



Int. J. Mol. Sci. 2017, 18(1), 82; https://doi.org/10.3390/ijms18010082 - 3 Jan 2017
Cited by 32 | Viewed by 8459
Abstract
►
Show Figures
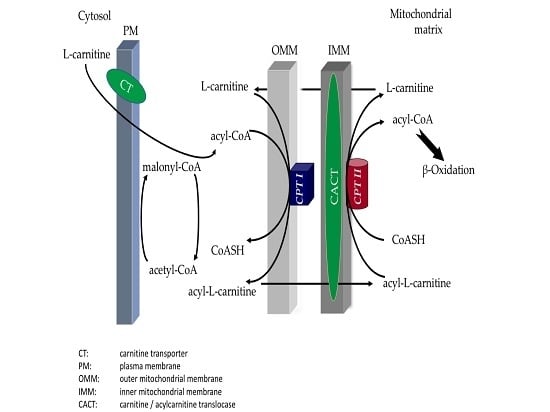
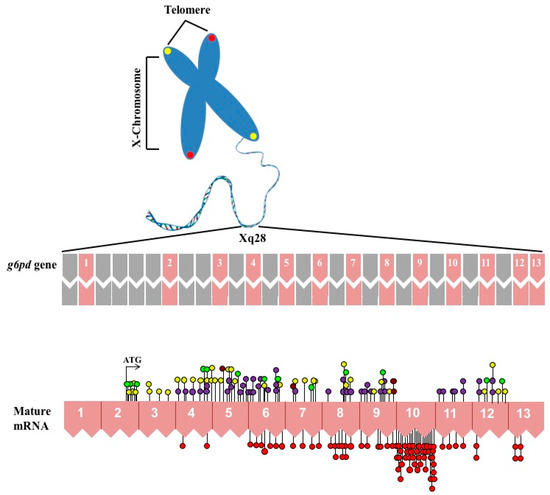
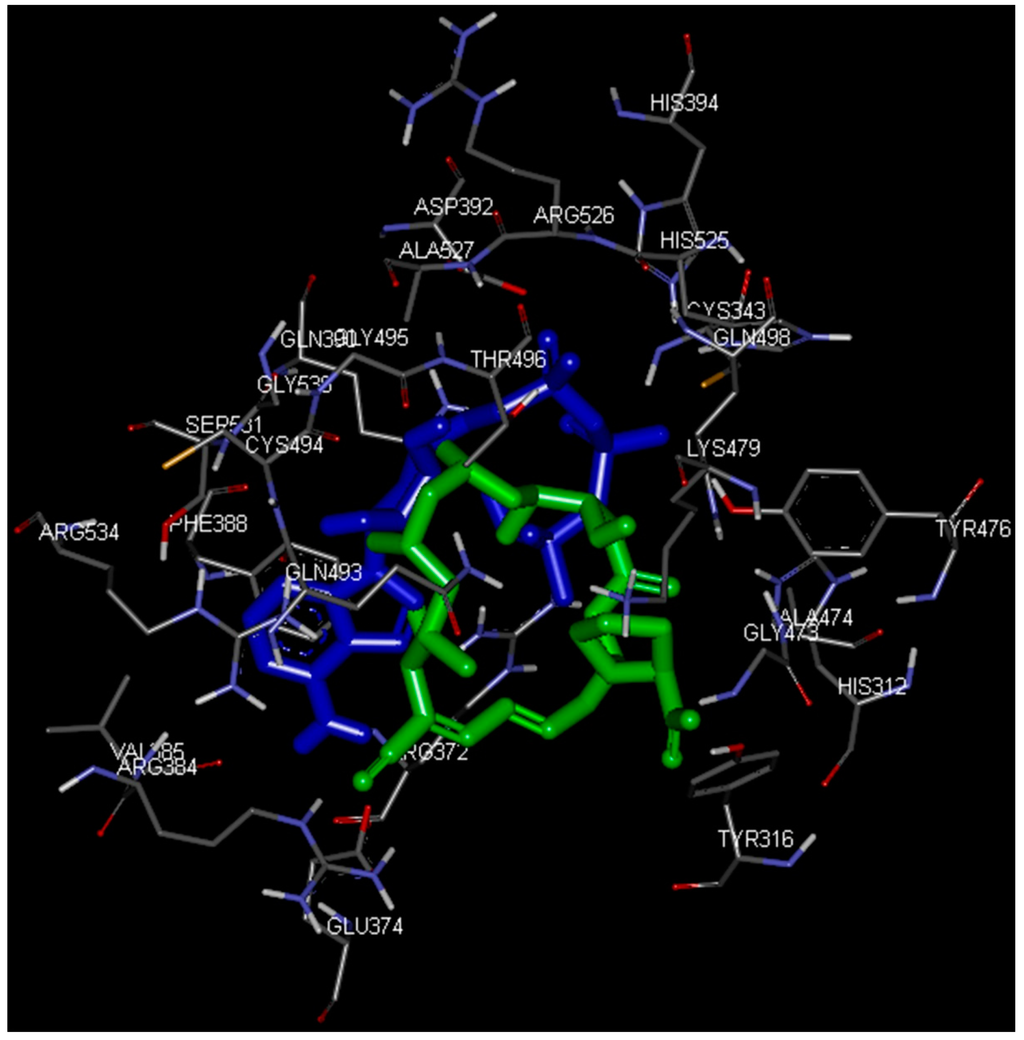
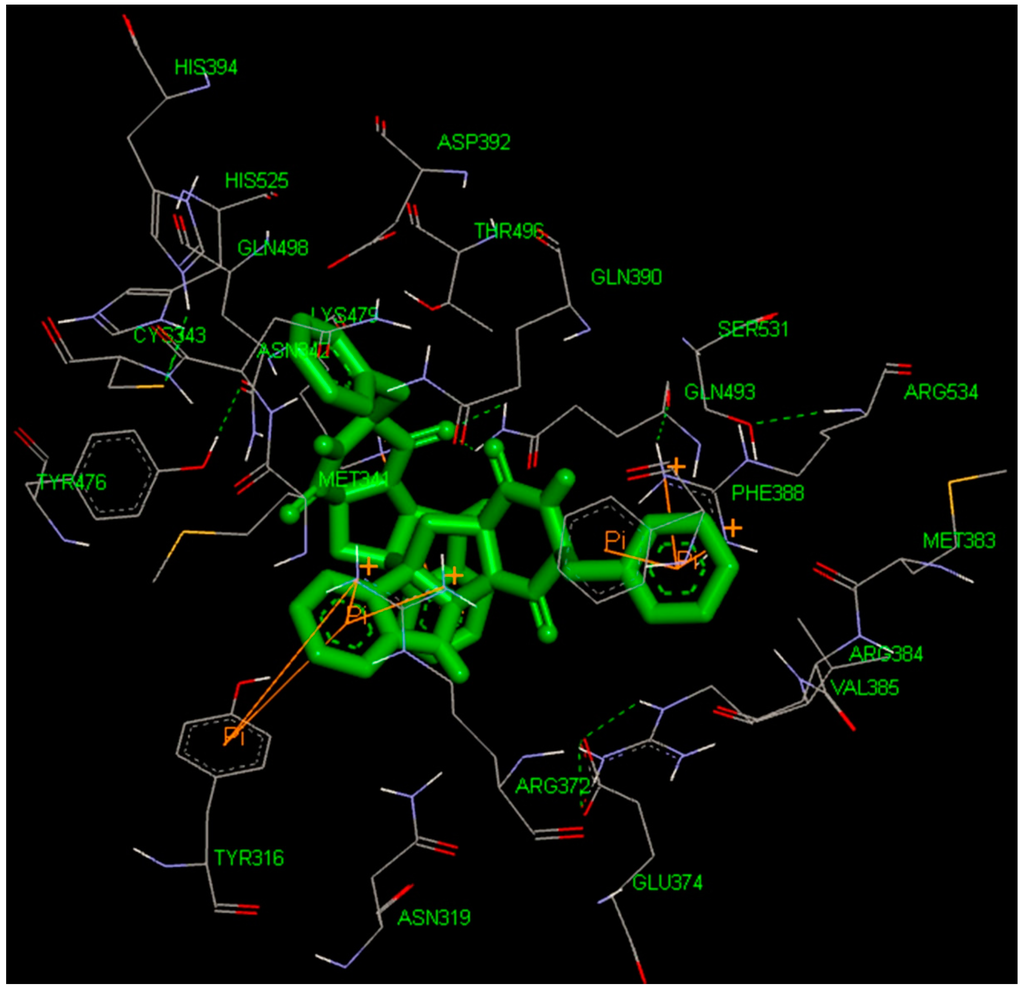
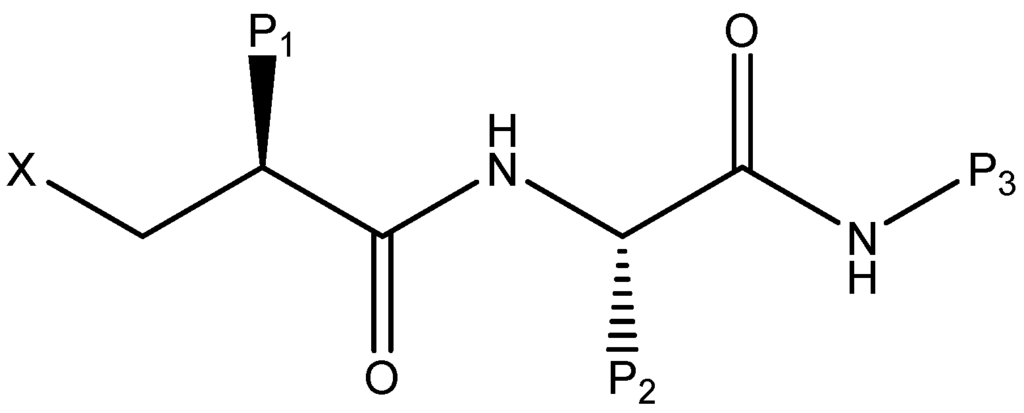
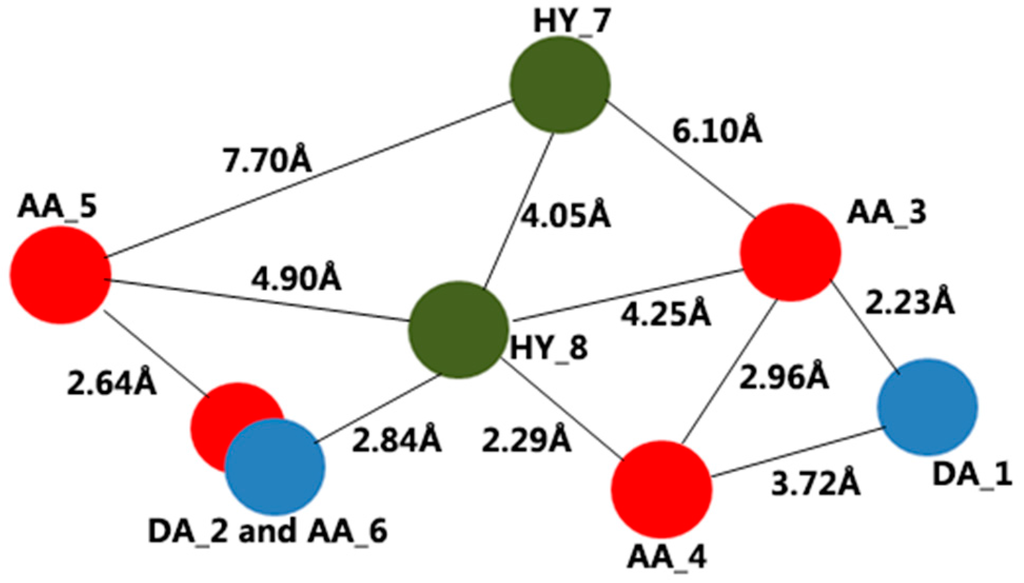
