A Combined Experimental and Computational Study of Vam3, a Derivative of Resveratrol, and Syk Interaction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction


2. Results and Discussion
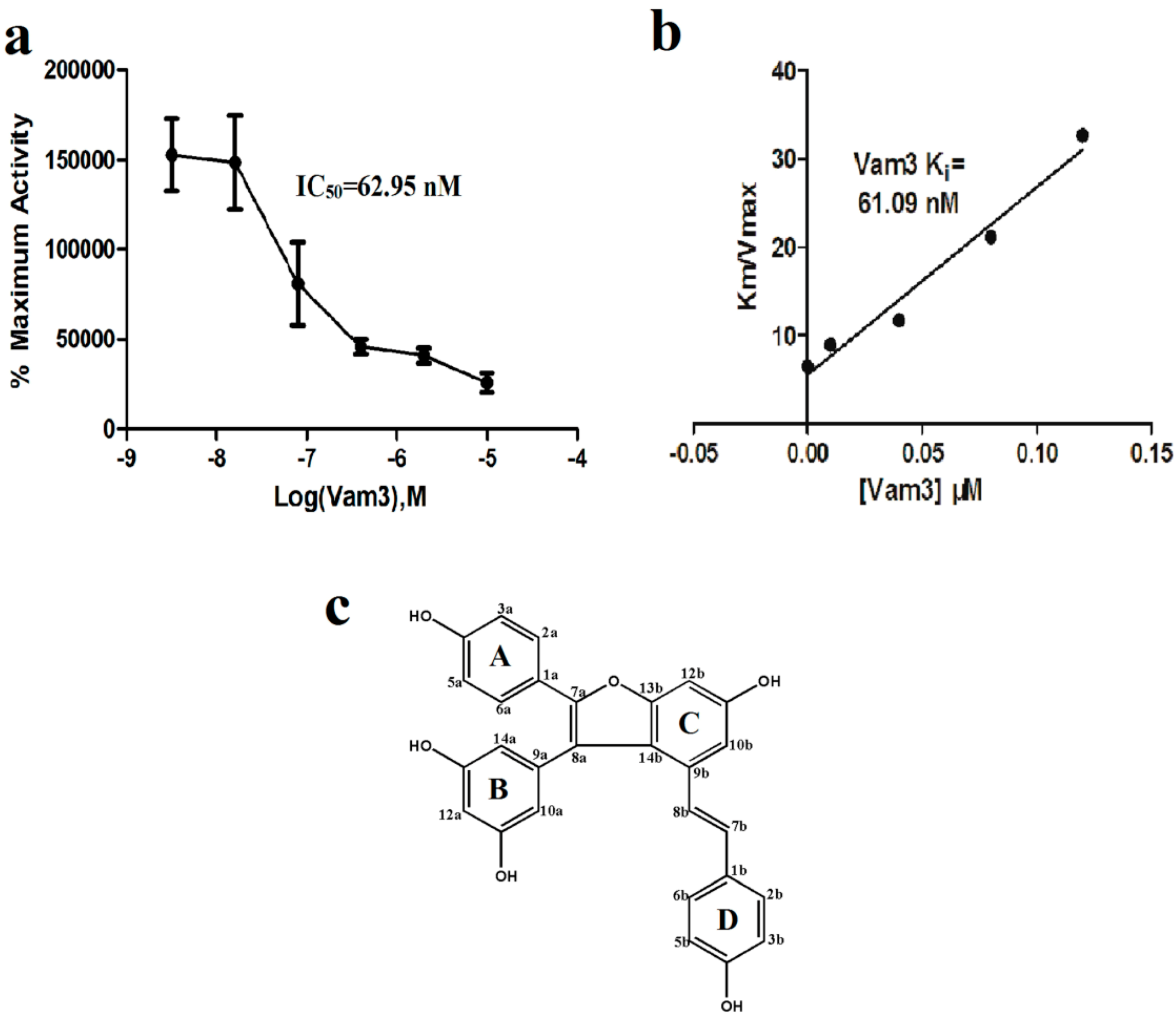
2.1. Vam3 Inhibited Syk Kinase Activity in Vitro
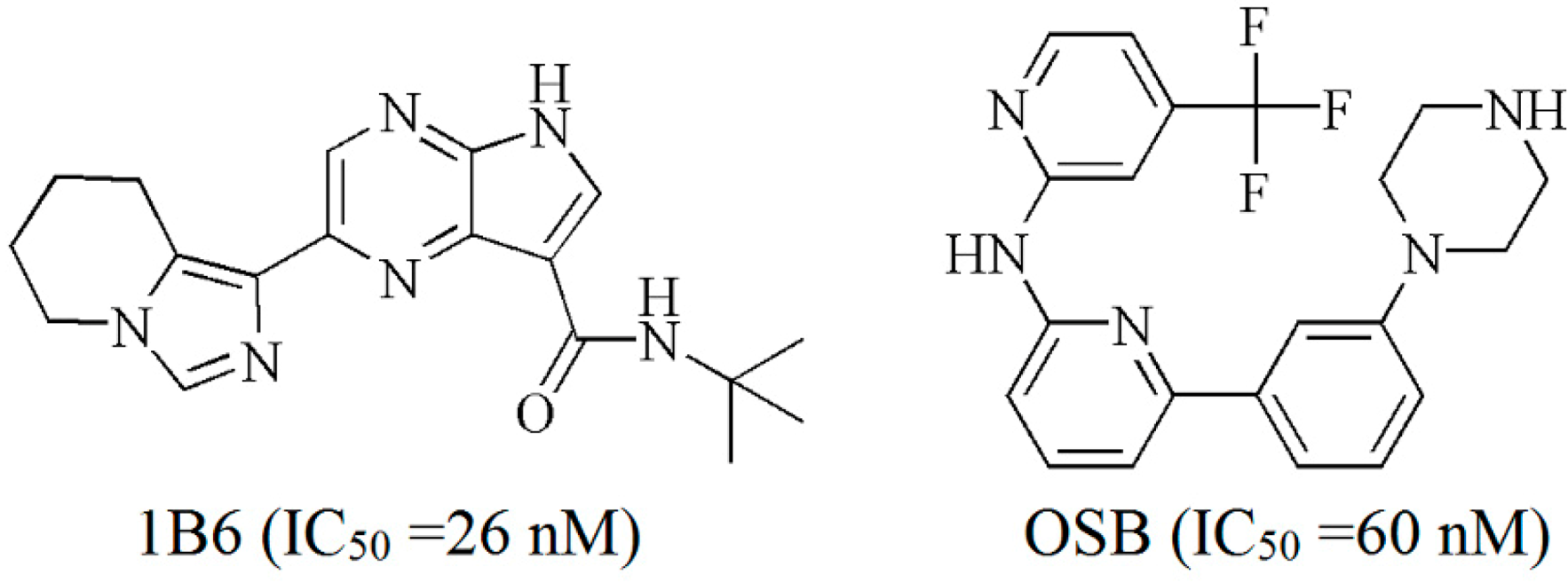
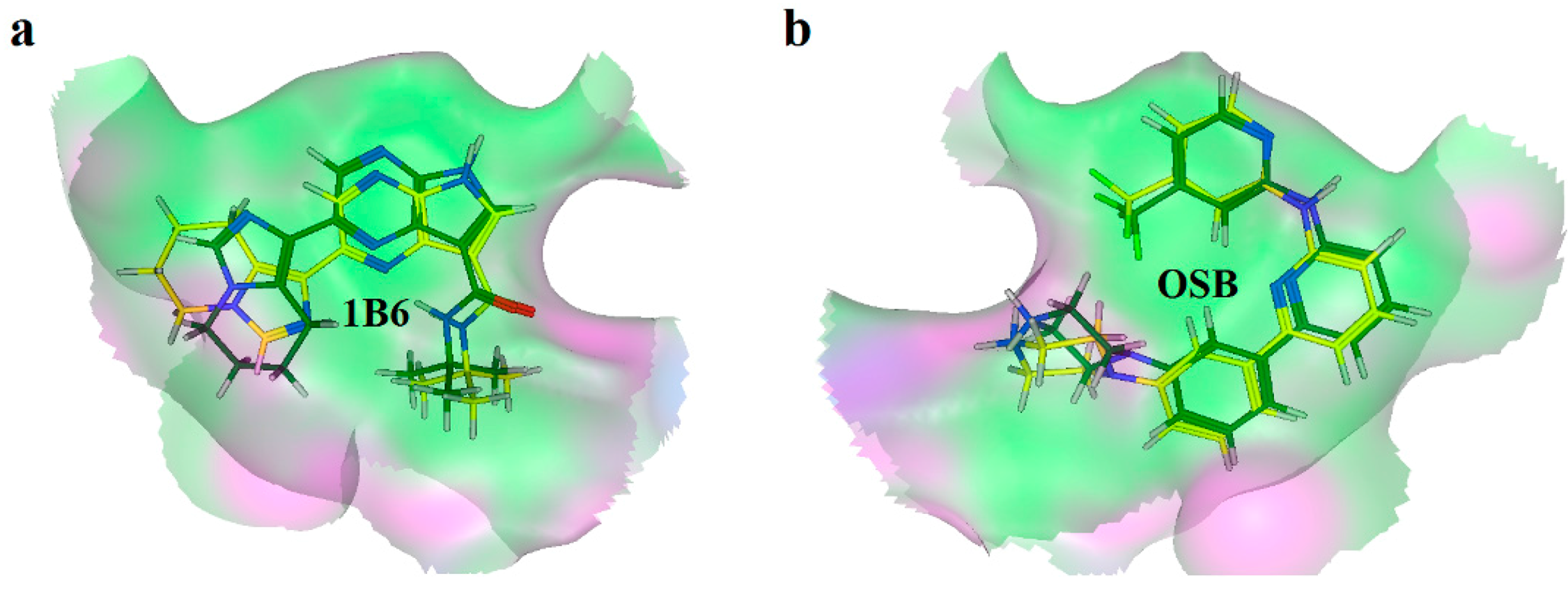
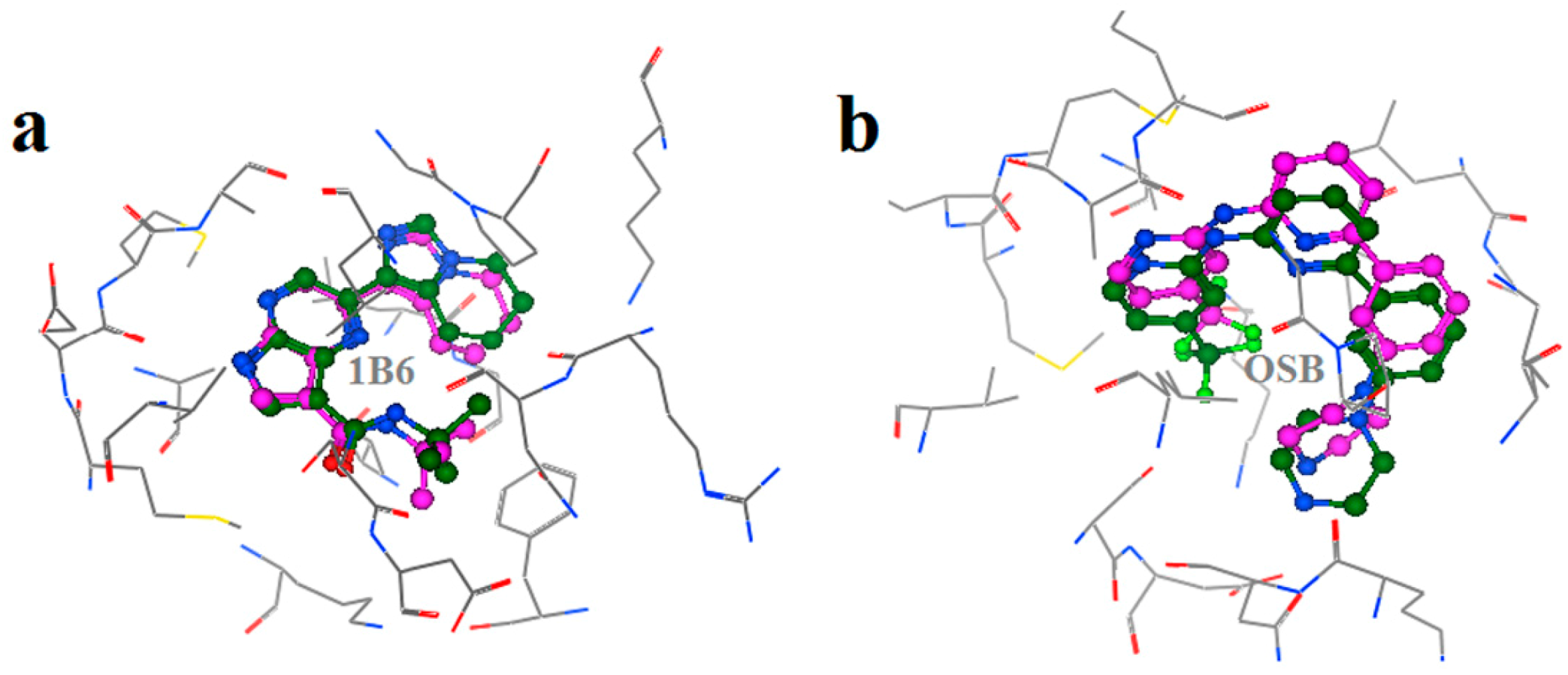
2.2. Extra Precision Docking Studies


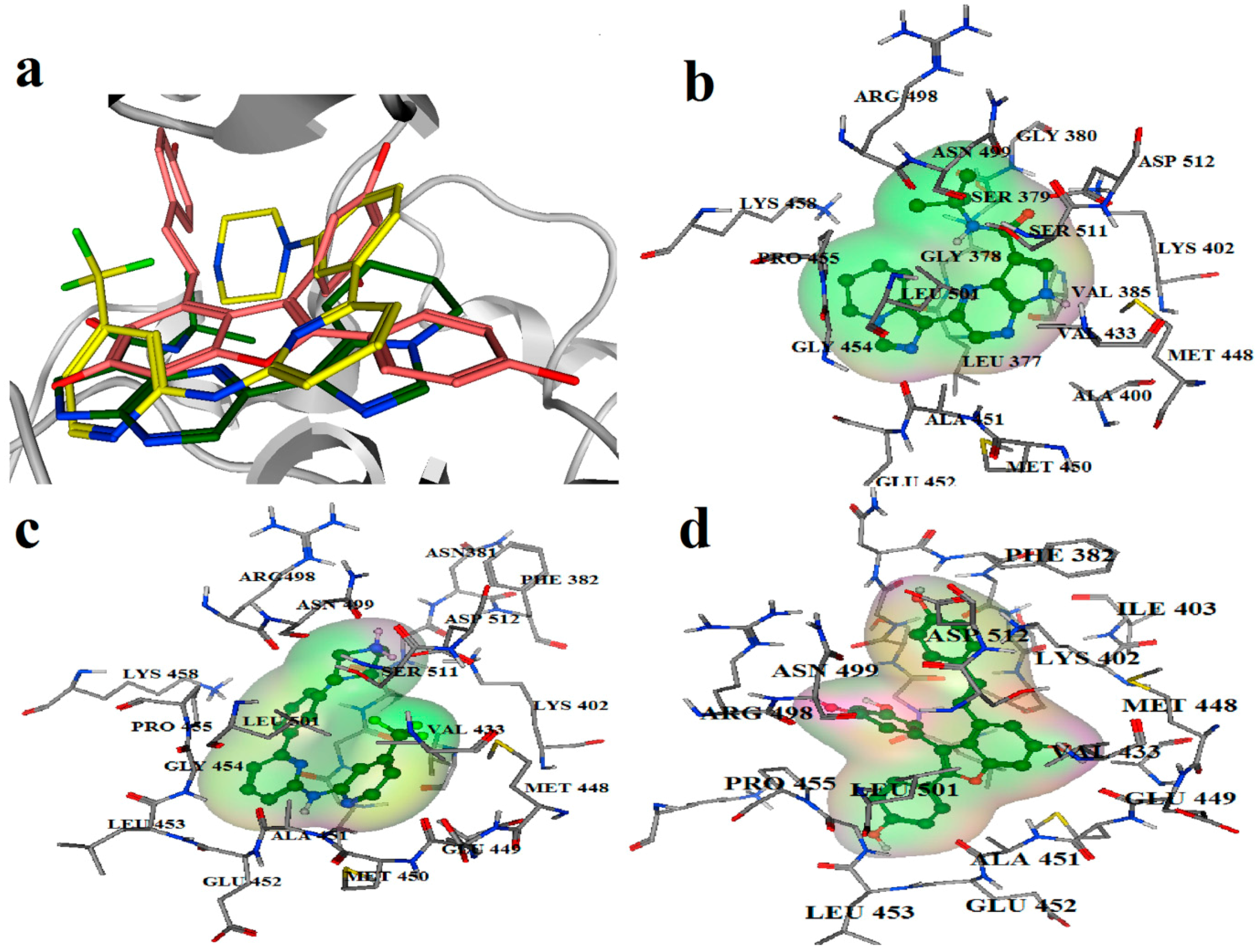
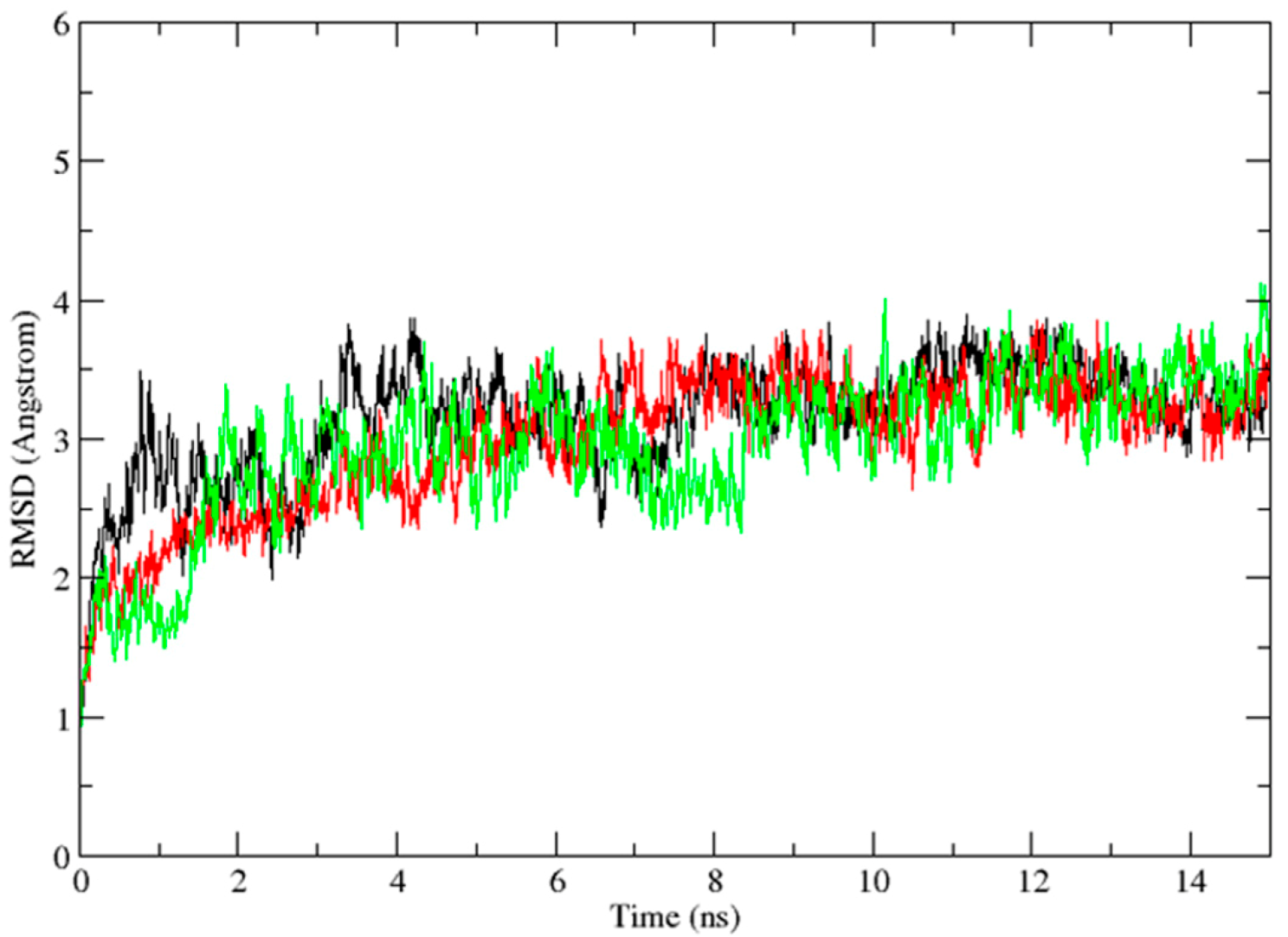
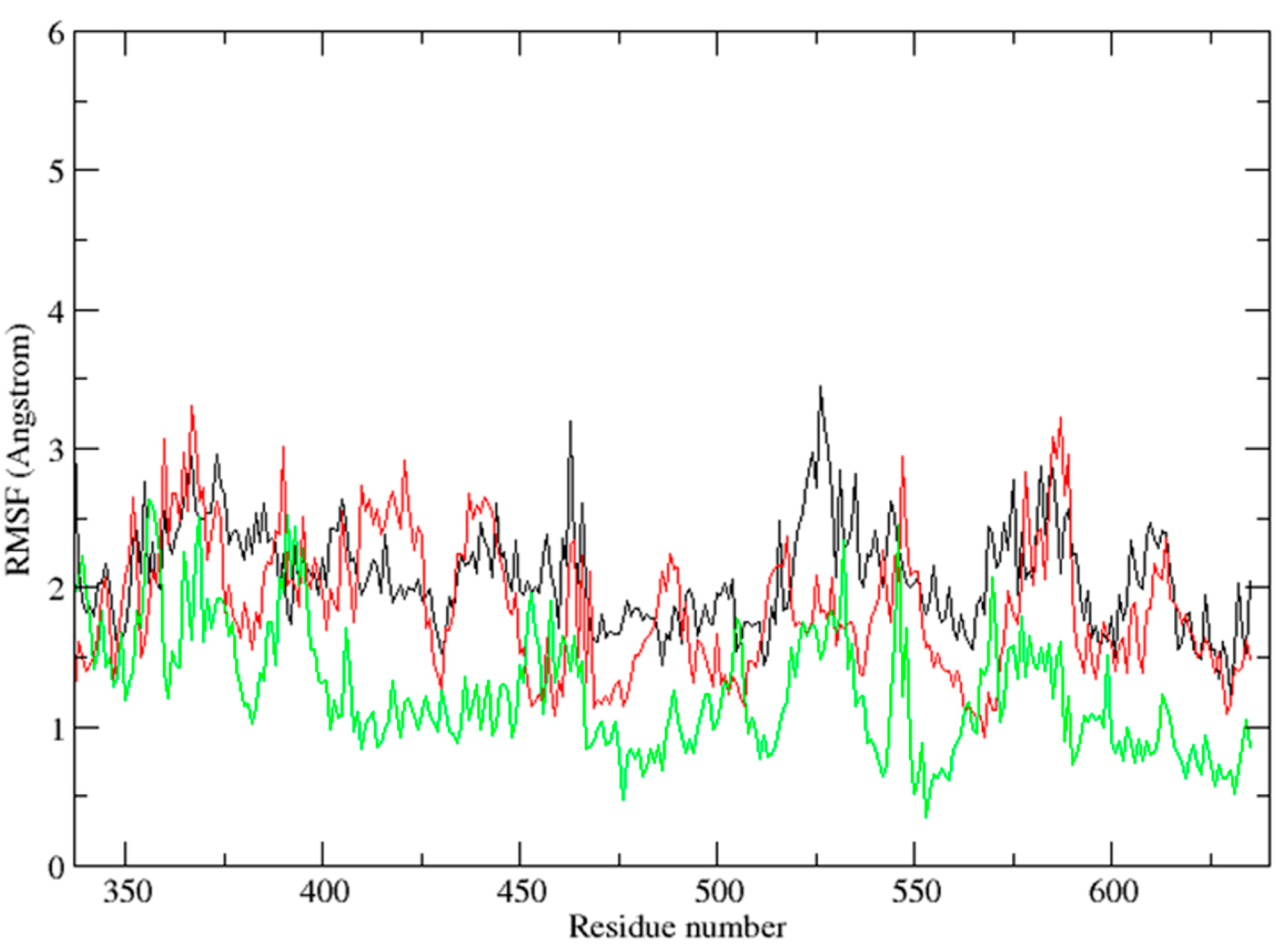
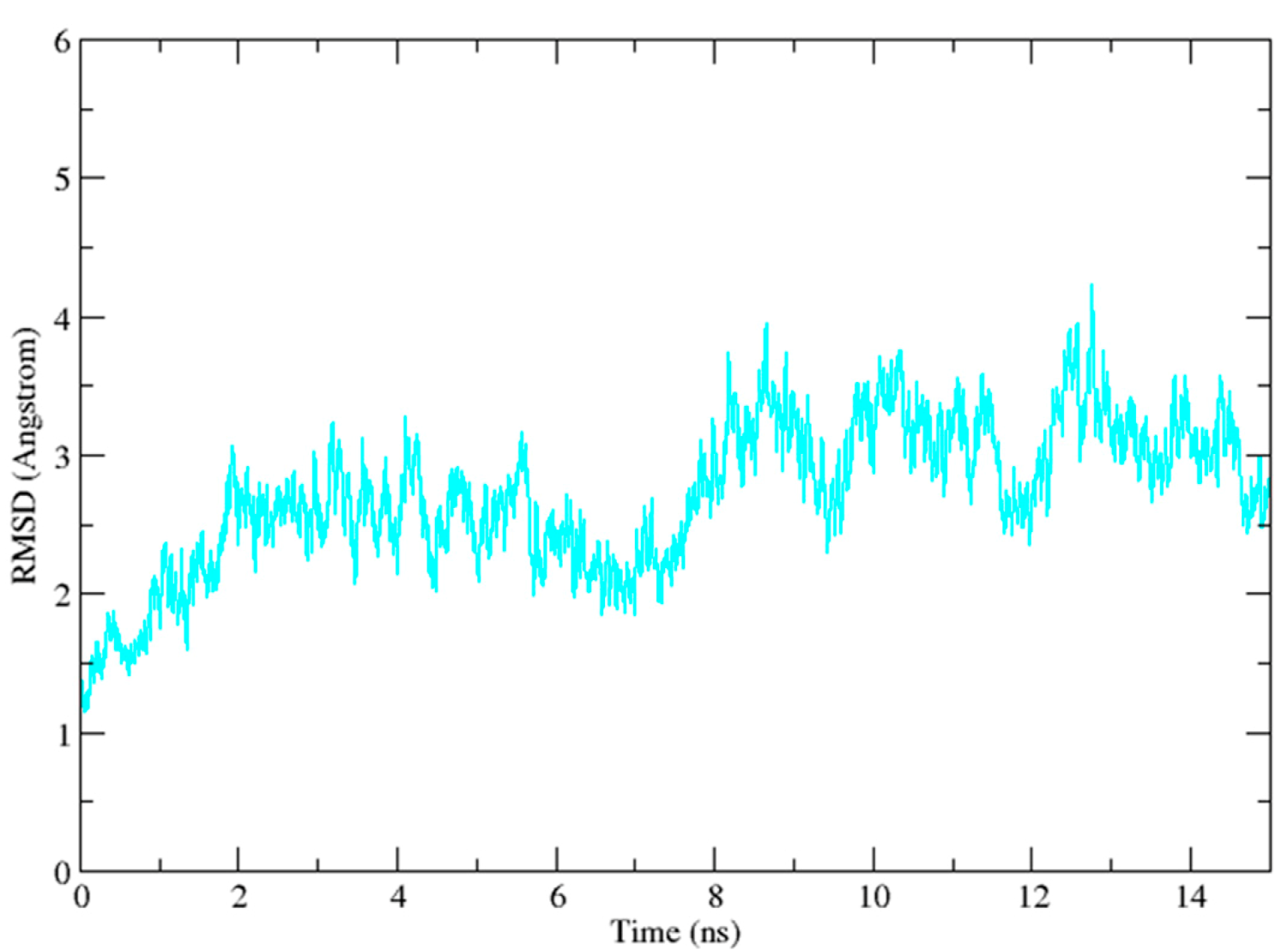
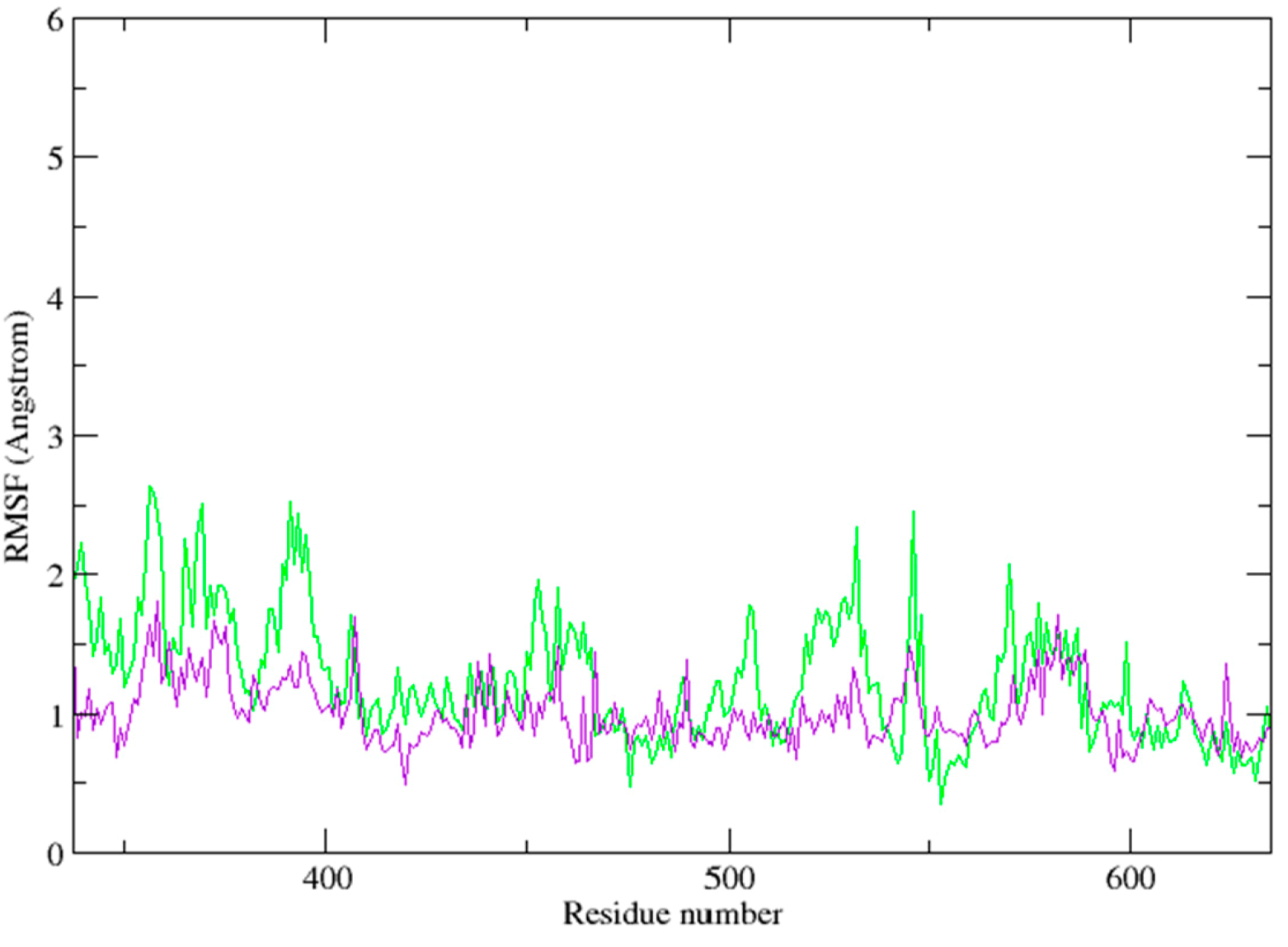
2.3. Molecular Dynamics Simulation Studies




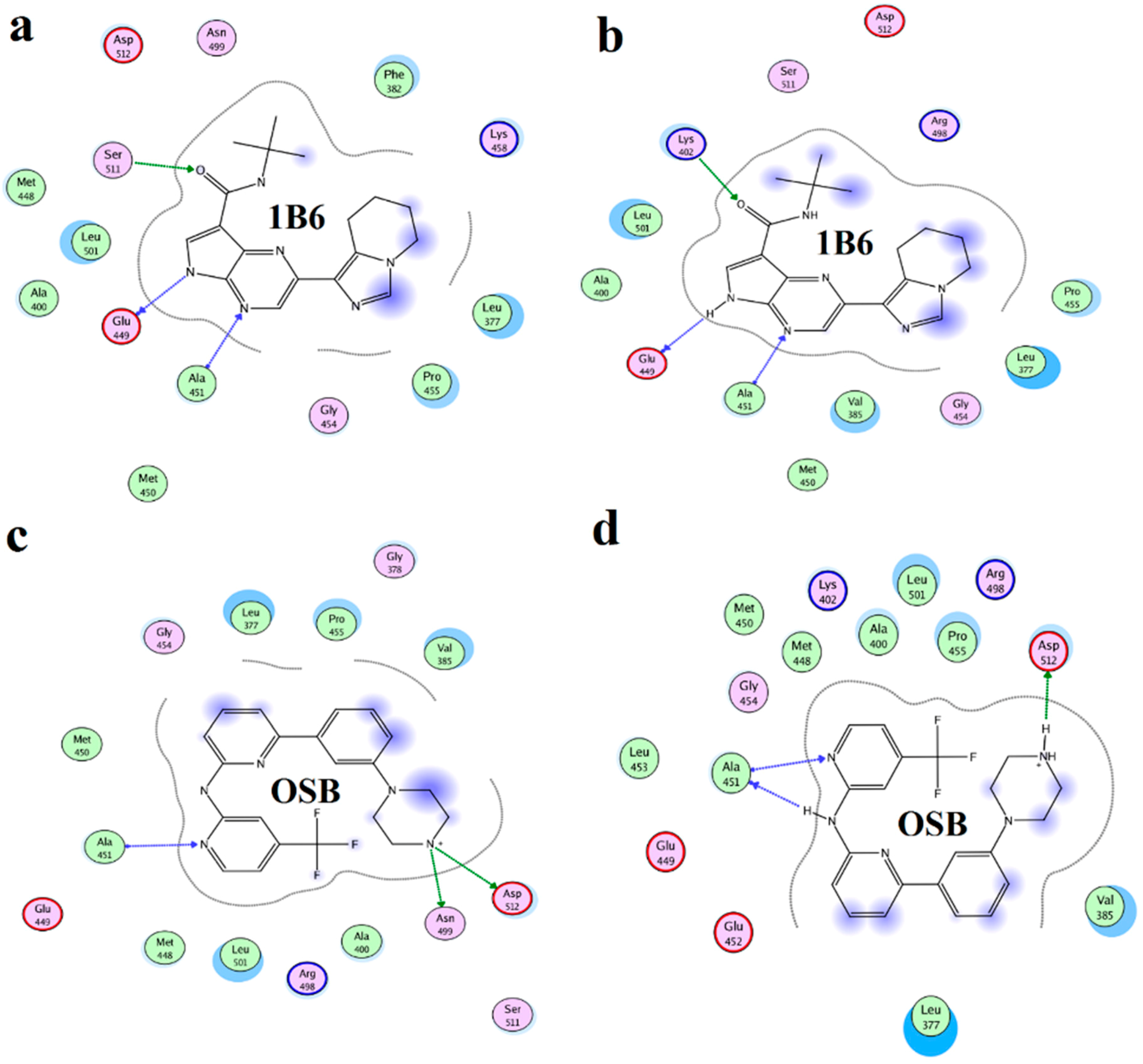
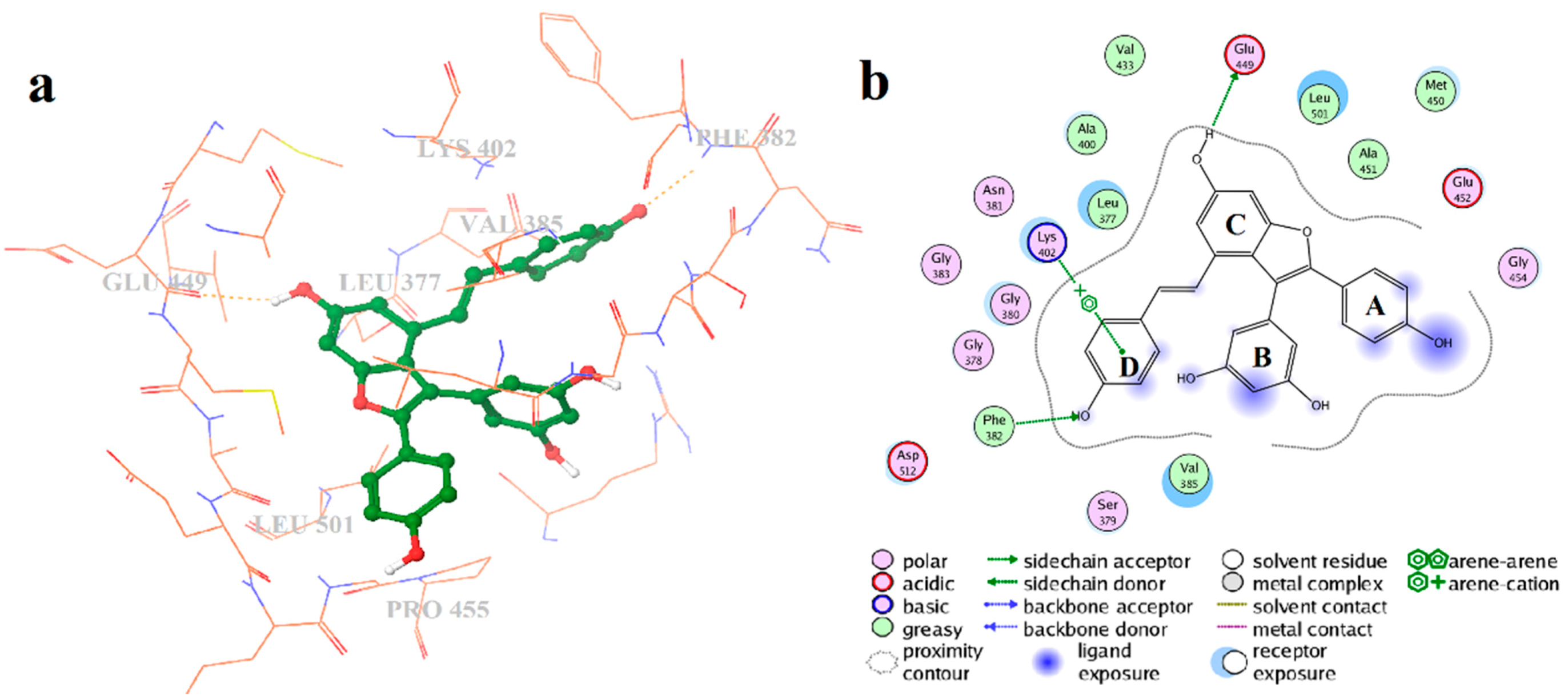
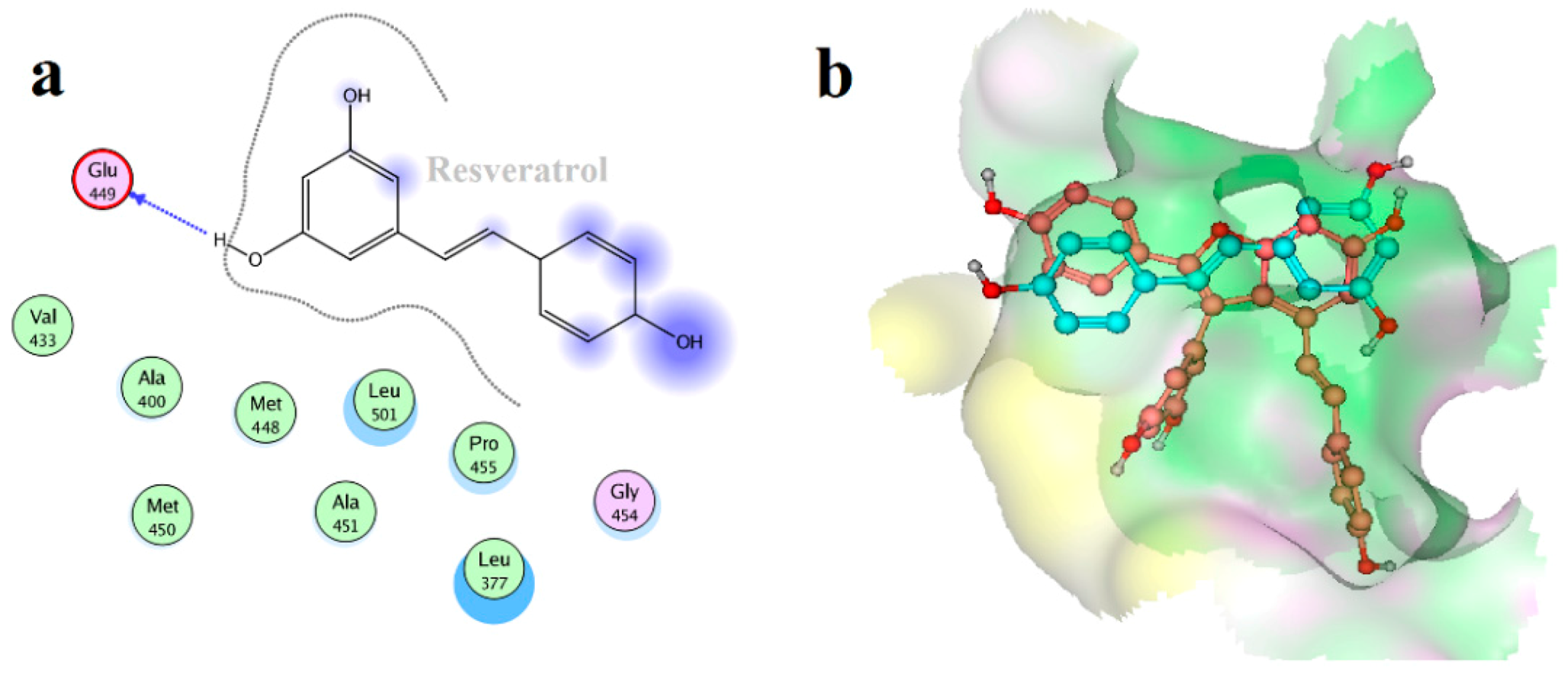
2.4. Binding Mode Analysis of Vam3–Syk Complex




3. Materials and Methods
3.1. Experimental Studies
3.1.1. Plant Material
3.1.2. In Vitro Fluorescence Polarization Kinase Assay
3.2. Computational Studies
3.2.1. Preparation of Protein Target Structure
