Molecular Modeling and MM-PBSA Free Energy Analysis of Endo-1,4-β-Xylanase from Ruminococcus albus 8


Abstract
:1. Introduction
2. Results and Discussion
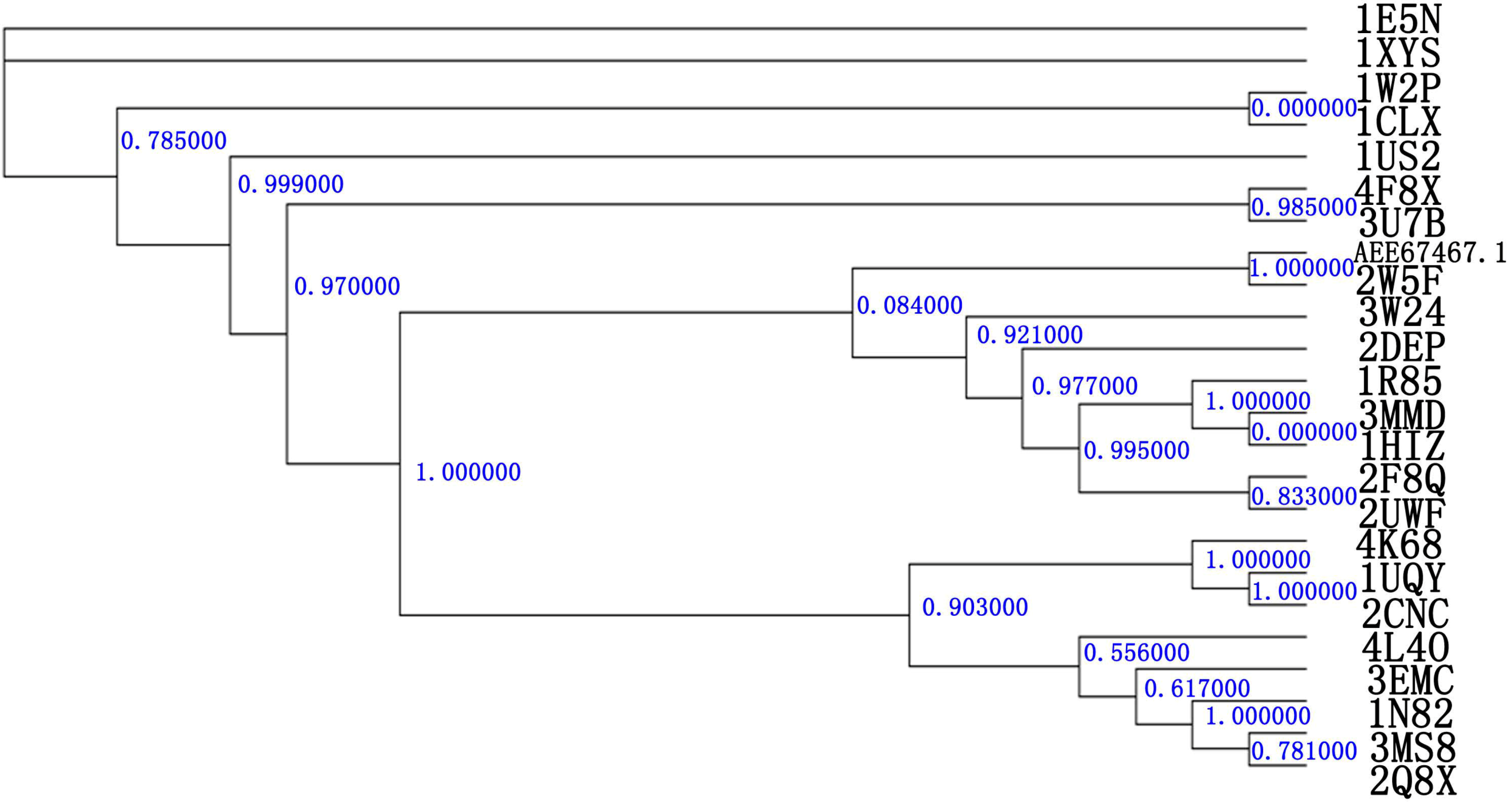
2.1. Relatedness of the GH10 Family
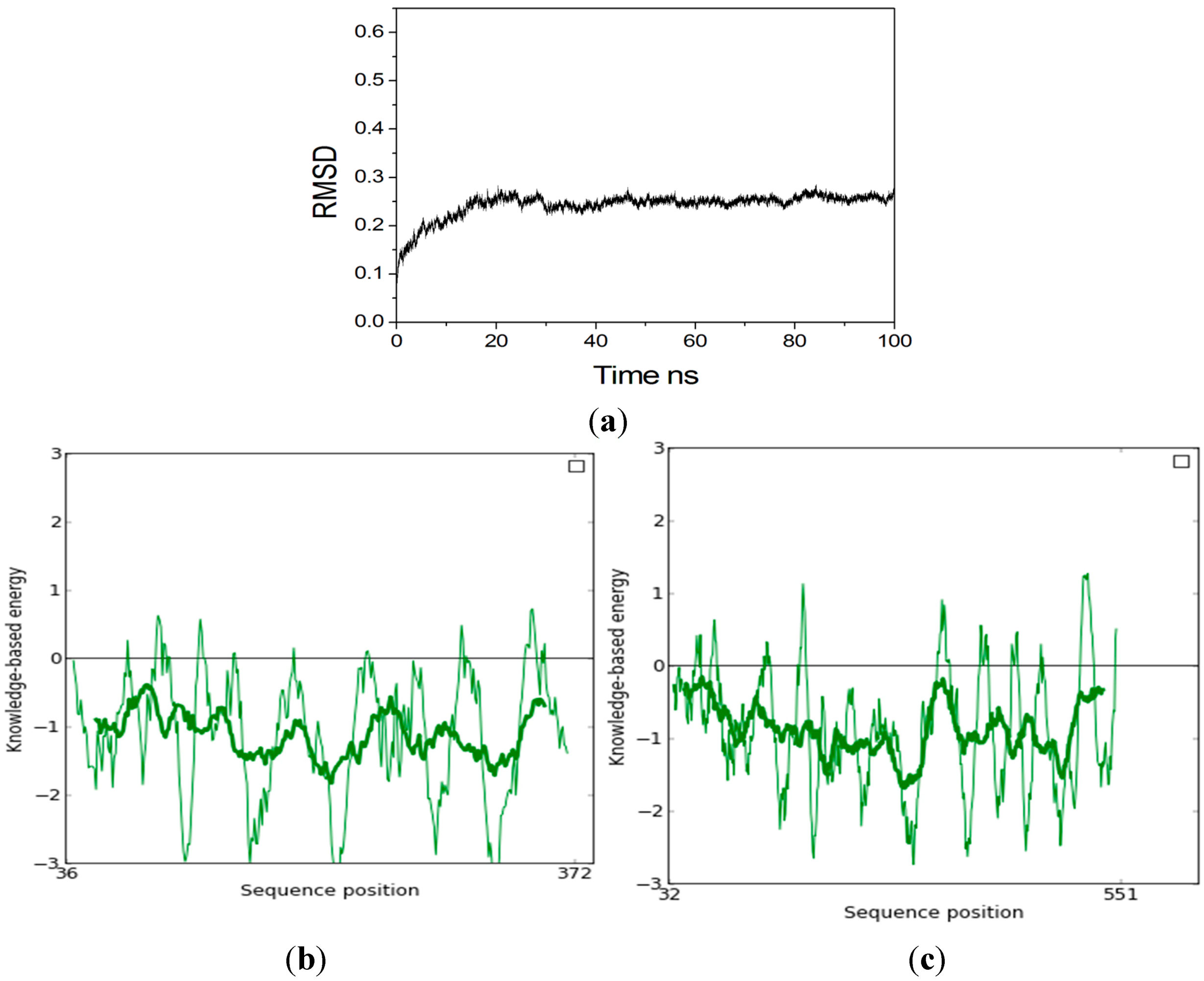
2.2. Homology Modeling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Template (PDB Id) | Sequence Identity | Resolution | Organism | Global Model Quality Estimate | Procheck | Verify_3D |
|---|---|---|---|---|---|---|
| 2W5F A | 39% | 1.90 | C. Thermocellum | 0.66 | 86.6% core 11.8% allow 1.0% gener 0.7% disall | 86.09% residues > 0.2 |
| 2WZE A | 39% | 2.50 | C. Thermocellum | 0.63 | 82.3% core 14.7% allow 2.0% gener 1.0% disall | 83.33% |
| 3W24 A | 38% | 1.32 | Thermoanaerobacterium saccharolyticum | 0.63 | 87.0% core 11.0% allow 1.7% gener 0.3% disall | 82.53% |
| 2Q8X A | 38% | 1.45 | G. stearothermophilus | 0.61 | 84.2% core 13.4% allow 1.0% gener 1.3% disall | 73.94% |
| 3MS8 A | 39% | 1.70 | G. stearothermophilus | 0.60 | 81.2% core 18.1% allow 0.3% gener 0.3% disall | 69.70% |
| 3MUI A | 39% | 1.80 | G. stearothermophilus | 0.61 | 83.6% core 14.4% allow 1.7% gener 0.3% disall | 70.30% |
| 1N82 A | 38% | 1.45 | G. stearothermophilus | 0.60 | 81.7% core 16.6% allow 0.3% gener 1.3% disall | 83.70% |
| Protein | Procheck | Verify_3D |
|---|---|---|
| The initial model | 86.6% core 11.8% allow 1.0% gener 0.7% disall | 86.09% residues > 0.2 |
| The last conformation model | 88.5% core 10.0% allow 1.1% gener 0.4% disall | 97.66% |



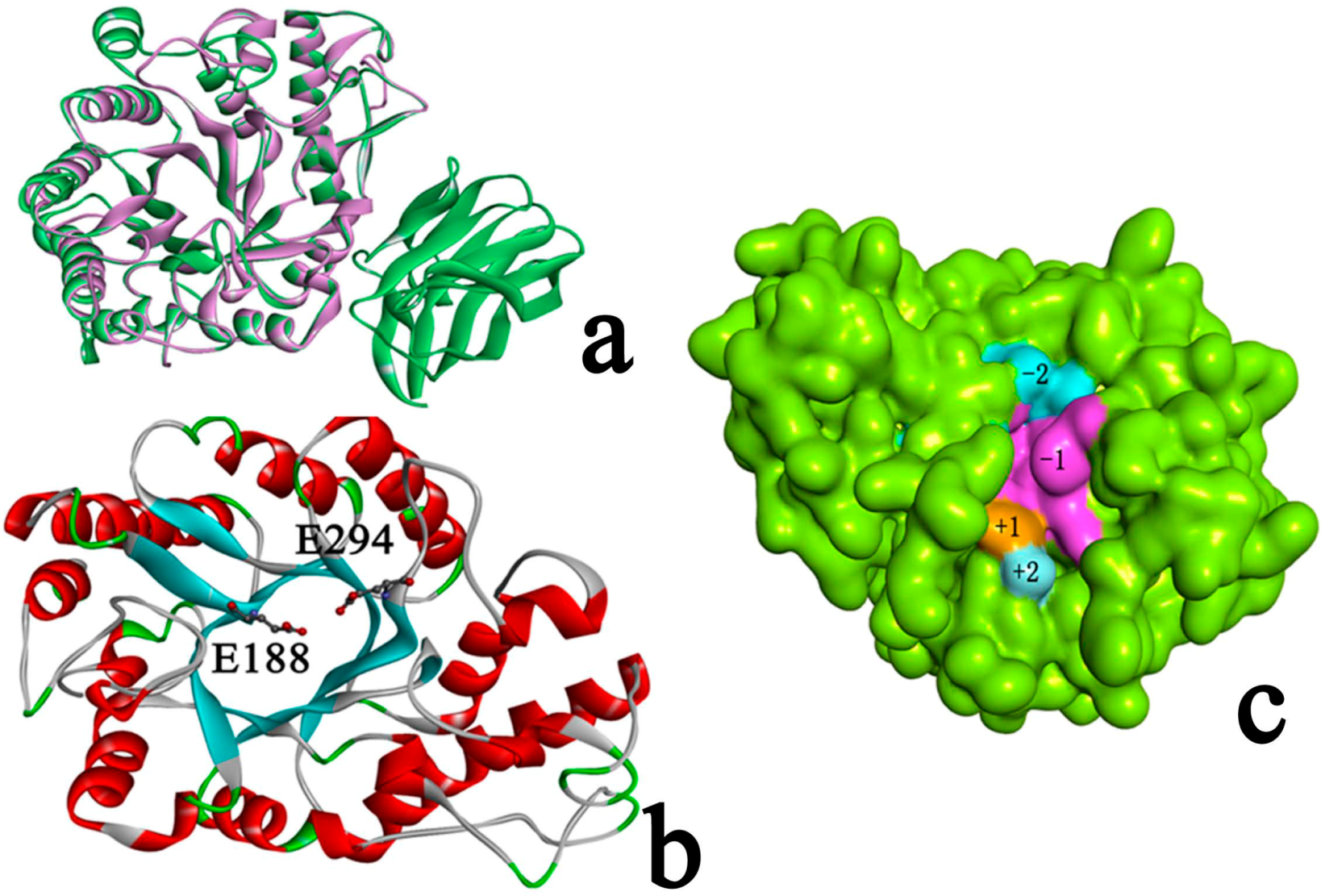
2.3. Identification of Binding Site in Xyn10A