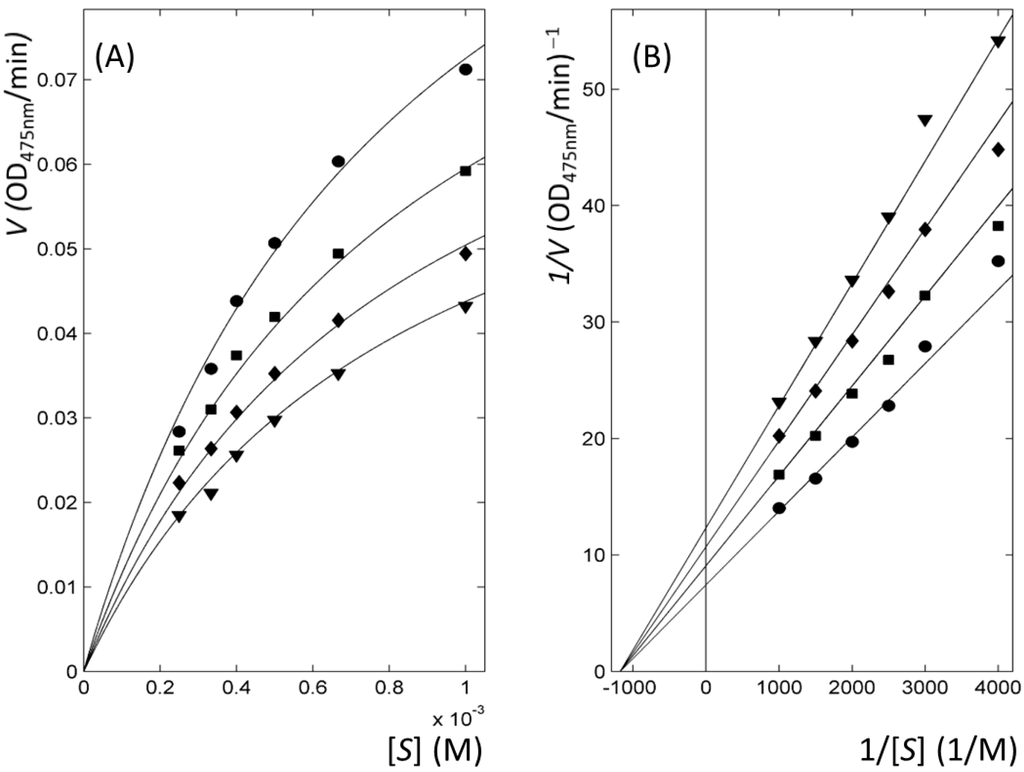
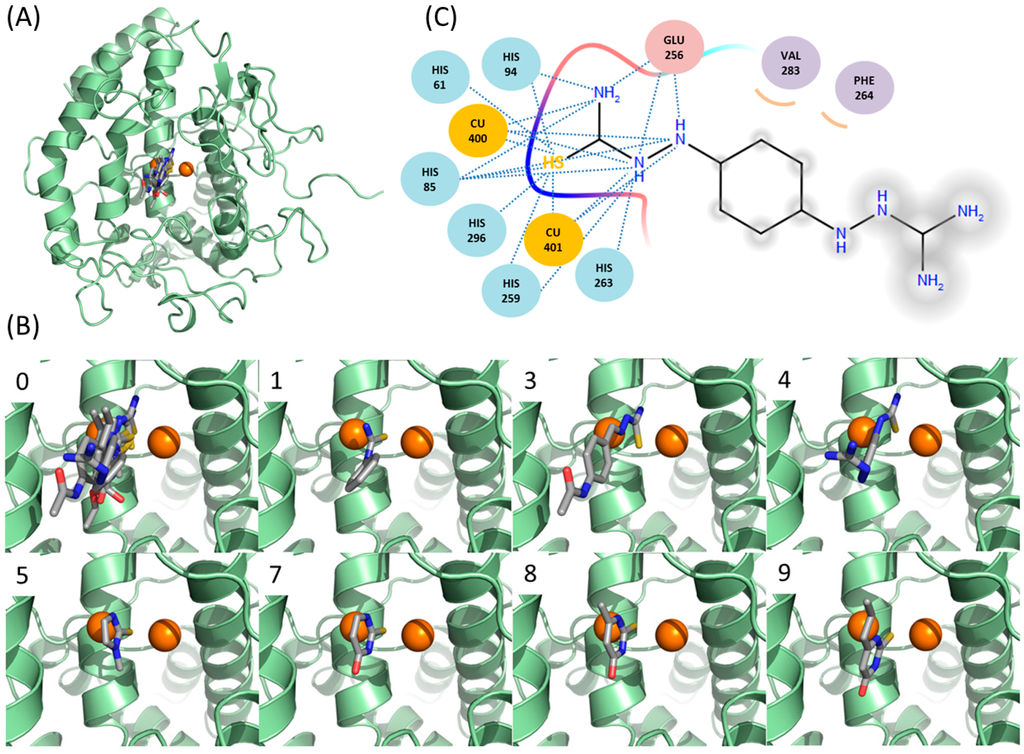
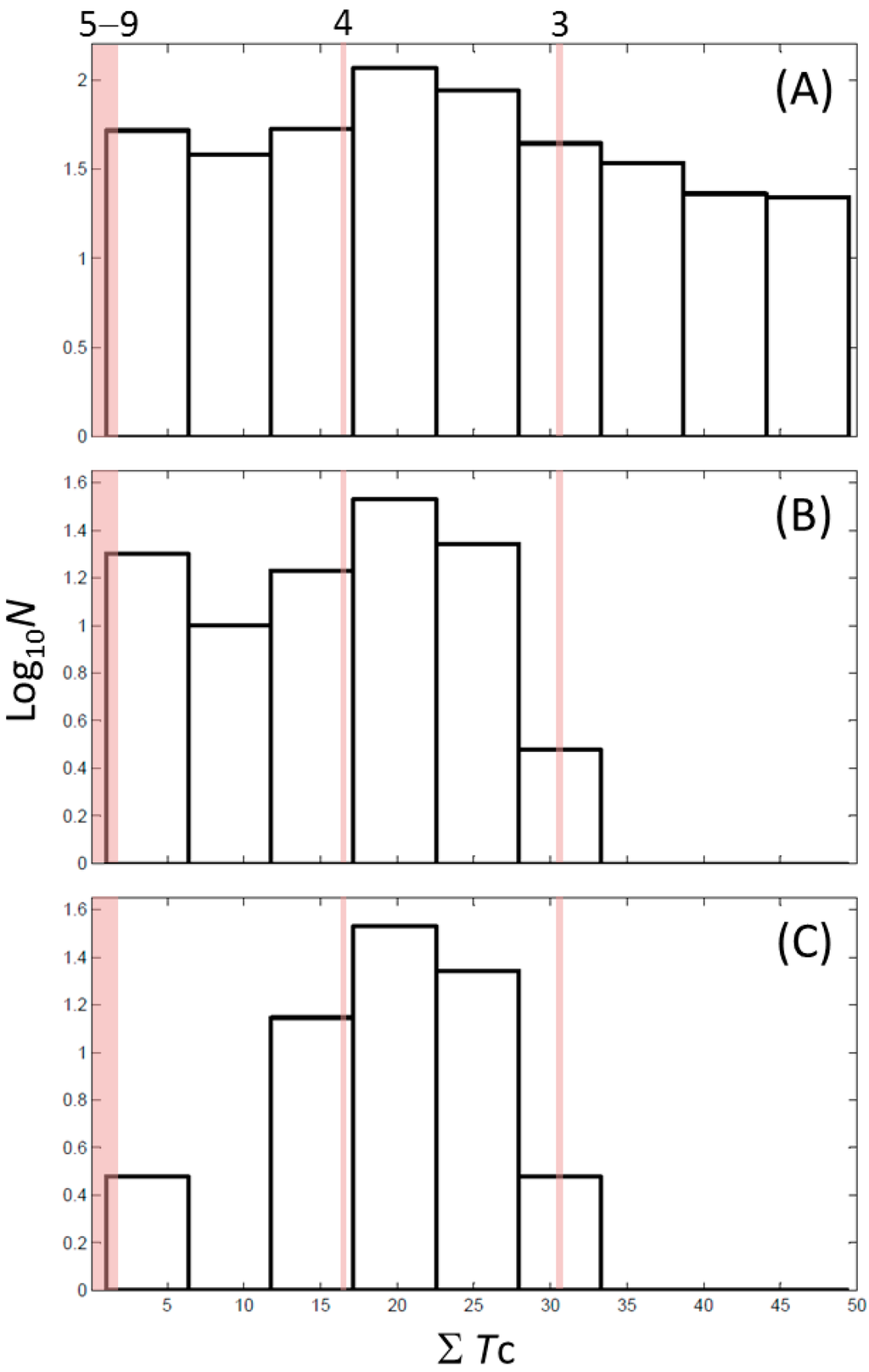
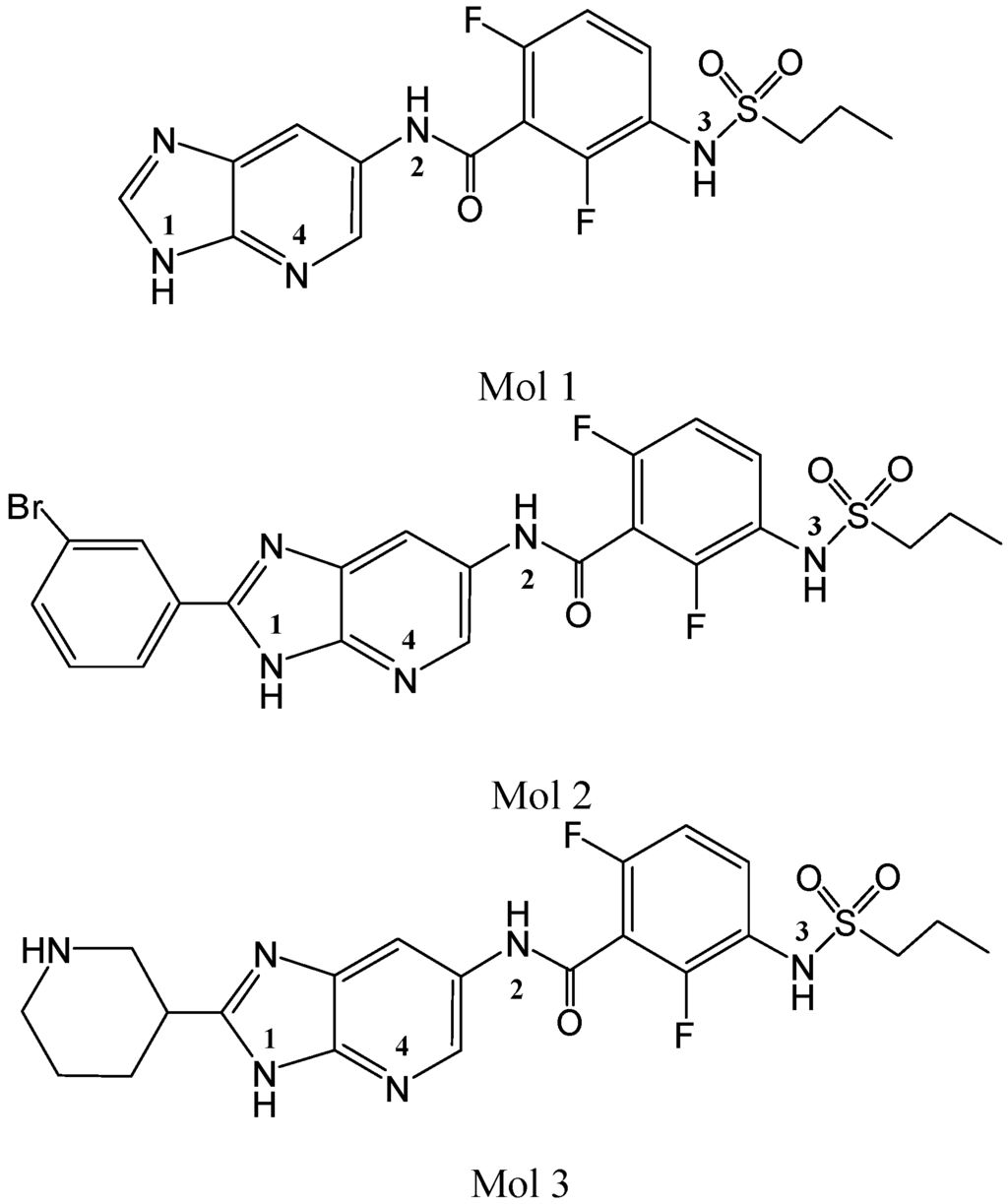
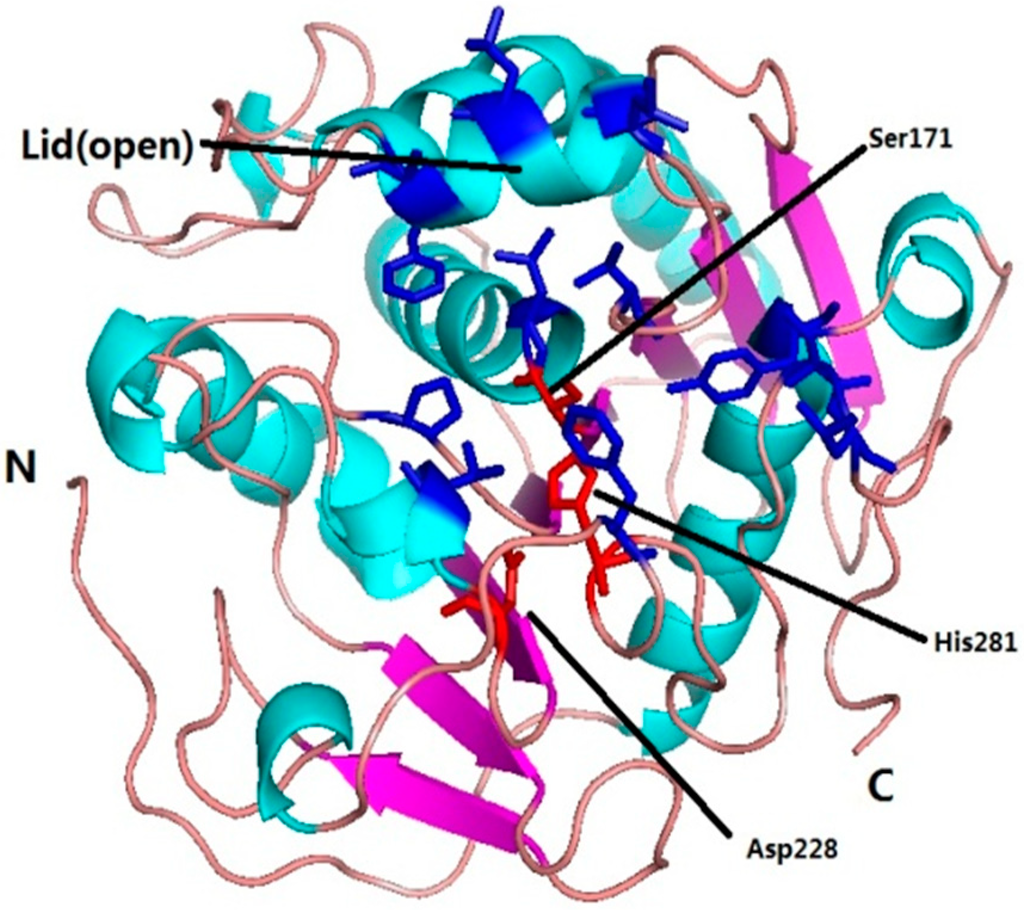
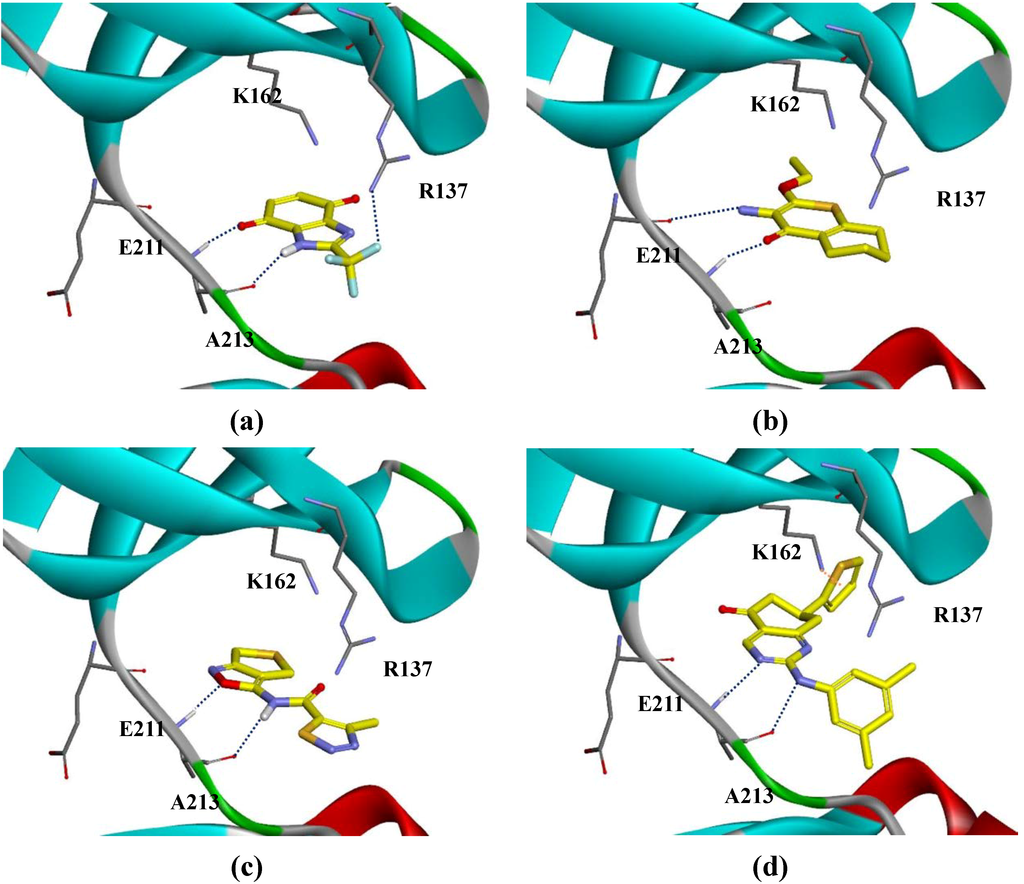
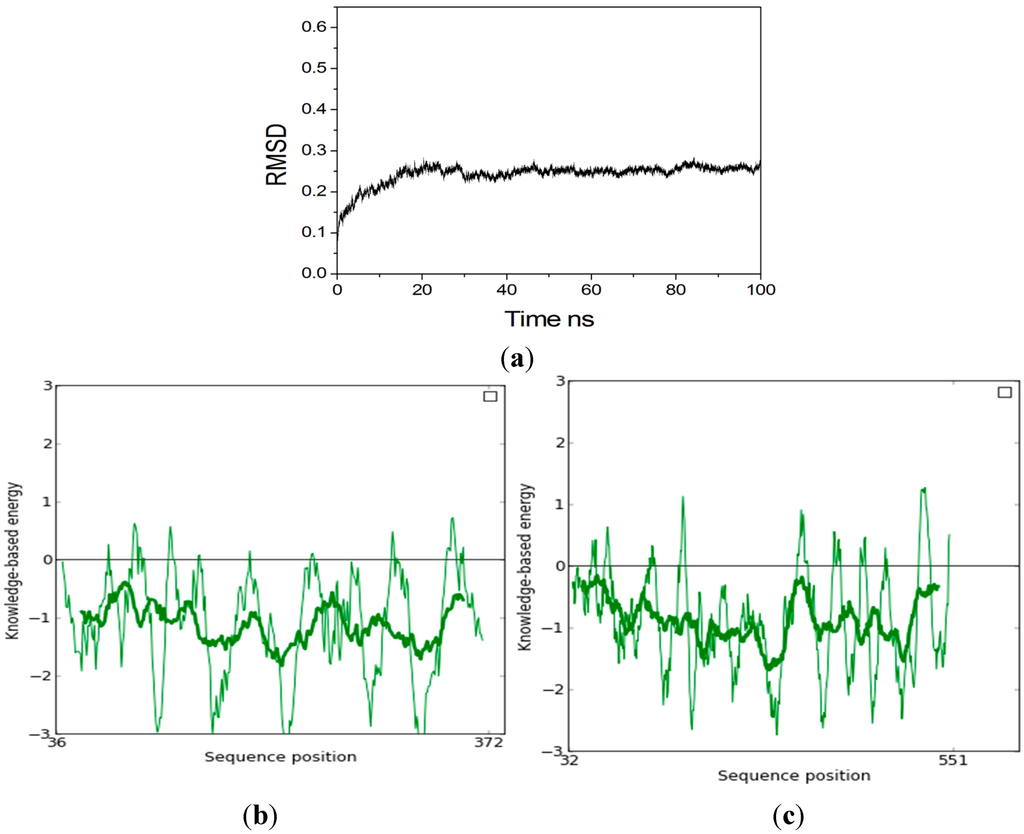
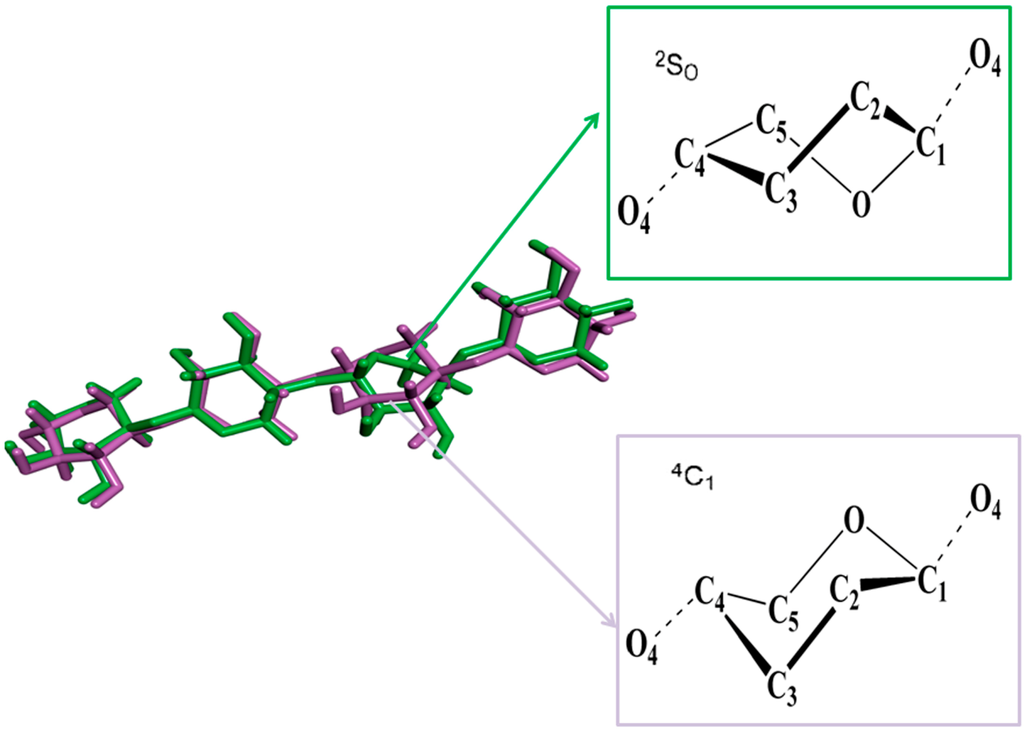
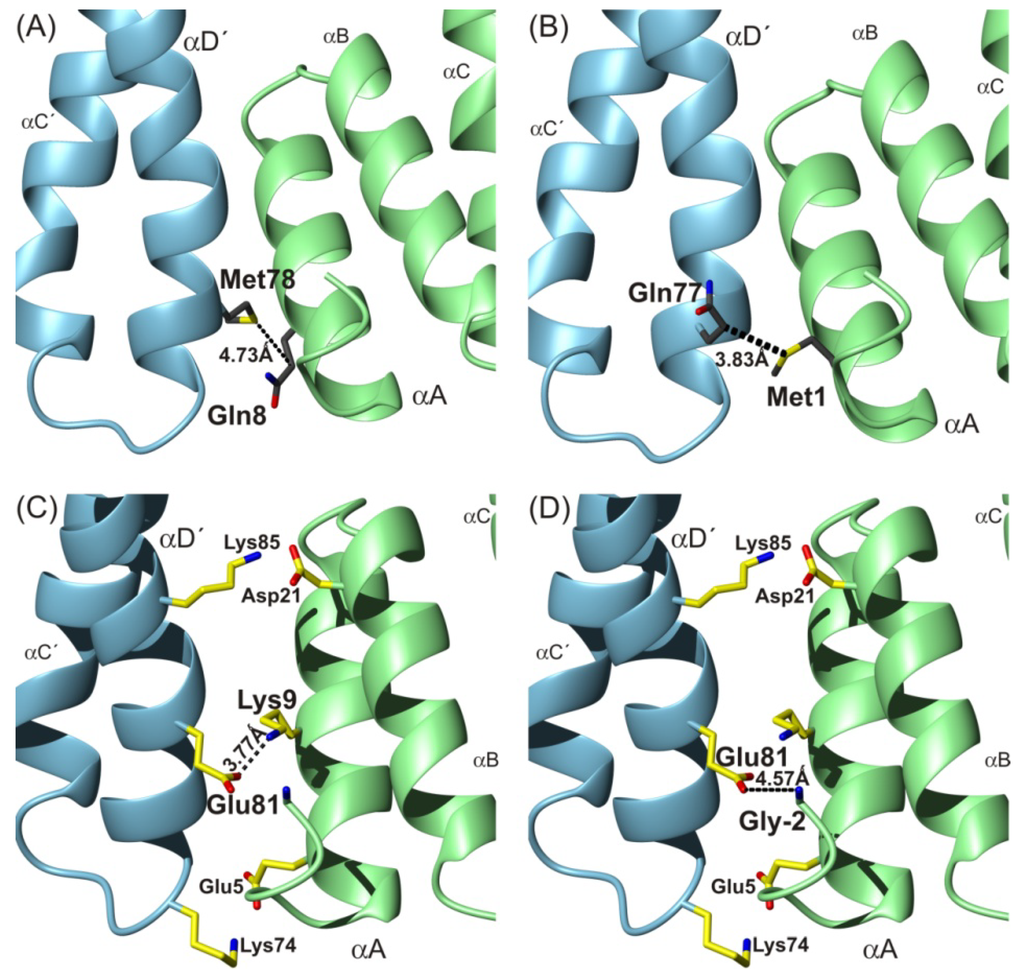
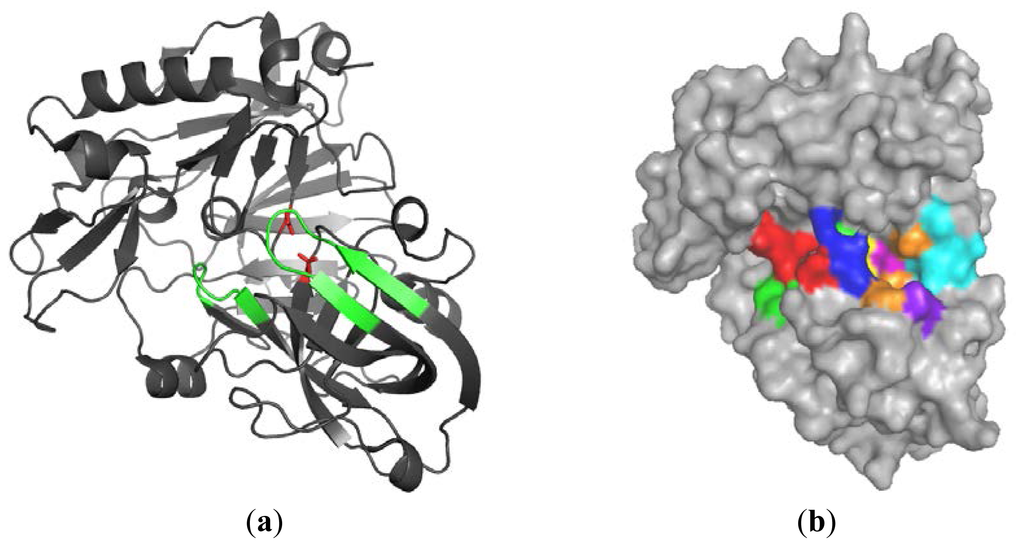