3.2.2. Ligand Preparation
3.2.3. Molecular Docking
3.2.4. Molecular Dynamics Stimulation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singh, R.; Masuda, E.S.; Payan, D.G. Discovery and development of spleen tyrosine kinase (SYK) inhibitors. J. Med. Chem. 2012, 55, 3614–3643. [Google Scholar]
- Bajpai, M.; Chopra, P.; Dastidar, S.G.; Ray, A. Spleen tyrosine kinase: A novel target for therapeutic intervention of rheumatoid arthritis. Expert Opin. Investig. Drugs 2008, 17, 641–659. [Google Scholar]
- Lucas, M.C.; Goldstein, D.M.; Hermann, J.C.; Kuglstatter, A.; Liu, W.; Luk, K.C.; Padilla, F.; Slade, M.; Villaseñor, A.G.; Wanner, J. Rational design of highly selective spleen tyrosine kinase inhibitors. J. Med. Chem. 2012, 55, 10414–10423. [Google Scholar]
- Zou, W.; Kitaura, H.; Reeve, J.; Long, F.; Tybulewicz, V.L.; Shattil, S.J.; Ginsberg, M.H.; Ross, F.P.; Teitelbaum, S.L. Syk, c-Src, the αvβ3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J. Cell Biol. 2007, 176, 877–888. [Google Scholar]
