 were defined as the regions that come within radius 15 Å from the geometric centroid of the ligands AZD7545, AZ12, respectively. The binding site spheres of PDK3 and PDK4 were defined by structural alignment with PDK1. During the docking process, the ligands were allowed to bind to the residues within the binding site spheres. After being extracted from the binding site, the initial compound AZD7545 was re-docked into the crystal structure of PDK1. The RMSD between the docked pose and the crystal structure of the complex was 0.6 Å, indicating the CDOCKER module was highly reliable for reproducing the experimentally observed binding mode of PDKs. The structures of identified hits were prepared and docked into the lipoamide-binding pocket of PDKs. Different poses for each test molecule were generated and analyzed on the basis of CDOCKER interaction energy.
AutoDock 4.2 (The Scripps Research Institute, (La Jolla, CA, USA) was used to crosscheck the docking results of CDOCKER. The AutoDockTools 1.5.6 was used to prepare the protein and ligands for docking procedure. All solvent molecules, water molecules, and the cocrystallized ligand were removed from the structure; Kollman charges and polar hydrogens were added. AutoGrid (La Jolla, CA, USA) was used to generate the grid maps. Each grid was centered at the lipoamid binding site of the PDKs. The grid dimensions were 45 points in each dimension separated by 0.375 Å. The files were generated as PDBQT format. For all ligands, random starting positions, orientations and torsions were used. PDBQT file of the ligands was generated with all the default values accepted. The Genetic Algorithm was used with 2,500,000 energy evaluations and a population of 300 individuals; 100 runs were carried out. Following docking, the results were clustered into groups with RMSD <2.0 Å. The ranking of the clusters was performed based on the lowest energy representative of each cluster. Visualization and analysis of interactions between protein and ligand were performed using DS3.5.
4.4. Molecular Dynamics Simulation
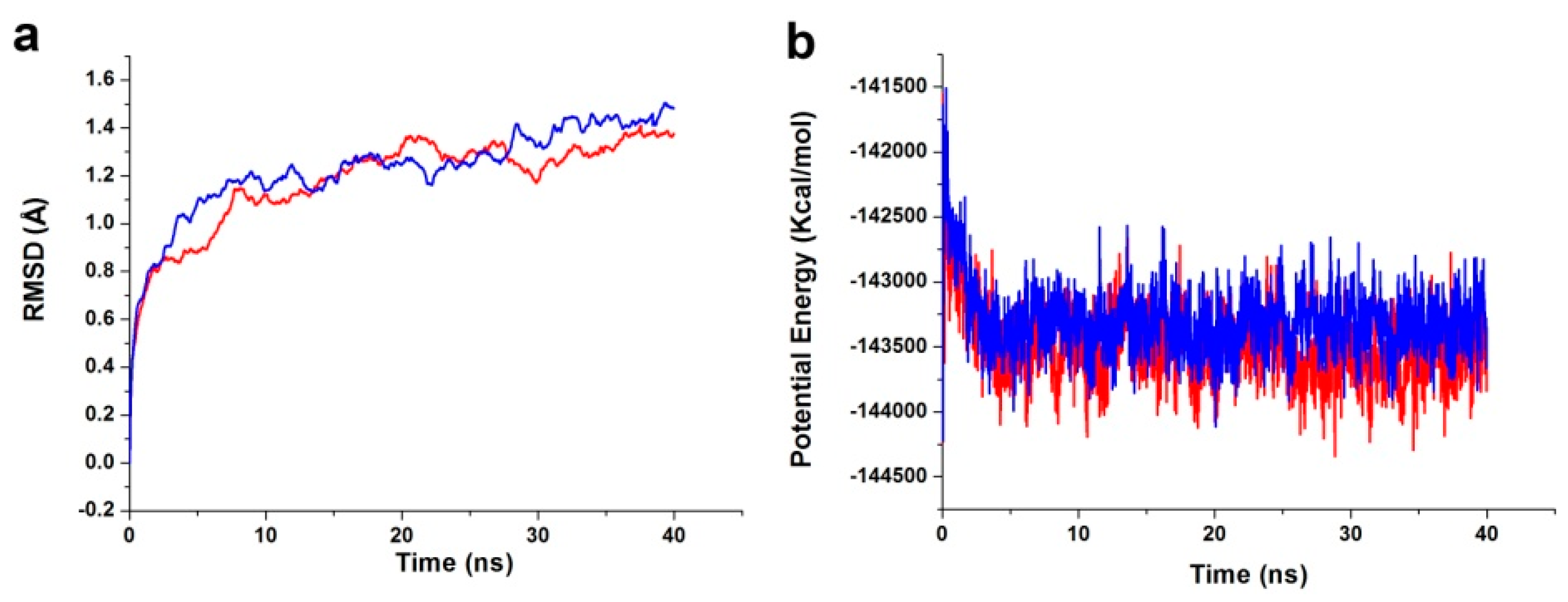
The best binding conformations of the PDK1-inhibitor complexes among the poses predicted by the molecular docking program were selected and used as MD simulation starting points. The ligand-receptor complex was put into an orthorhombic box and solvated with an Explicit Periodic Boundary solvation water model. The size of the box was 88 × 75 × 72 Å, containing 13,802 water molecules. In order to simulate the physiological environment, sodium chloride were added to the system with the ionic strength of 0.145. Then, the system was subjected to the CHARMm force-field and relaxed by energy minimization (1000 steps of steepest descent and 1000 steps of conjugated gradient), with the final RMS gradient of 0.08326. The system was slowly driven from an initial temperature of 50 K to the target temperature of 300 K for 2 ns and equilibration simulations were run for 1 ns. The MD simulations (production) were performed for 40 ns with time step of 2 fs. The simulation was performed with the NPT (normal pressure and temperature) system at a constant temperature of 300 K and the results were saved at a frequency of 0.02 ns. The Particle Mesh Ewald (PME) algorithm was used to calculate long range electrostatics, and the linear constraint solver (LINCS) algorithm was adapted to fix all bonds involving hydrogen. The initial complex was set as a reference. The MD trajectory was determined for structural properties, root mean-square deviation (RMSD), and potential energy by using the Discovery Studio 3.5 analyze trajectory protocol (San Diego, CA, USA).
4.5. ADME and Toxicity Prediction
The ADMET and TOPKAT modules of DS were employed to calculate the ADMET properties of the compounds, such as their aqueous solubility, blood-brain barrier (BBB) penetration, cytochrome P450 2D6 (CYP2D6) inhibition, hepatotoxicity, human intestinal absorption, plasma protein binding (PPB) level, rodent carcinogenicity, Ames mutagenicity and developmental toxicity potential.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}