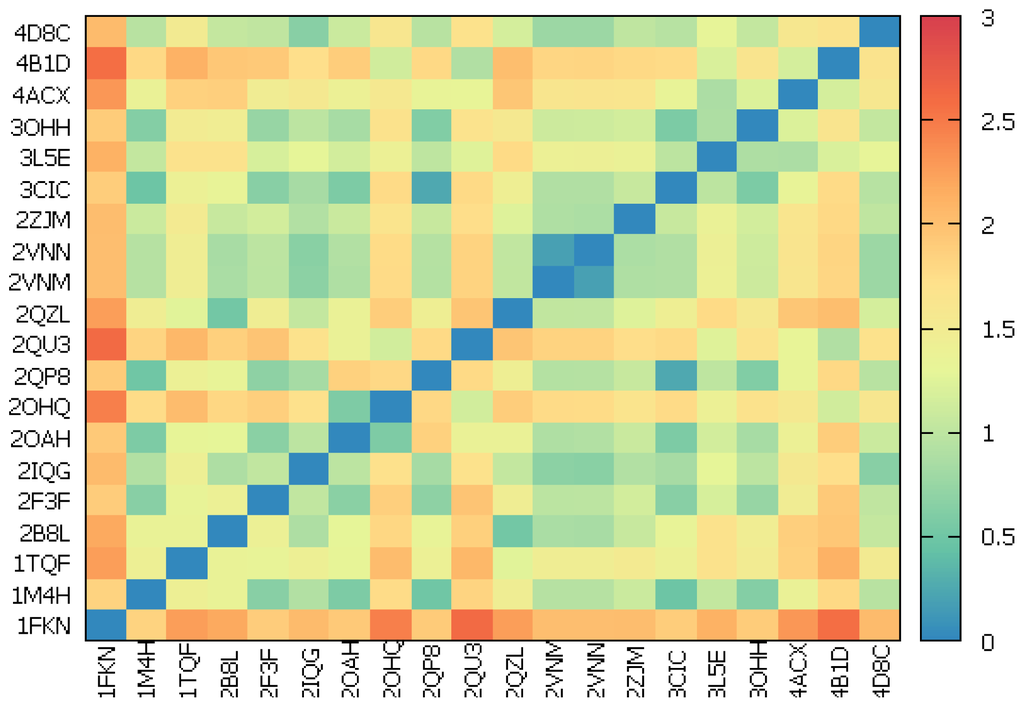
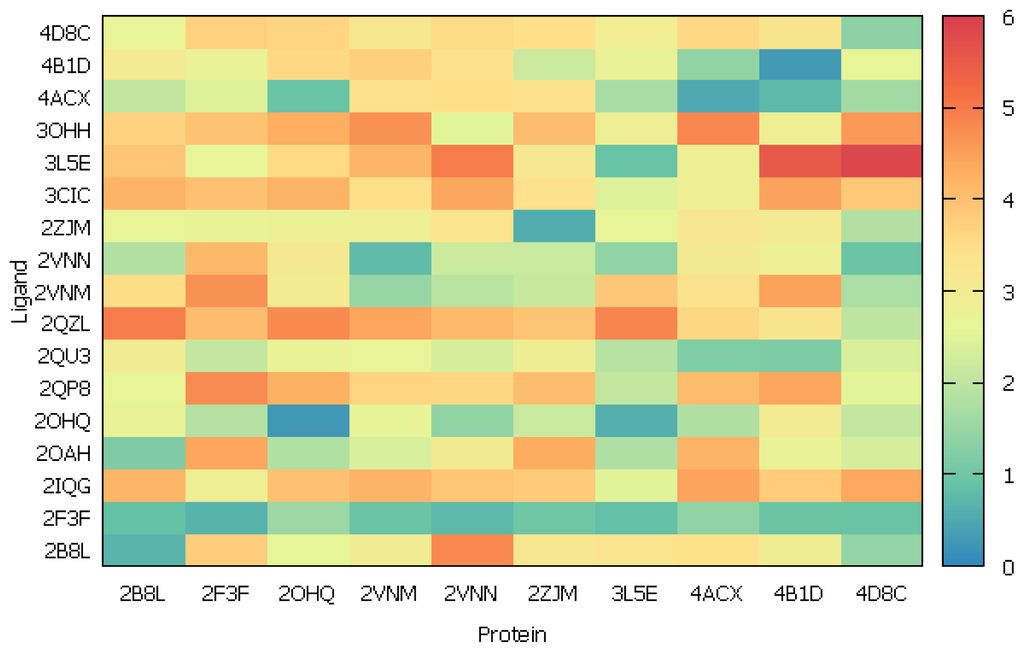
CPT (carnitine palmitoyltransferase) II muscle deficiency is the most common form of muscle fatty acid metabolism disorders. In contrast to carnitine deficiency, it is clinically characterized by attacks of myalgia and rhabdomyolysis without persistent muscle weakness and lipid accumulation in muscle fibers. The
[...] Read more.