- Young, R.M.; Hardy, I.R.; Clarke, R.L.; Lundy, N.; Pine, P.; Turner, B.C.; Potter, T.A.; Refaeli, Y. Mouse models of non-Hodgkin lymphoma reveal Syk as an important therapeutic target. Blood 2009, 113, 2508–2516. [Google Scholar]
- Gradler, U.; Schwarz, D.; Dresing, V.; Musil, D.; Bomke, J.; Frech, M.; Greiner, H.; Jakel, S.; Rysiok, T.; Muller-Pompalla, D.; et al. Structural and biophysical characterization of the Syk activation switch. J. Mol. Biol. 2013, 425, 309–333. [Google Scholar]
- Siraganian, R.P.; Zhang, J.; Suzuki, K.; Sada, K. Protein tyrosine kinase Syk in mast cell signaling. Mol. Immunol. 2002, 38, 1229–1233. [Google Scholar]
- Van Oers, N.S.; Weiss, A. The Syk/ZAP-70 protein tyrosine kinase connection to antigen receptor signalling processes. Semin. Immunol. 1995, 7, 227–236. [Google Scholar]
- Tsang, E.; Giannetti, A.M.; Shaw, D.; Dinh, M.; Joyce, K.; Gandhi, S.; Ho, H.; Wang, S.; Papp, E.; Bradshaw, J.M. Molecular mechanism of the Syk activation switch. J. Biol. Chem. 2008, 283, 32650–32659. [Google Scholar]
- Castillo, M.; Forns, P.; Erra, M.; Mir, M.; Lopez, M.; Maldonado, M.; Orellana, A.; Carreno, C.; Ramis, I.; Miralpeix, M.; et al. Highly potent aminopyridines as Syk kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5419–5423. [Google Scholar]
- Forns, P.; Esteve, C.; Taboada, L.; Alonso, J.A.; Orellana, A.; Maldonado, M.; Carreno, C.; Ramis, I.; Lopez, M.; Miralpeix, M.; et al. Pyrazine-based Syk kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2784–2788. [Google Scholar]
- Padilla, F.; Bhagirath, N.; Chen, S.; Chiao, E.; Goldstein, D.M.; Hermann, J.C.; Hsu, J.; Kennedy-Smith, J.J.; Kuglstatter, A.; Liao, C.; et al. Pyrrolopyrazines as selective spleen tyrosine kinase inhibitors. J. Med. Chem. 2013, 56, 1677–1692. [Google Scholar]
- Atwell, S.; Adams, J.M.; Badger, J.; Buchanan, M.D.; Feil, I.K.; Froning, K.J.; Gao, X.; Hendle, J.; Keegan, K.; Leon, B.C. A novel mode of Gleevec binding is revealed by the structure of spleen tyrosine kinase. J. Biol. Chem. 2004, 279, 55827–55832. [Google Scholar]