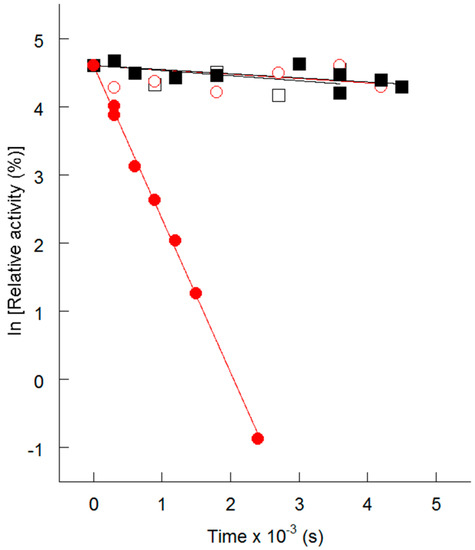
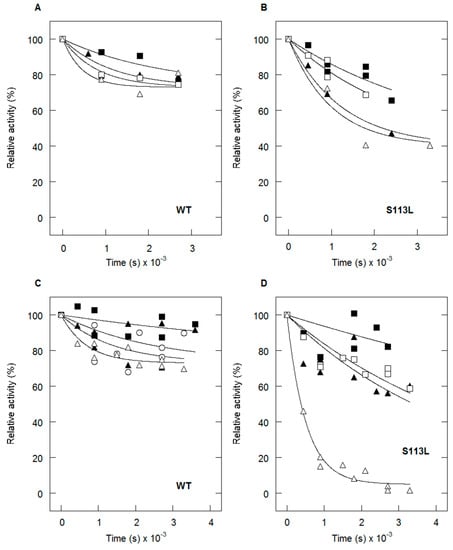
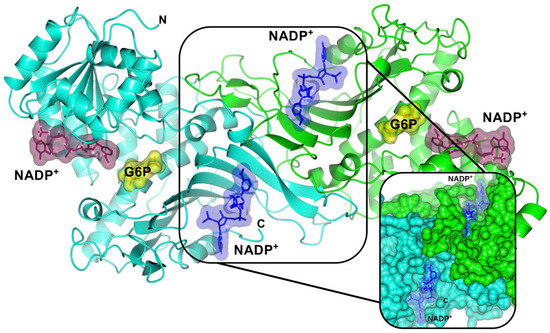
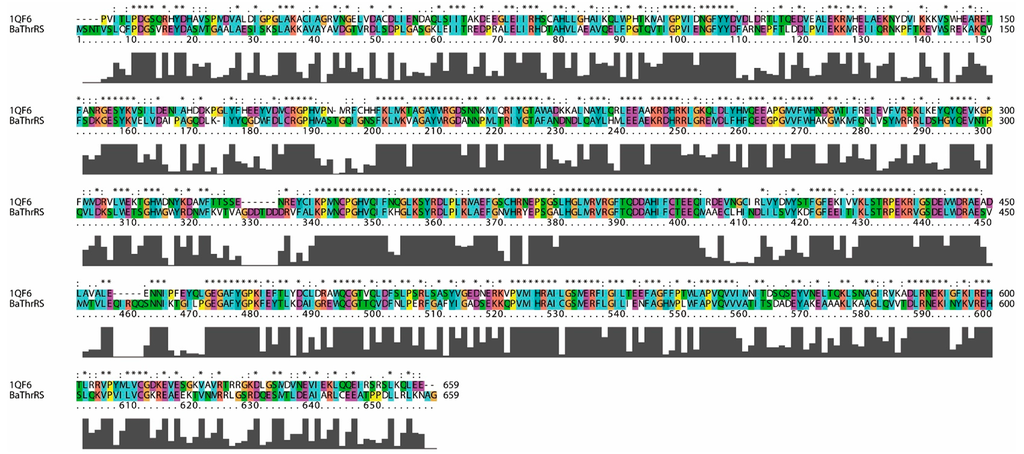
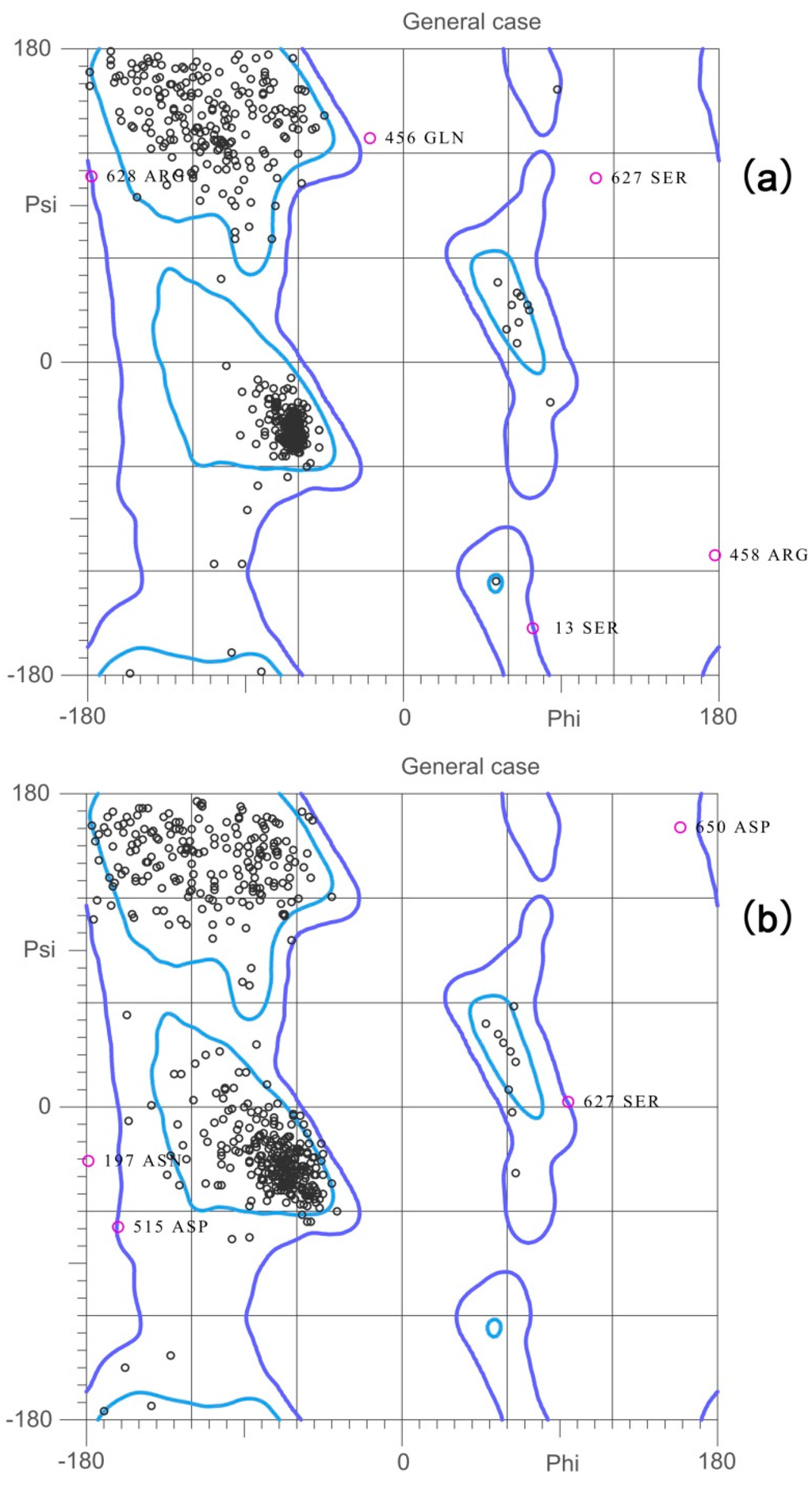
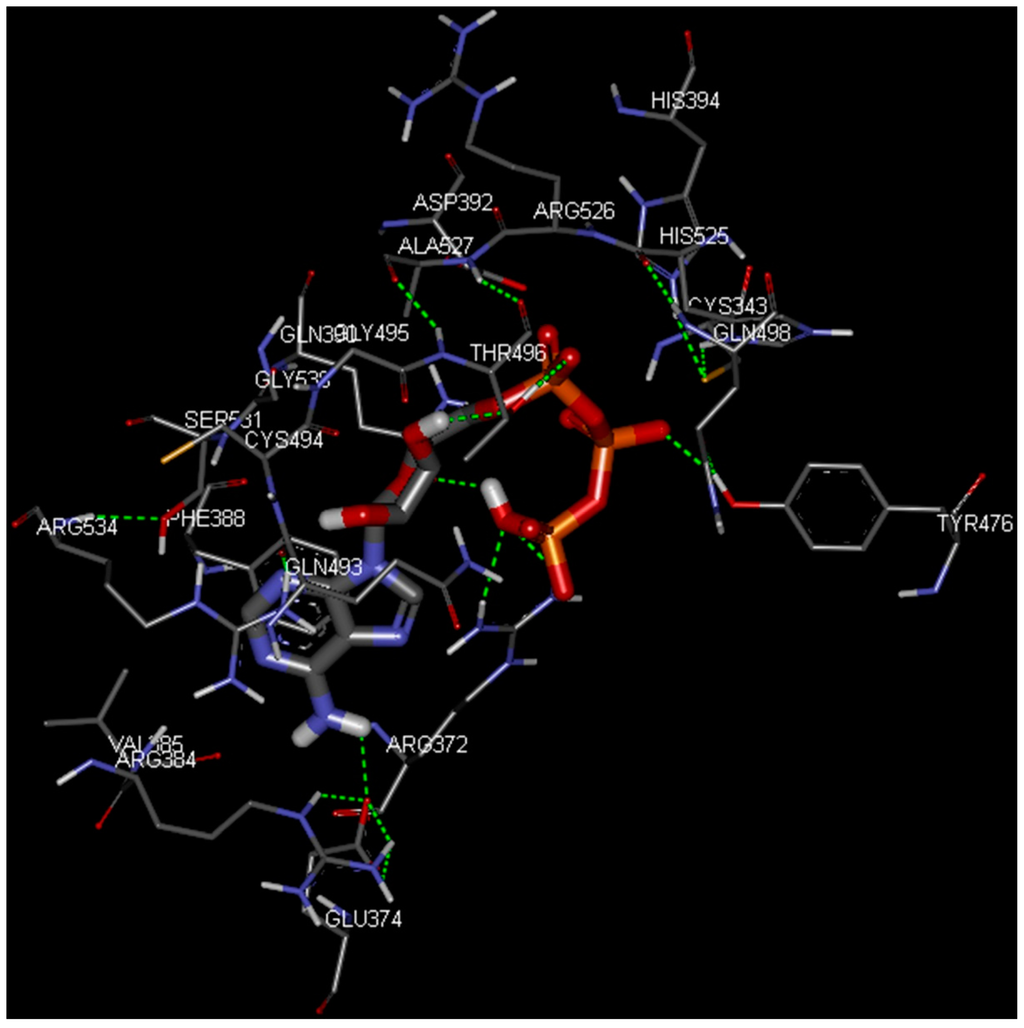
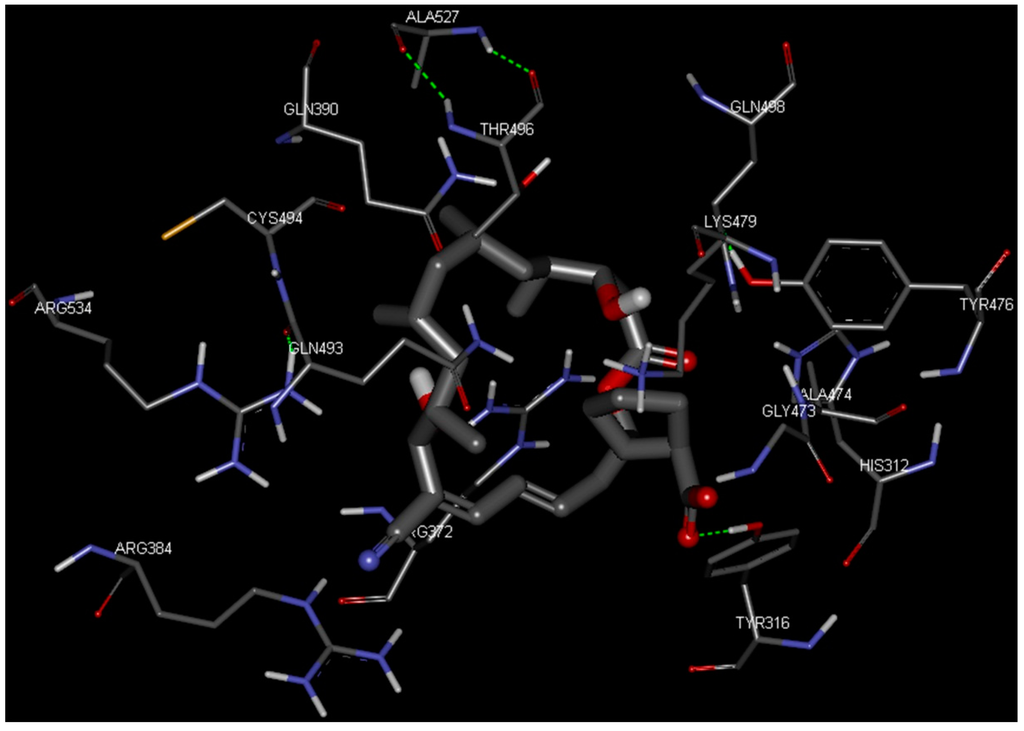
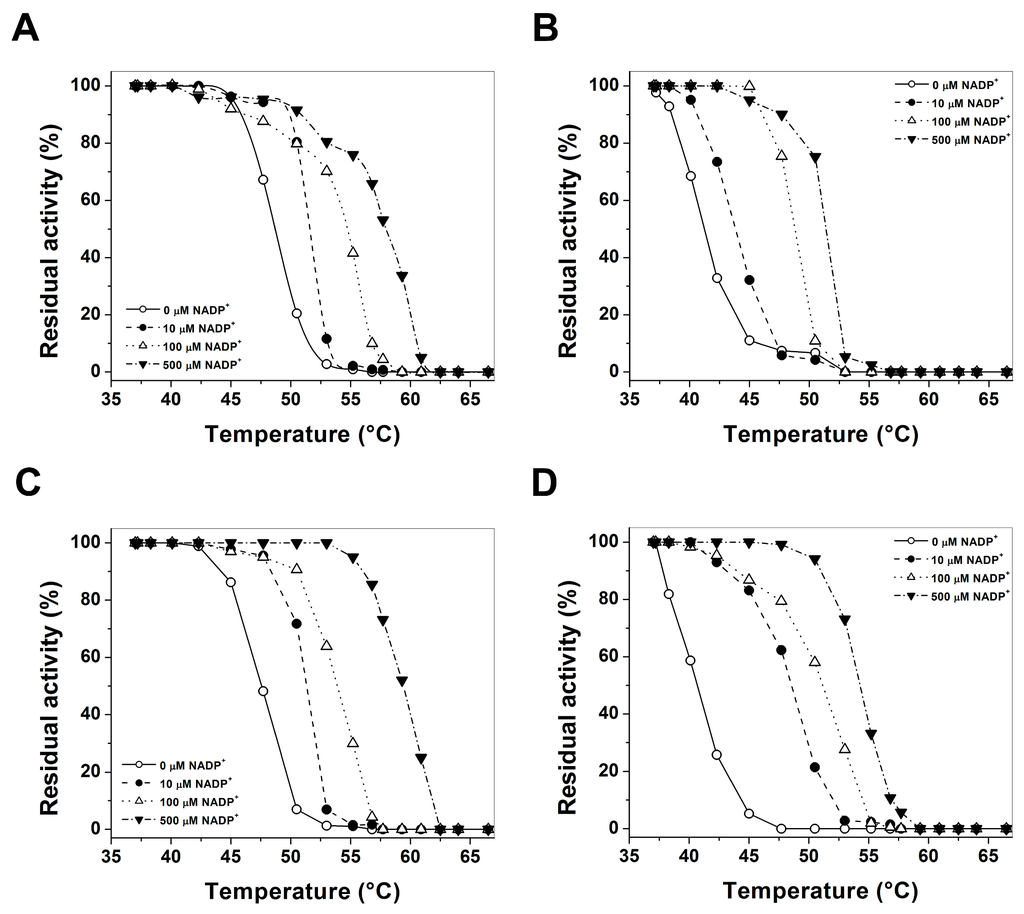
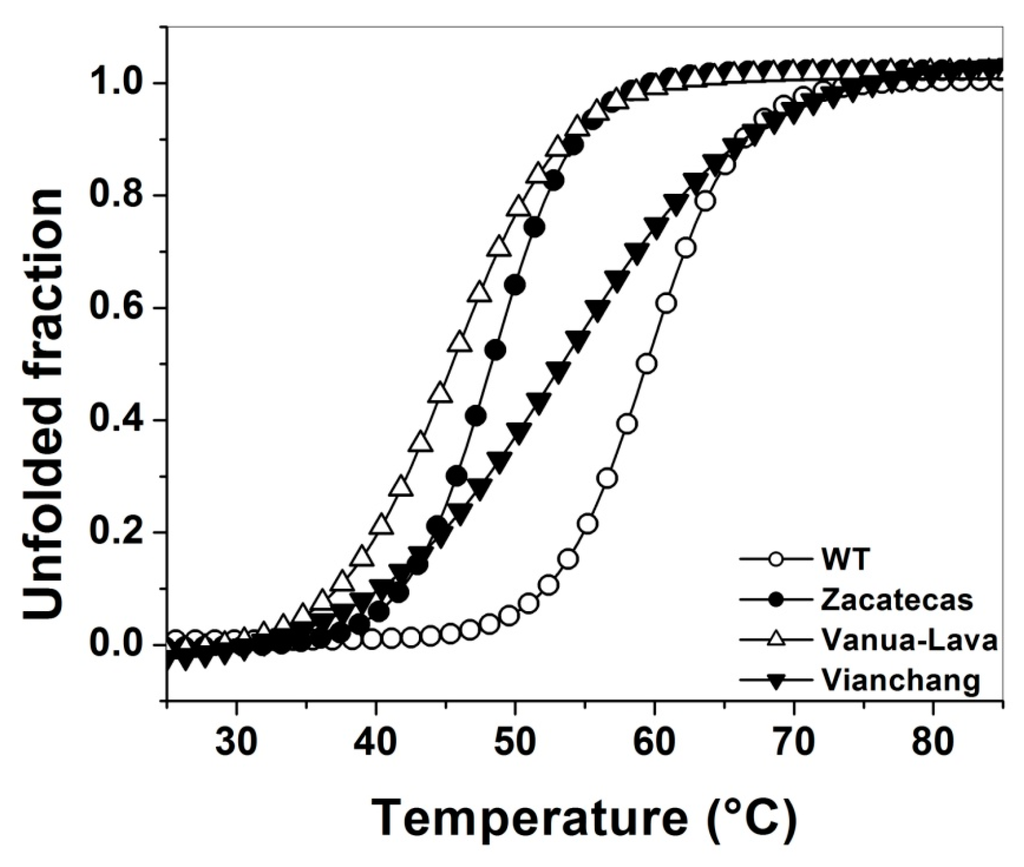
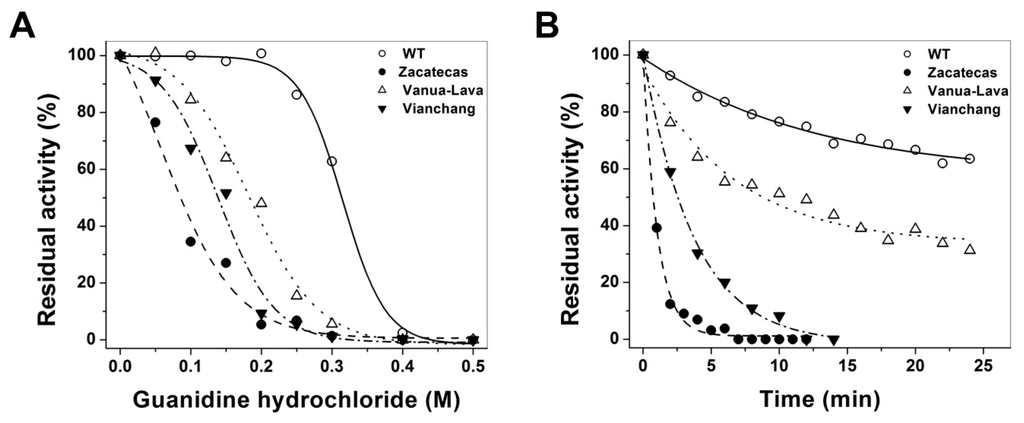
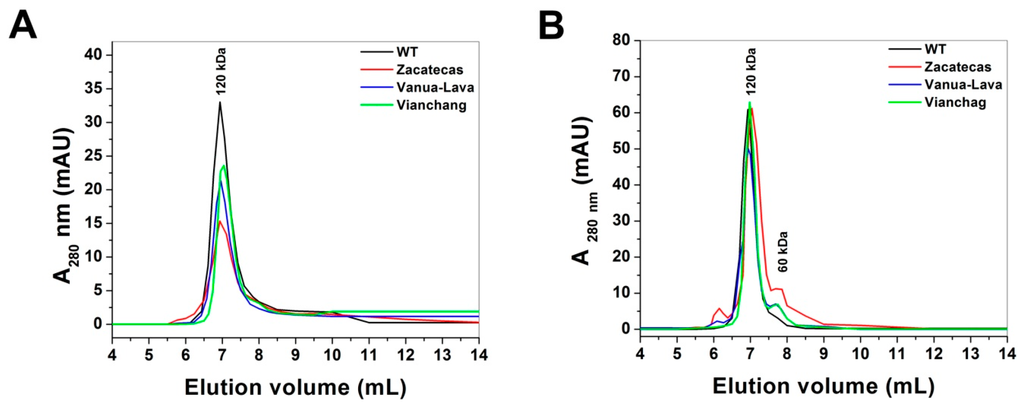
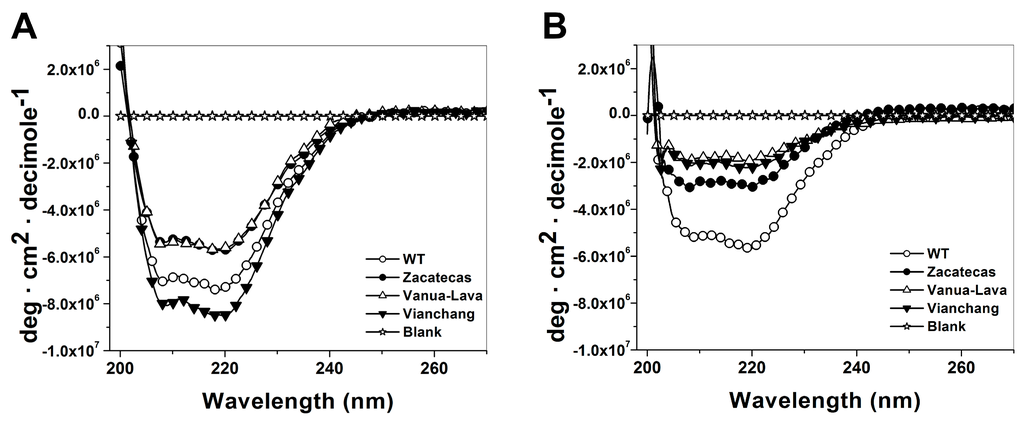
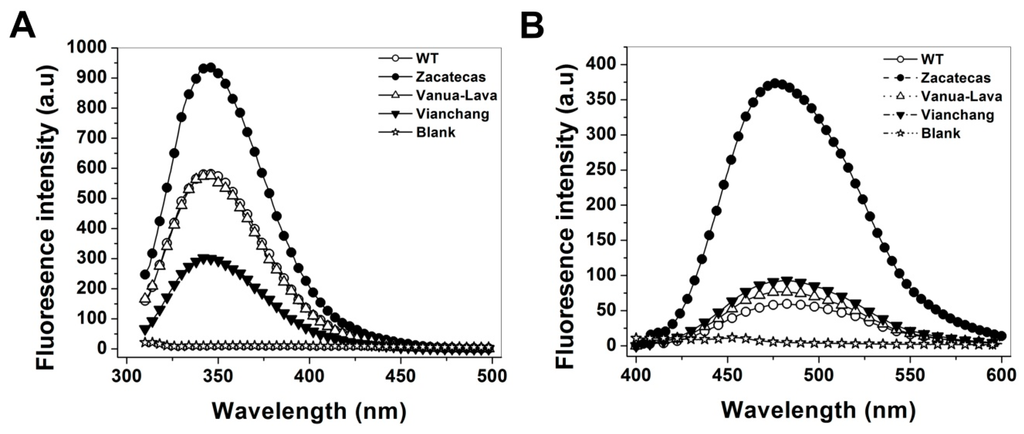
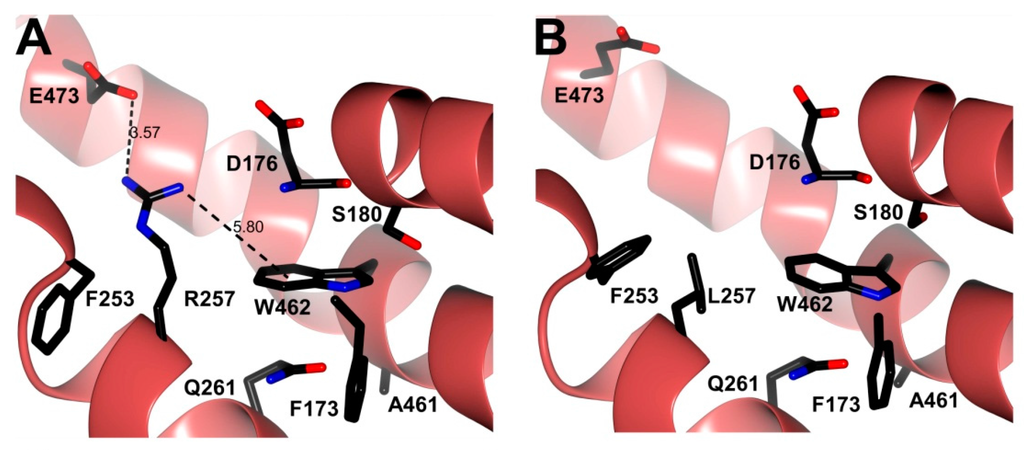
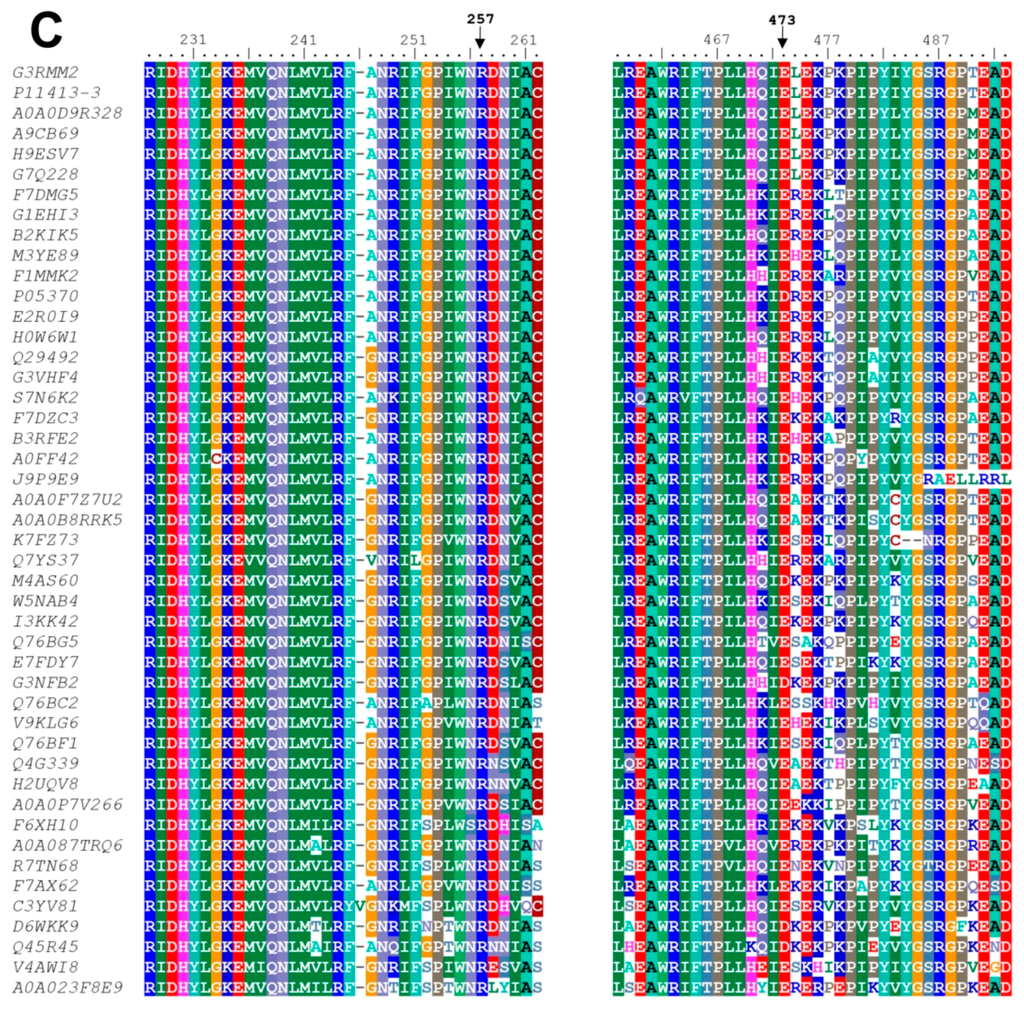
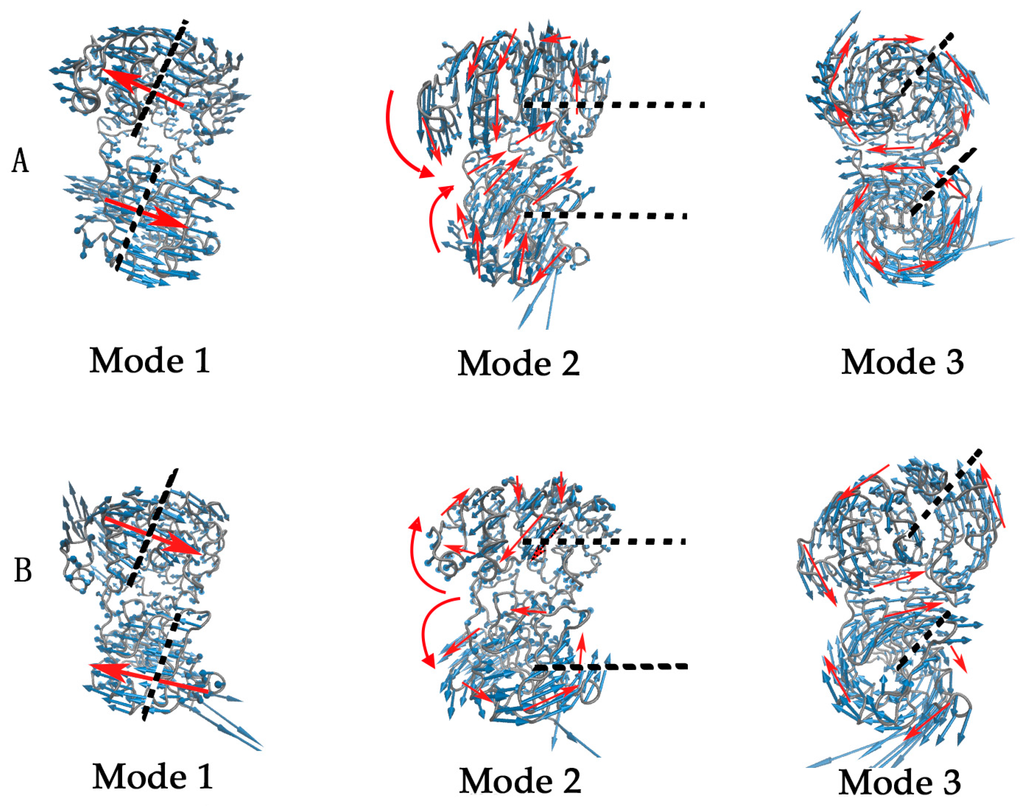