- Braselmann, S.; Taylor, V.; Zhao, H.; Wang, S.; Sylvain, C.; Baluom, M.; Qu, K.; Herlaar, E.; Lau, A.; Young, C. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J. Pharmacol. Exp. Ther. 2006, 319, 998–1008. [Google Scholar]
- Cherry, M.; Lamers, M.; Smith, S.; Williams, D. Spleen Tyrosine Kinase Catalytic Domain: Crystal Structure and Binding Pockets Thereof. U.S. Patent 20070020253 A1, 25 January 2007. [Google Scholar]
- Villaseñor, A.G.; Kondru, R.; Ho, H.; Wang, S.; Papp, E.; Shaw, D.; Barnett, J.W.; Browner, M.F.; Kuglstatter, A. Structural insights for design of potent spleen tyrosine kinase inhibitors from crystallographic analysis of three inhibitor complexes. Chem. Biol. Drug Des. 2009, 73, 466–470. [Google Scholar]
- Shi, J.; Yin, N.; Xuan, L.L.; Yao, C.S.; Meng, A.M.; Hou, Q. Vam3, a derivative of resveratrol, attenuates cigarette smoke-induced autophagy. Acta Pharmacol. Sin. 2012, 33, 888–896. [Google Scholar]
- Yang, L.; Yao, C.; Wu, Z.; Xuan, L.; Bai, J.; Cheng, G.; Lin, M.; Wen, M.; Hou, Q. Effects of dihydroxy-stilbene compound Vam3 on airway inflammation, expression of ICAM-1, activities of NF-κB and MMP-9 in asthmatic mice. Yao Xue Xue Bao 2010, 45, 1503–1508. (In Chinese) [Google Scholar]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. [Google Scholar]
- Norberg, J.; Nilsson, L. Advances in biomolecular simulations: Methodology and recent applications. Q. Rev. Biophys. 2003, 36, 257–306. [Google Scholar]
- Lu, Y.; Li, Y.; Jahng, Y.; Son, J.K.; Chang, H.W. Citreorosein inhibits degranulation and leukotriene C4 generation through suppression of Syk pathway in mast cells. Mol. Cell. Biochem. 2012, 365, 333–341. [Google Scholar]
- Han, S.Y.; Bae, J.Y.; Park, S.H.; Kim, Y.H.; Park, J.H.Y.; Kang, Y.H. Resveratrol inhibits IgE-mediated basophilic mast cell degranulation and passive cutaneous anaphylaxis in mice. J. Nutr. 2013, 143, 632–639. [Google Scholar]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar]
- Maestro 8.5, Schrödinger, LLC: New York, NY, USA, 2008.
- LigPrep 2.2, Schrödinger, LLC: New York, NY, USA, 2008.