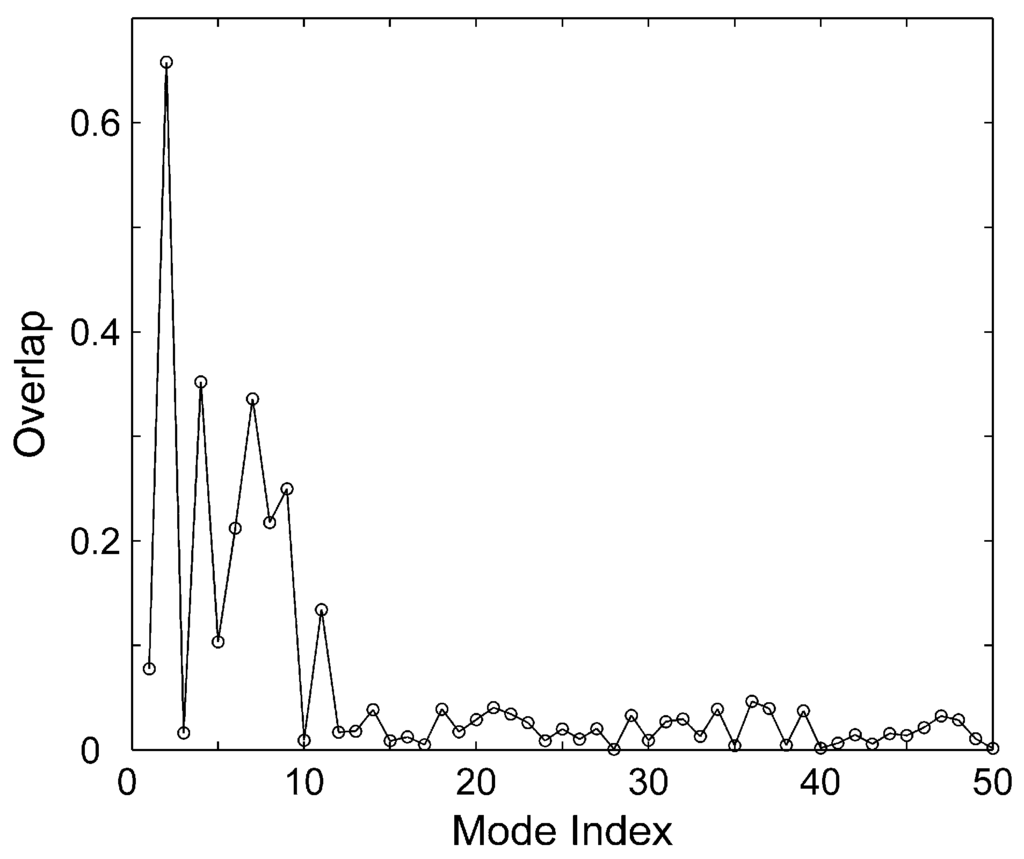
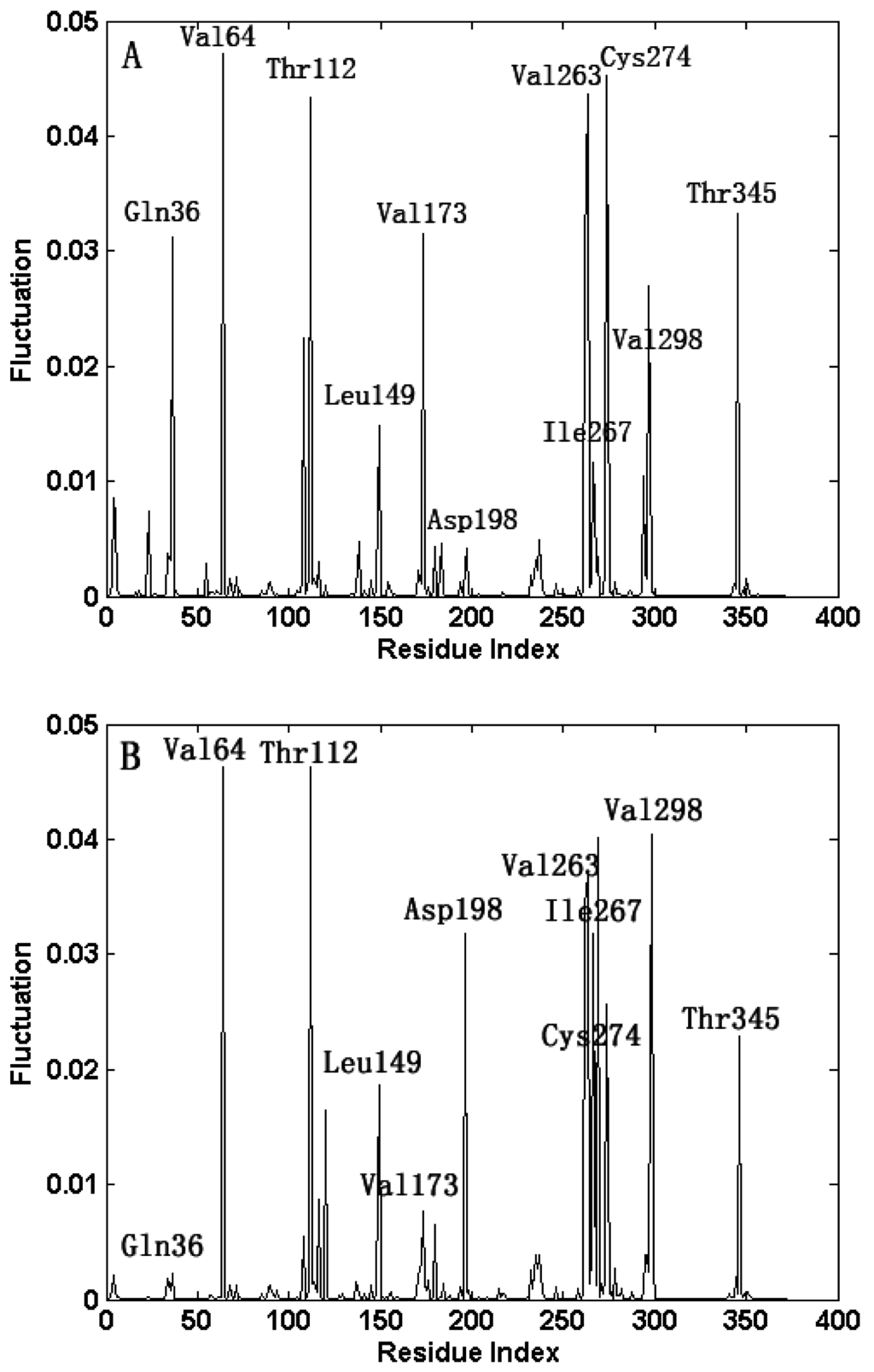
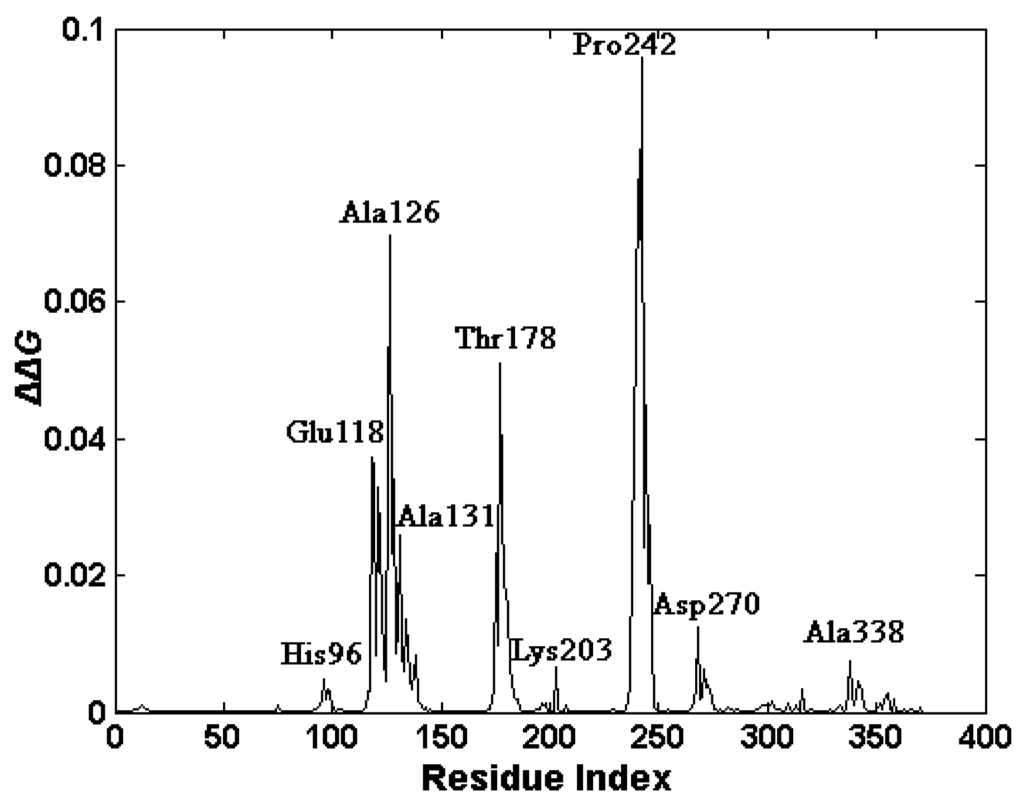
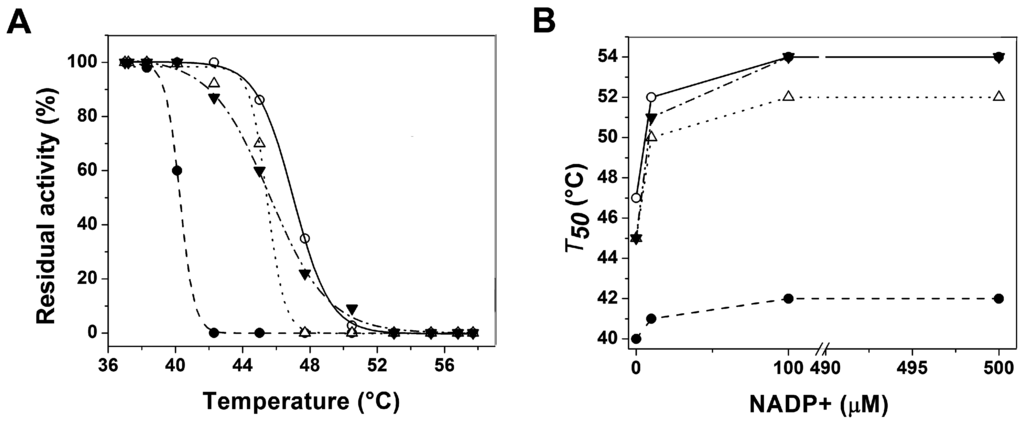
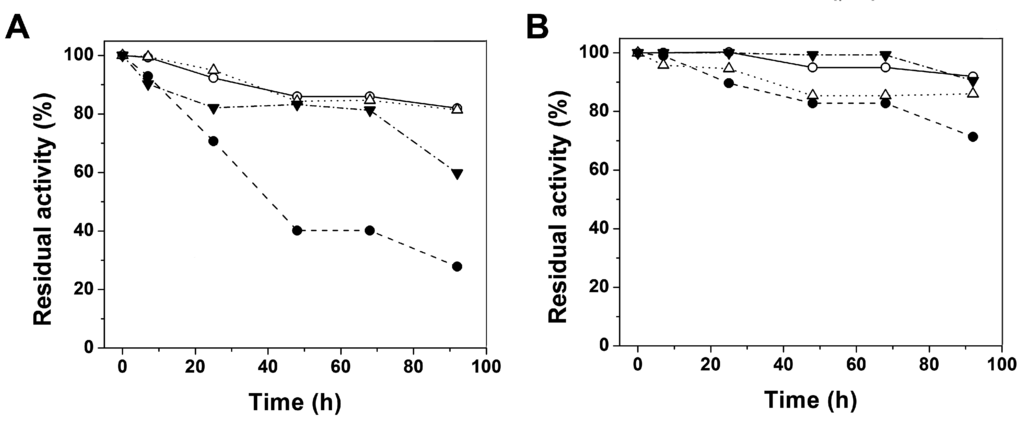
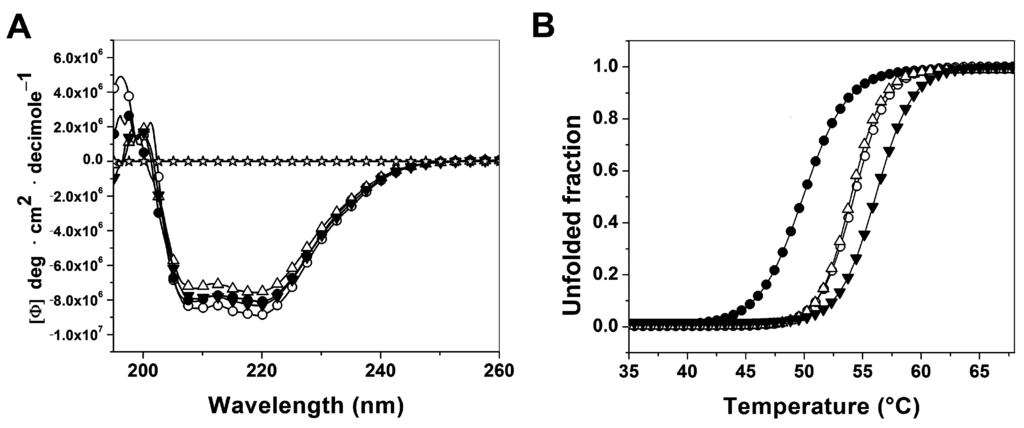
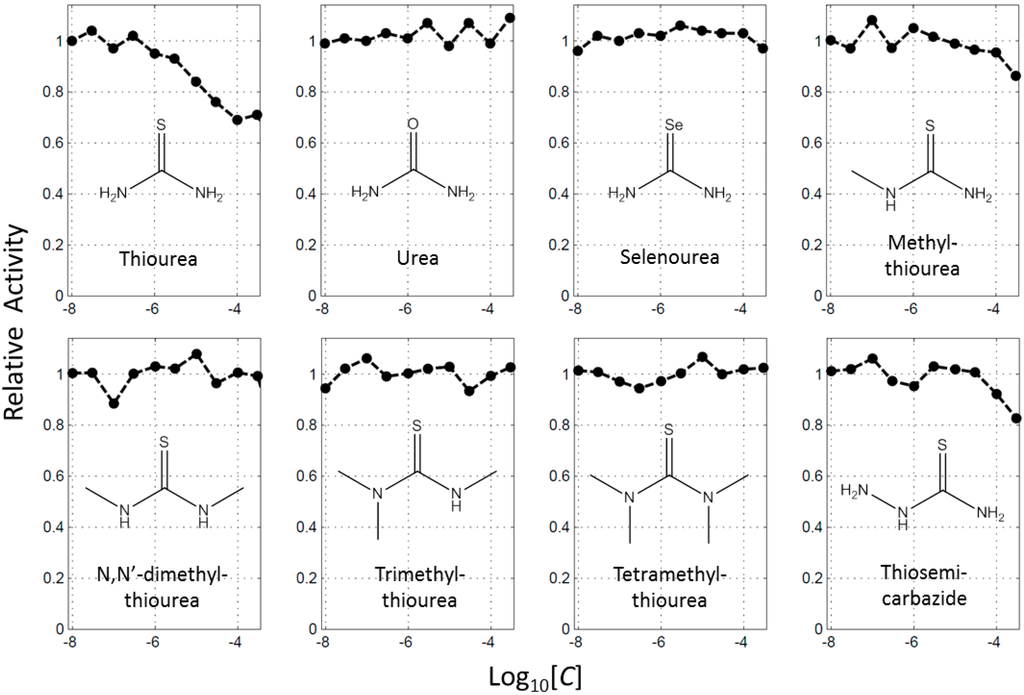
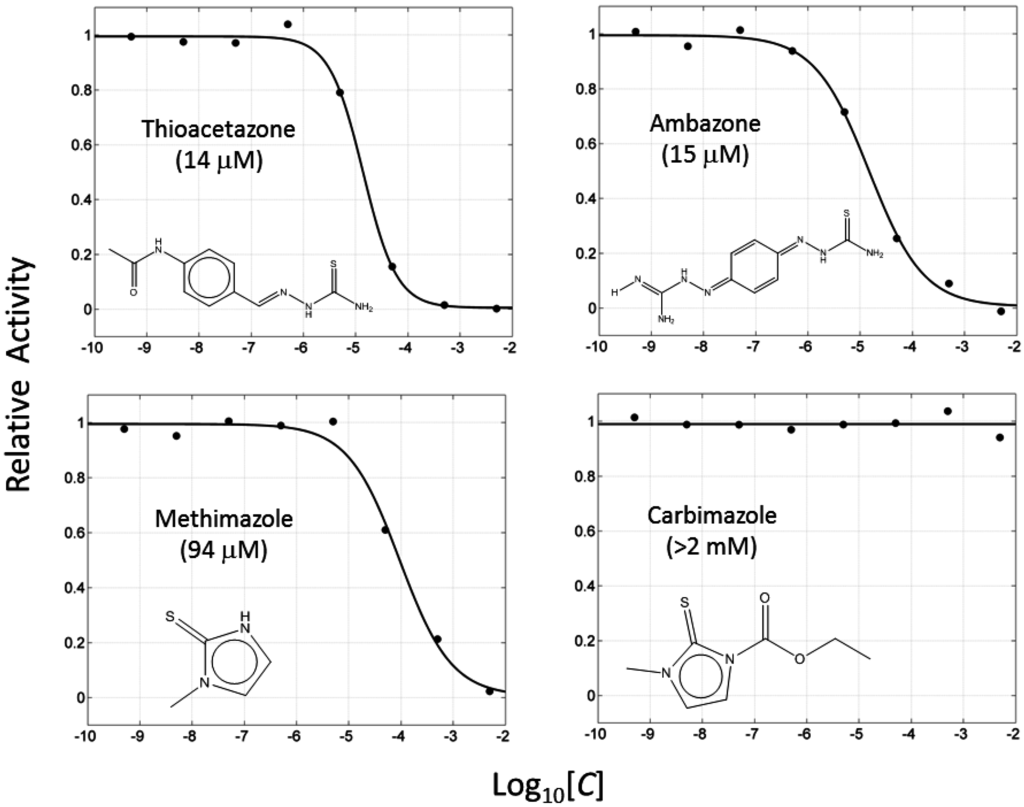
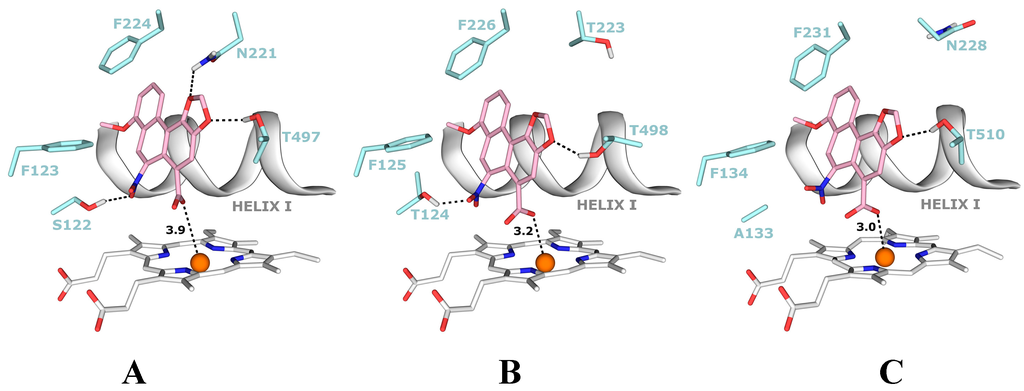
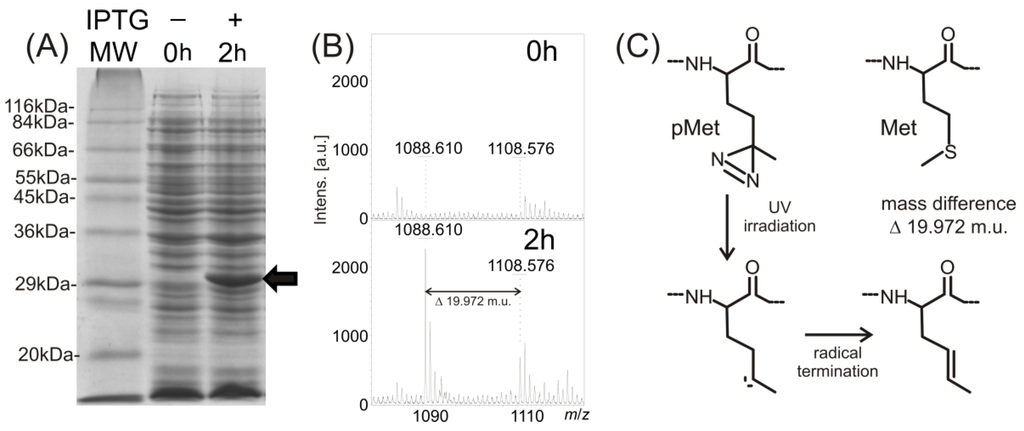
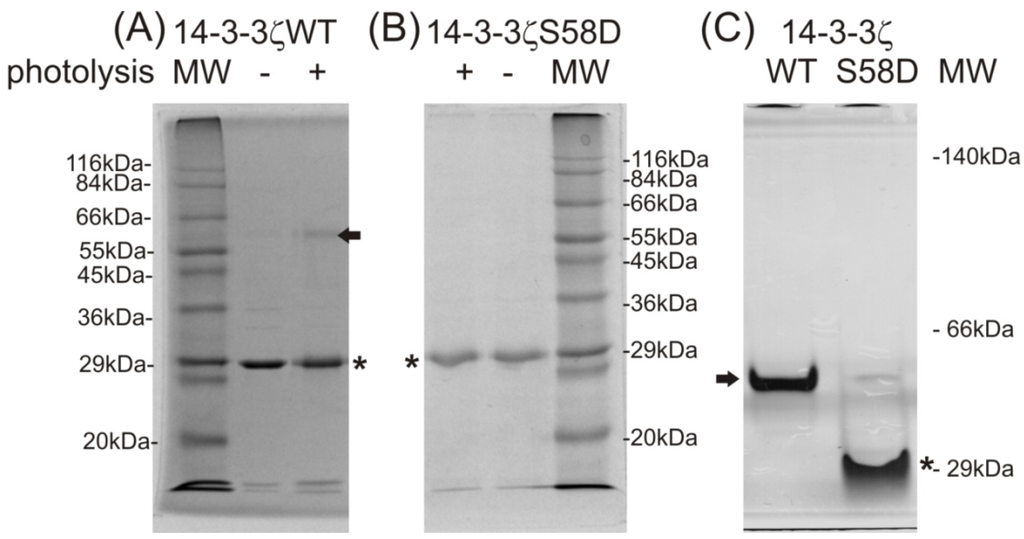
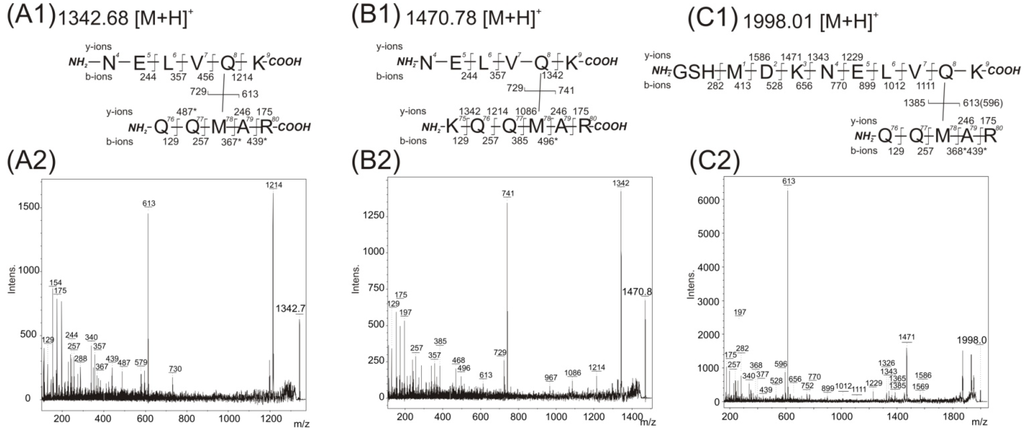
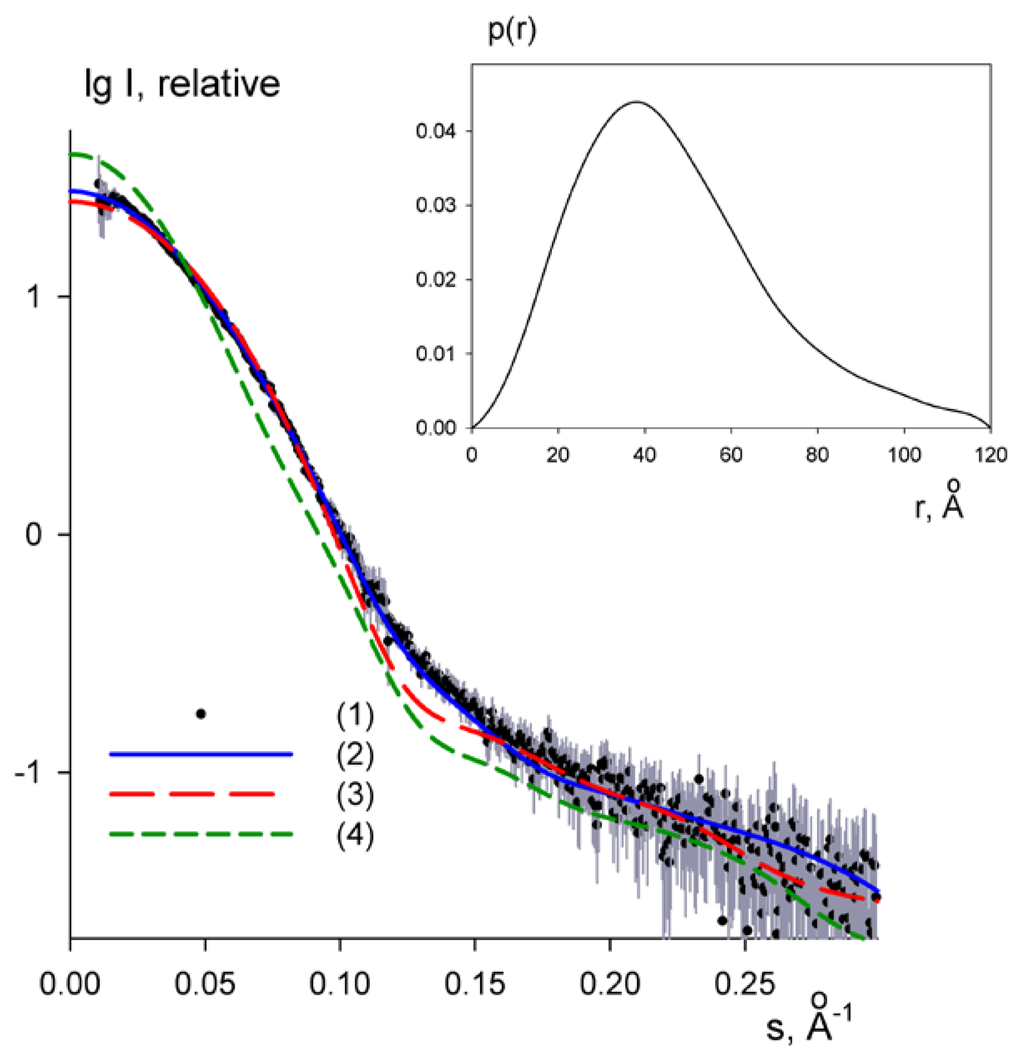
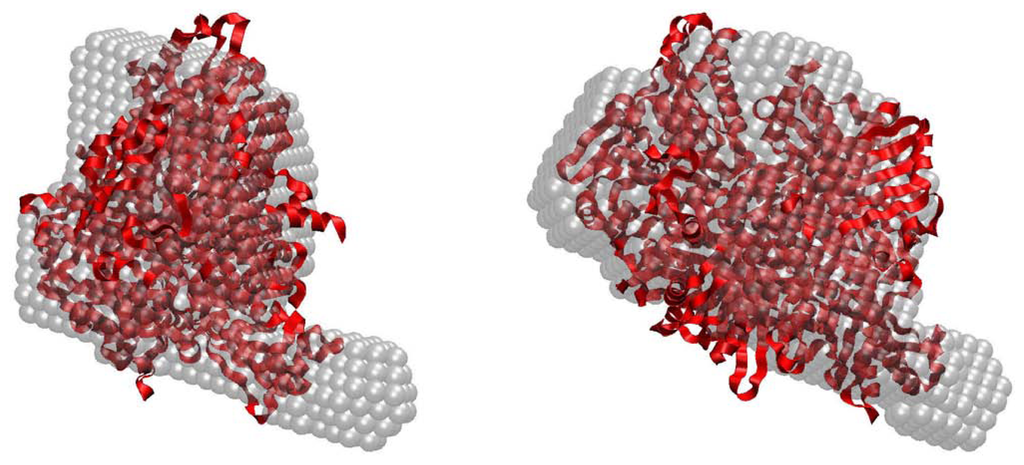
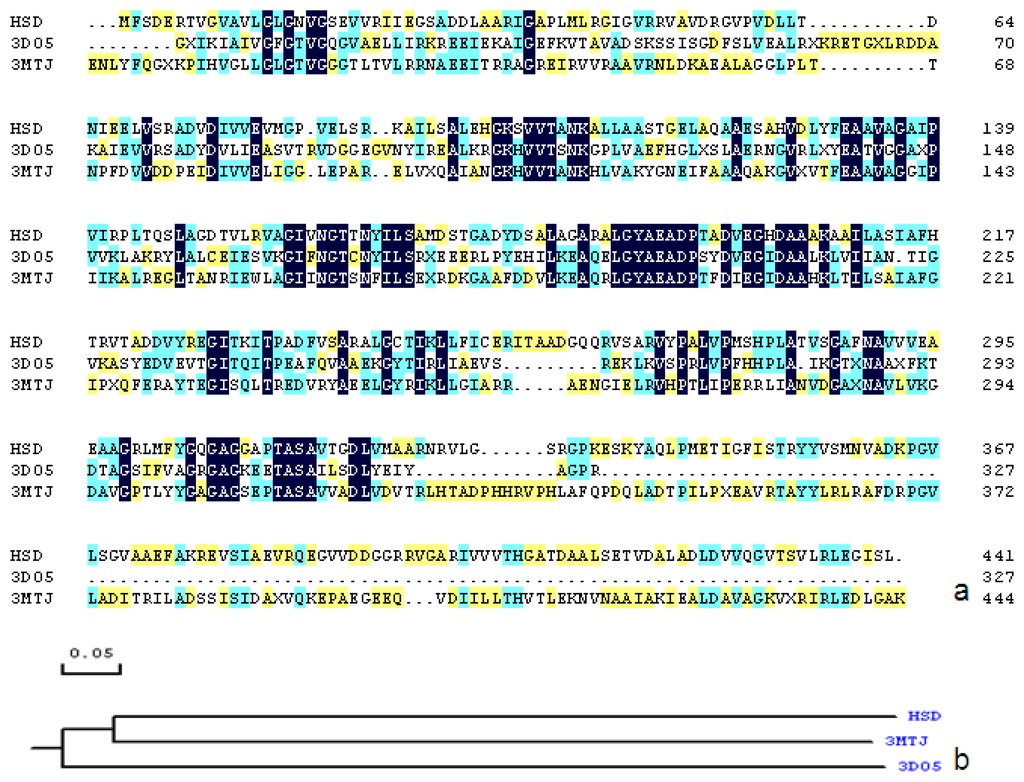
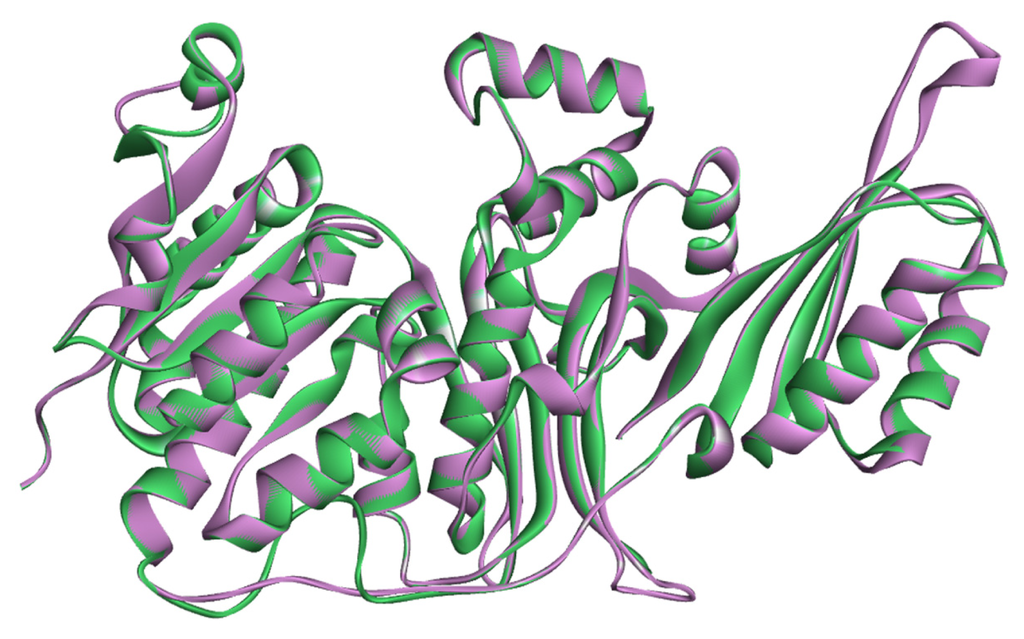