- Hayes, J.M.; Stein, M.; Weiser, J. Accurate calculations of ligand binding free energies: Chiral separation with enantioselective receptors. J. Phys. Chem. A 2004, 108, 3572–3580. [Google Scholar]
- Glide 5.0, Schrödinger, LLC: New York, NY, USA, 2008.
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Desmond 2.0, D.E. Shaw Research/Schrödinger, LLC: New York, NY, USA,, 2008.
- MacroModel 9.6, Schrödinger, LLC: New York, NY, USA, 2008.
- Andersen, H.C. RATTLE: A “Velocity” version of the SHAKE algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar]
- Strahan, G.D.; Keniry, M.A.; Shafer, R.H. NMR structure refinement and dynamics of the K+–[d(G3T4G3)] 2 quadruplex via particle mesh Ewald molecular dynamics simulations. Biophys. J. 1998, 75, 968–981. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar]
- Knapp, B.; Lederer, N.; Omasits, U.; Schreiner, W. vmdICE: A plug-in for rapid evaluation of molecular dynamics simulations using VMD. J. Comput. Chem. 2010, 31, 2868–2873. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, M.; Liu, R.; Chen, Y.; Zheng, Q.; Fan, S.; Liu, P. A Combined Experimental and Computational Study of Vam3, a Derivative of Resveratrol, and Syk Interaction. Int. J. Mol. Sci. 2014, 15, 17188-17203. https://doi.org/10.3390/ijms150917188
Jiang M, Liu R, Chen Y, Zheng Q, Fan S, Liu P. A Combined Experimental and Computational Study of Vam3, a Derivative of Resveratrol, and Syk Interaction. International Journal of Molecular Sciences. 2014; 15(9):17188-17203. https://doi.org/10.3390/ijms150917188
Chicago/Turabian StyleJiang, Ming, Renping Liu, Ying Chen, Qisheng Zheng, Saijun Fan, and Peixun Liu. 2014. "A Combined Experimental and Computational Study of Vam3, a Derivative of Resveratrol, and Syk Interaction" International Journal of Molecular Sciences 15, no. 9: 17188-17203. https://doi.org/10.3390/ijms150917188
APA StyleJiang, M., Liu, R., Chen, Y., Zheng, Q., Fan, S., & Liu, P. (2014). A Combined Experimental and Computational Study of Vam3, a Derivative of Resveratrol, and Syk Interaction. International Journal of Molecular Sciences, 15(9), 17188-17203. https://doi.org/10.3390/ijms150917188