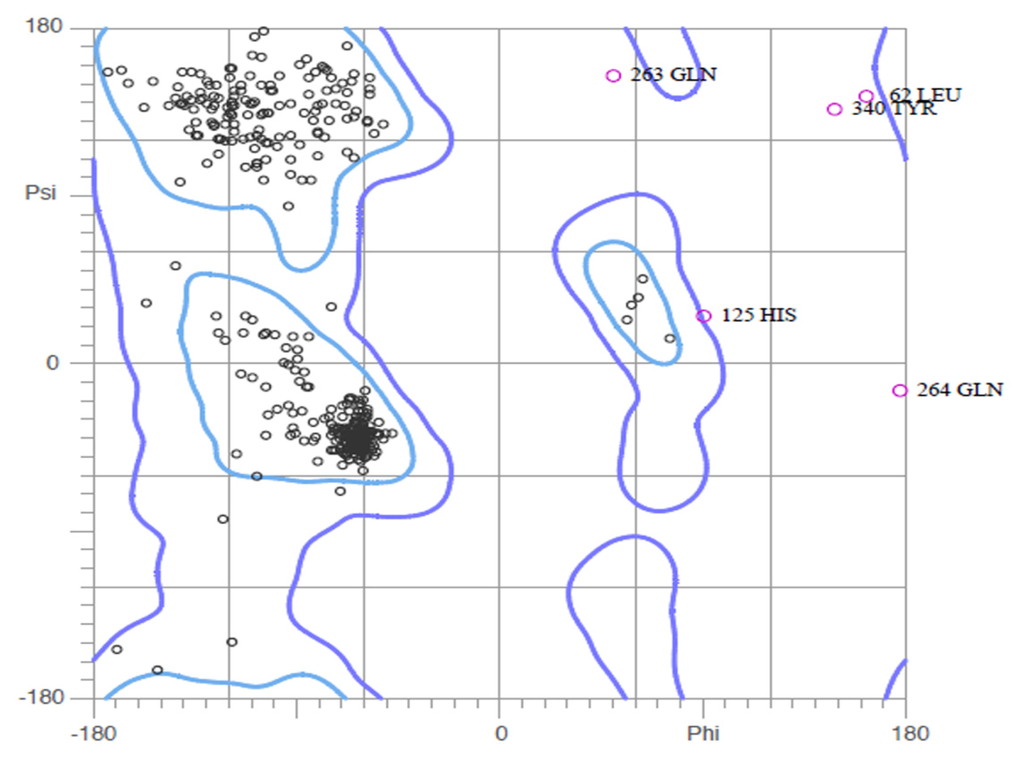
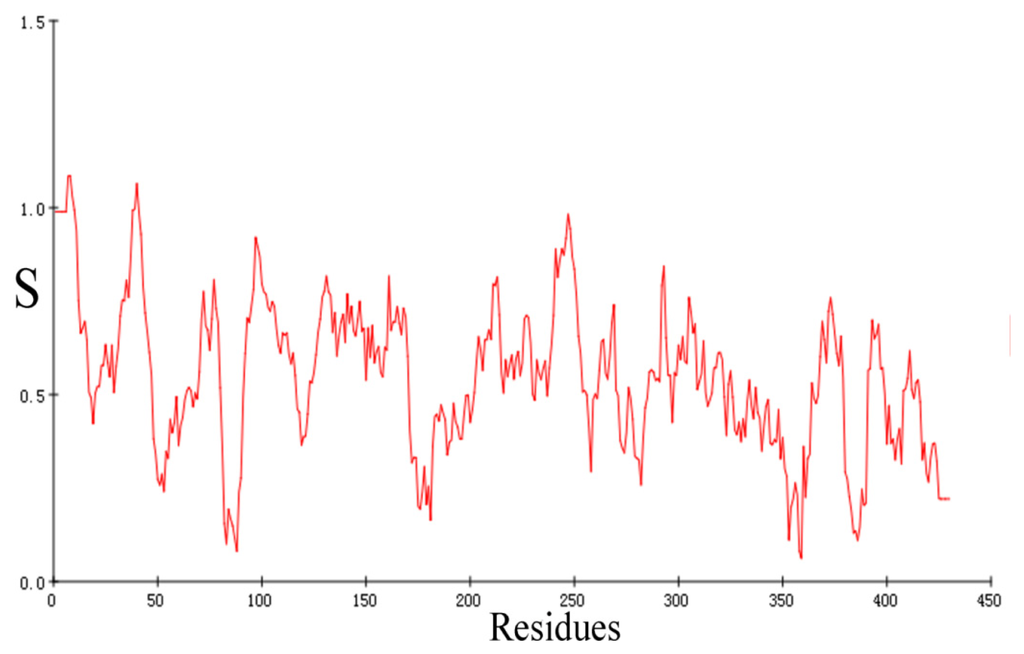
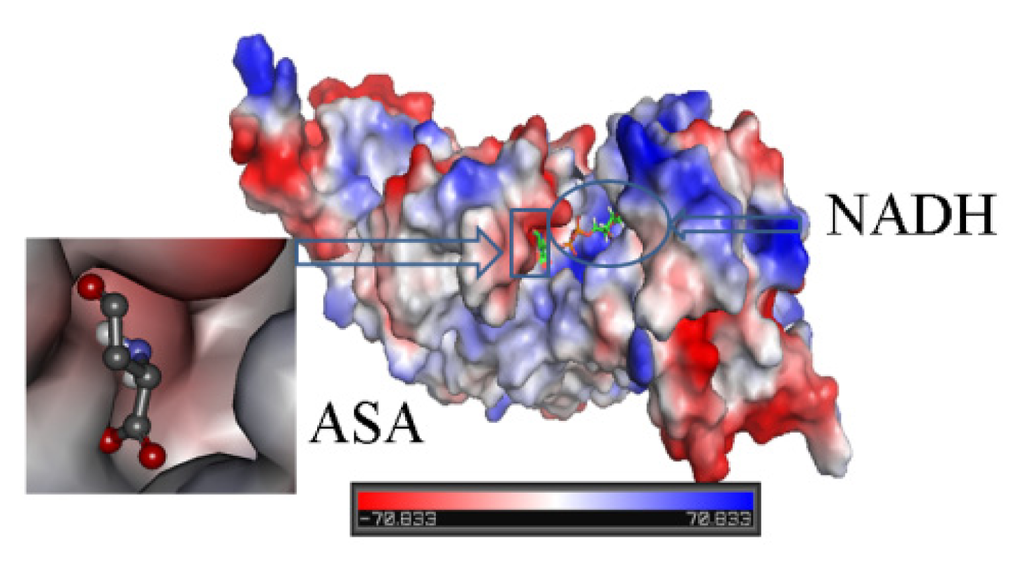
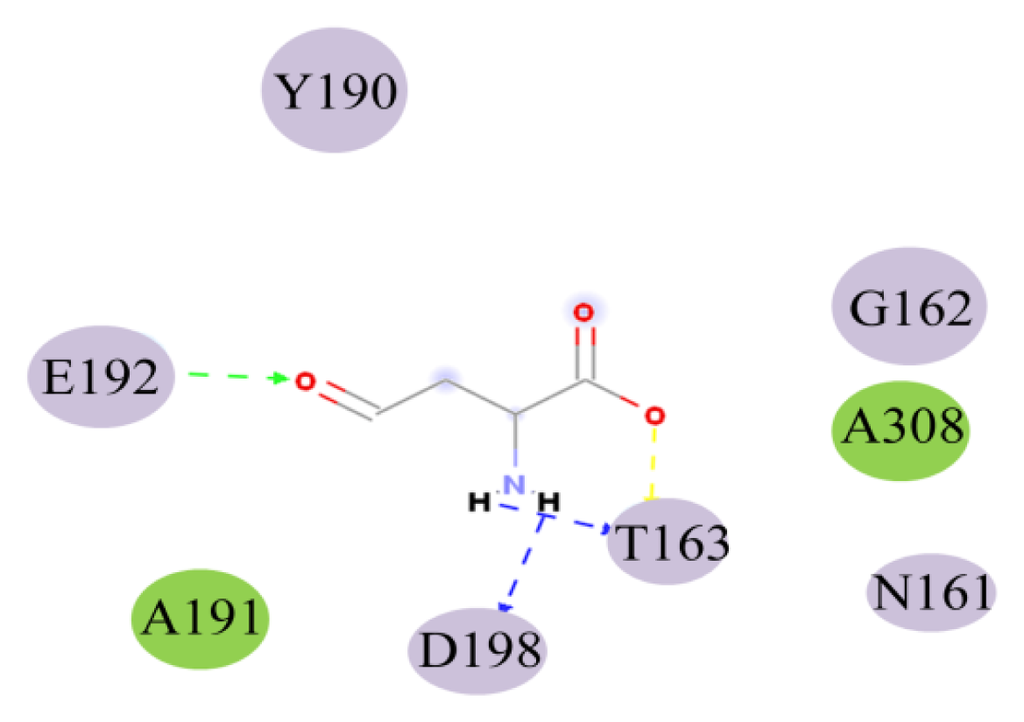
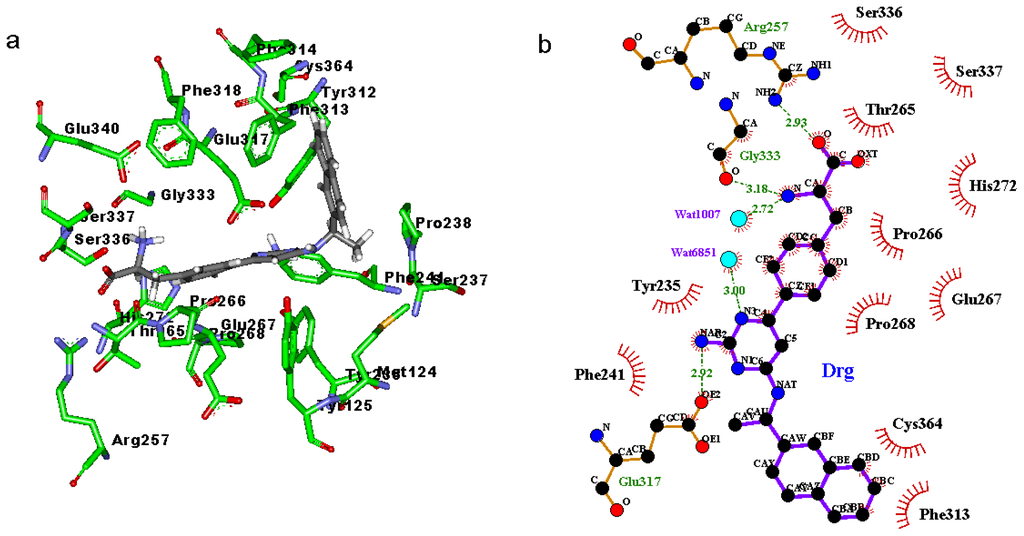
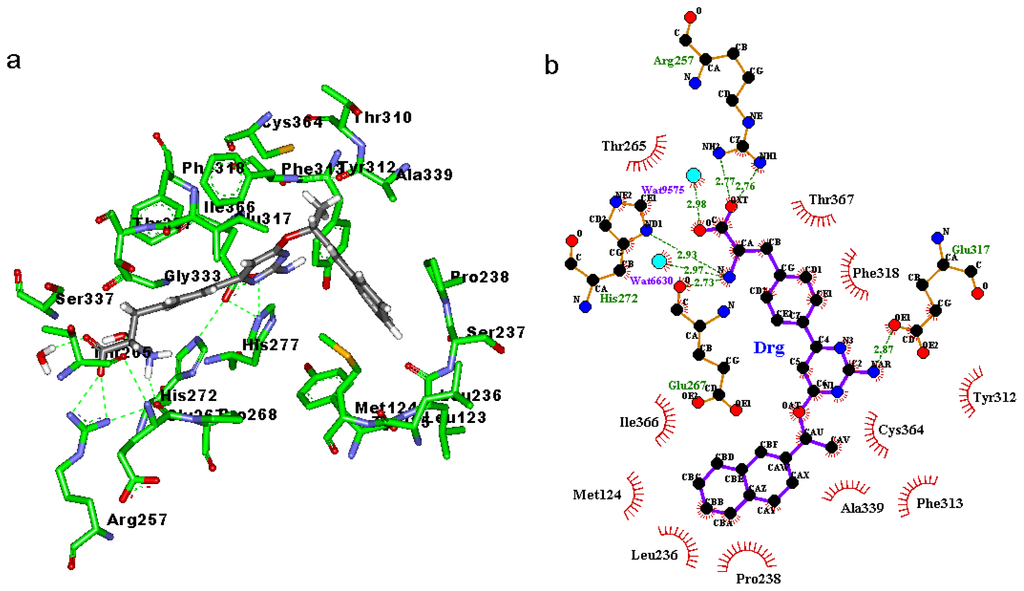
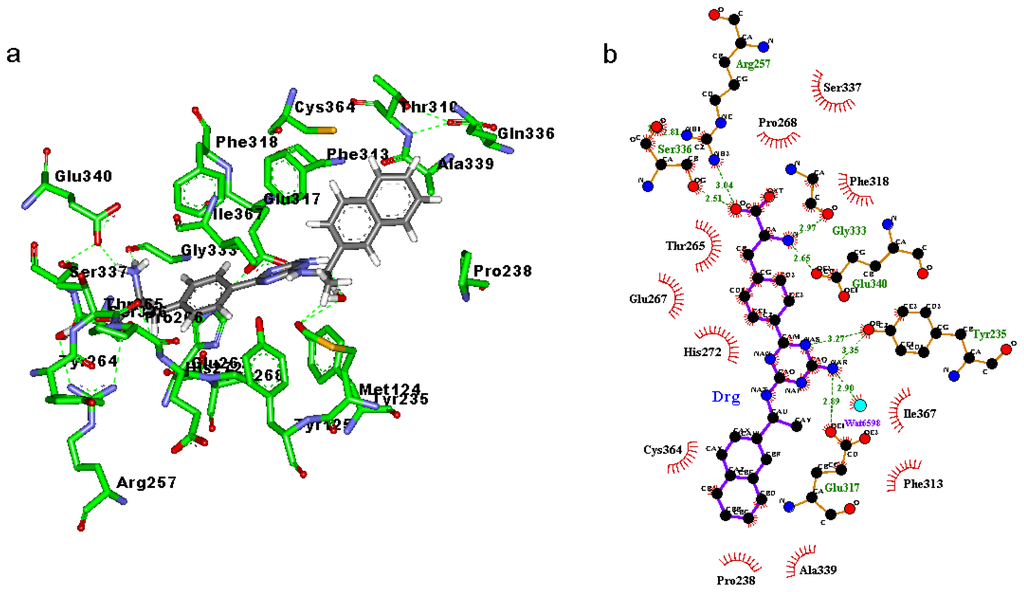
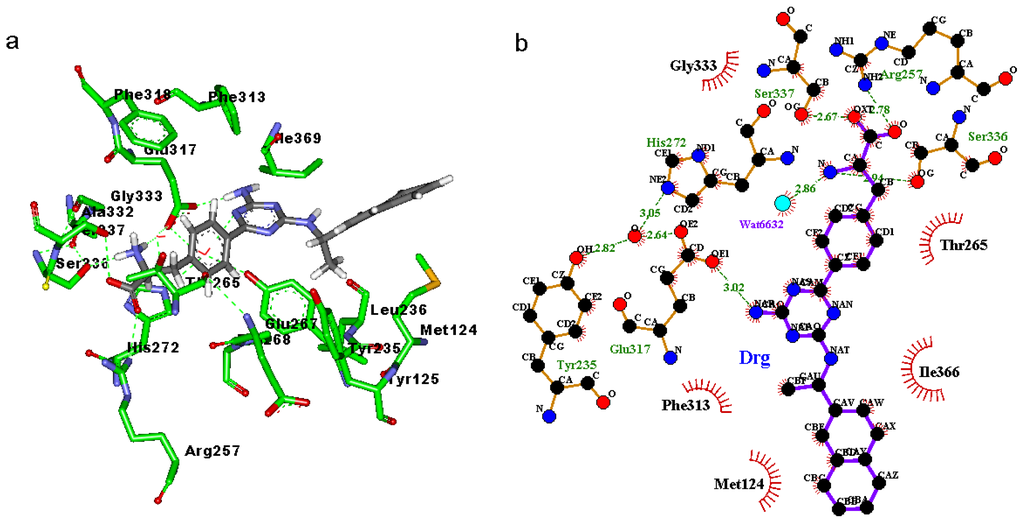
CPT (carnitine palmitoyltransferase) II muscle deficiency is the most common form of muscle fatty acid metabolism disorders. In contrast to carnitine deficiency, it is clinically characterized by attacks of myalgia and rhabdomyolysis without persistent muscle weakness and lipid accumulation in muscle fibers. The biochemical consequences of the disease-causing mutations are still discussed controversially. CPT activity in muscles of patients with CPT II deficiency ranged from not detectable to reduced to normal. Based on the observation that in patients, total CPT is completely inhibited by malony-CoA, a deficiency of malonyl-CoA-insensitive CPT II has been suggested. In contrast, it has also been shown that in muscle CPT II deficiency, CPT II protein is present in normal concentrations with normal enzymatic activity. However, CPT II in patients is abnormally sensitive to inhibition by malonyl-CoA, Triton X-100 and fatty acid metabolites. A recent study on human recombinant CPT II enzymes (His6-N-hCPT2 and His6-N-hCPT2/S113L) revealed that the wild-type and the S113L variants showed the same enzymatic activity. However, the mutated enzyme showed an abnormal thermal destabilization at 40 and 45 °C and an abnormal sensitivity to inhibition by malony-CoA. The thermolability of the mutant enzyme might explain why symptoms in muscle CPT II deficiency mainly occur during prolonged exercise, infections and exposure to cold. In addition, the abnormally regulated enzyme might be mostly inhibited when the fatty acid metabolism is stressed.
Full article
Graphical abstract




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}