| Protein/Residue | 263 | 265 | 128 | 187 | 344 | 336 | 83 |
|---|---|---|---|---|---|---|---|
| Xyn10A | Q | H | H | N | W | W | K |
| 2W5F | Q | H | H | N | W | W | K |
| 3W24 | Q | H | H | N | W | W | K |
| 2Q8X | Q | H | H | N | W | W | K |
2.4. Docking Study
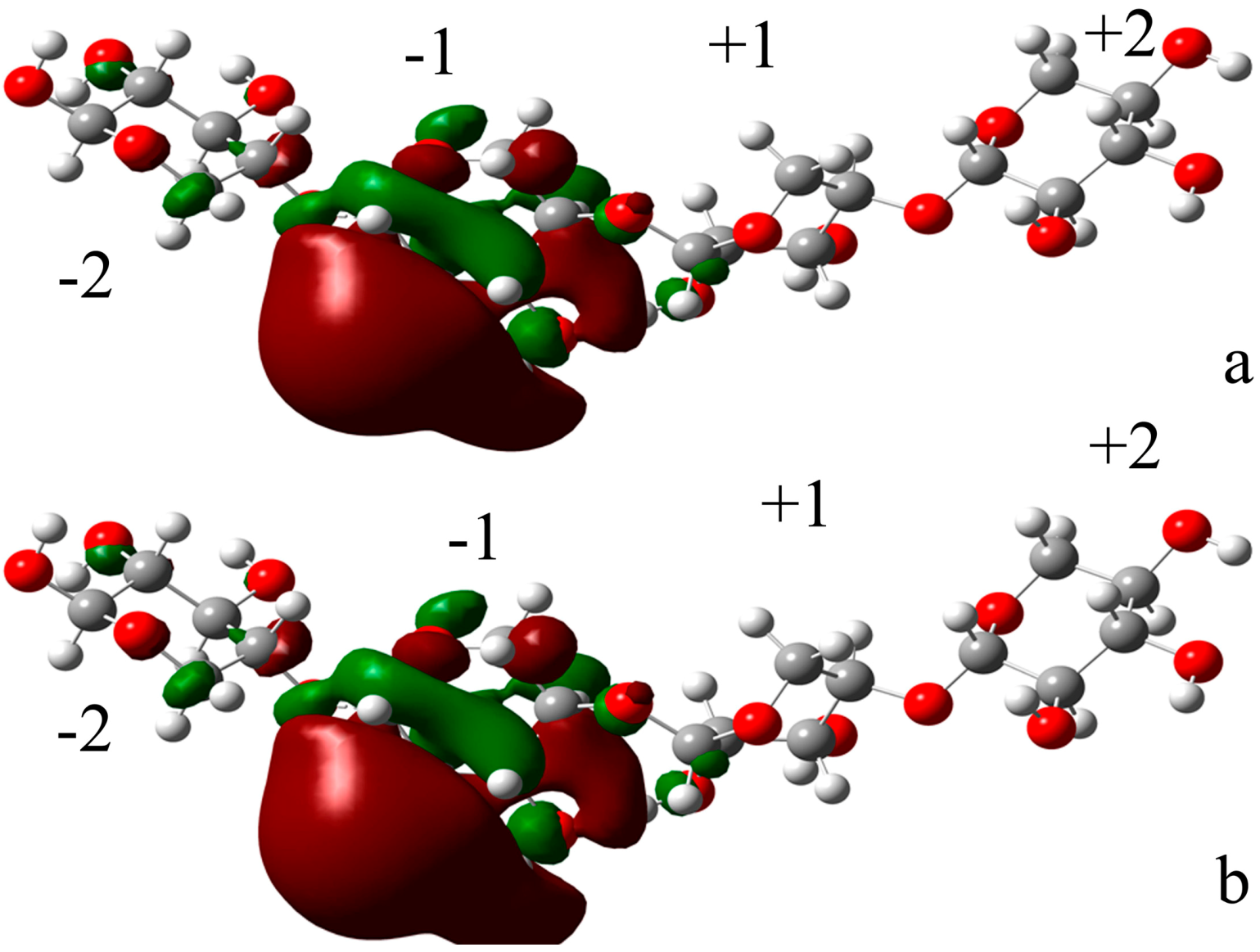
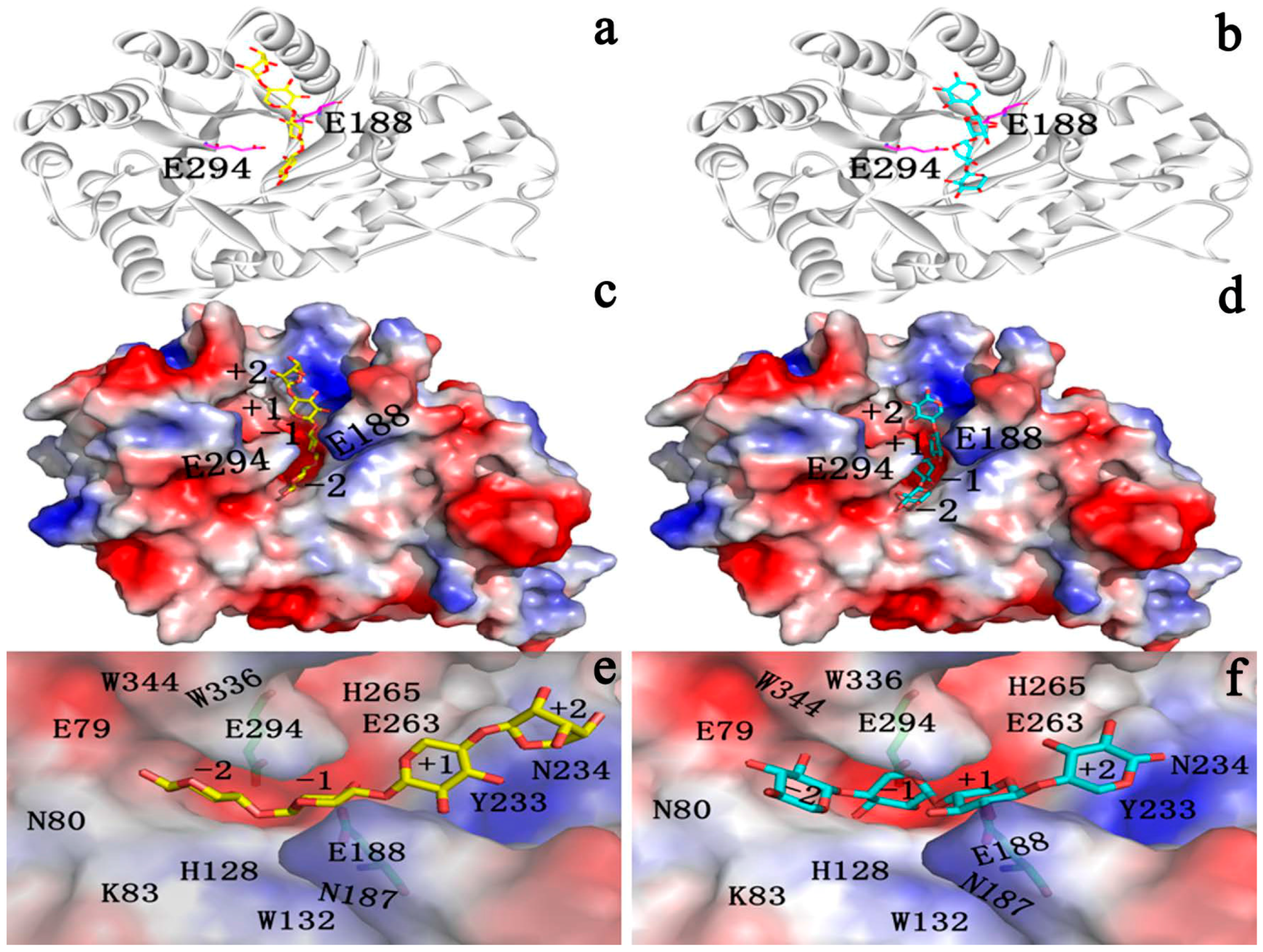
2.4.1. Structures of Ground State Complexes


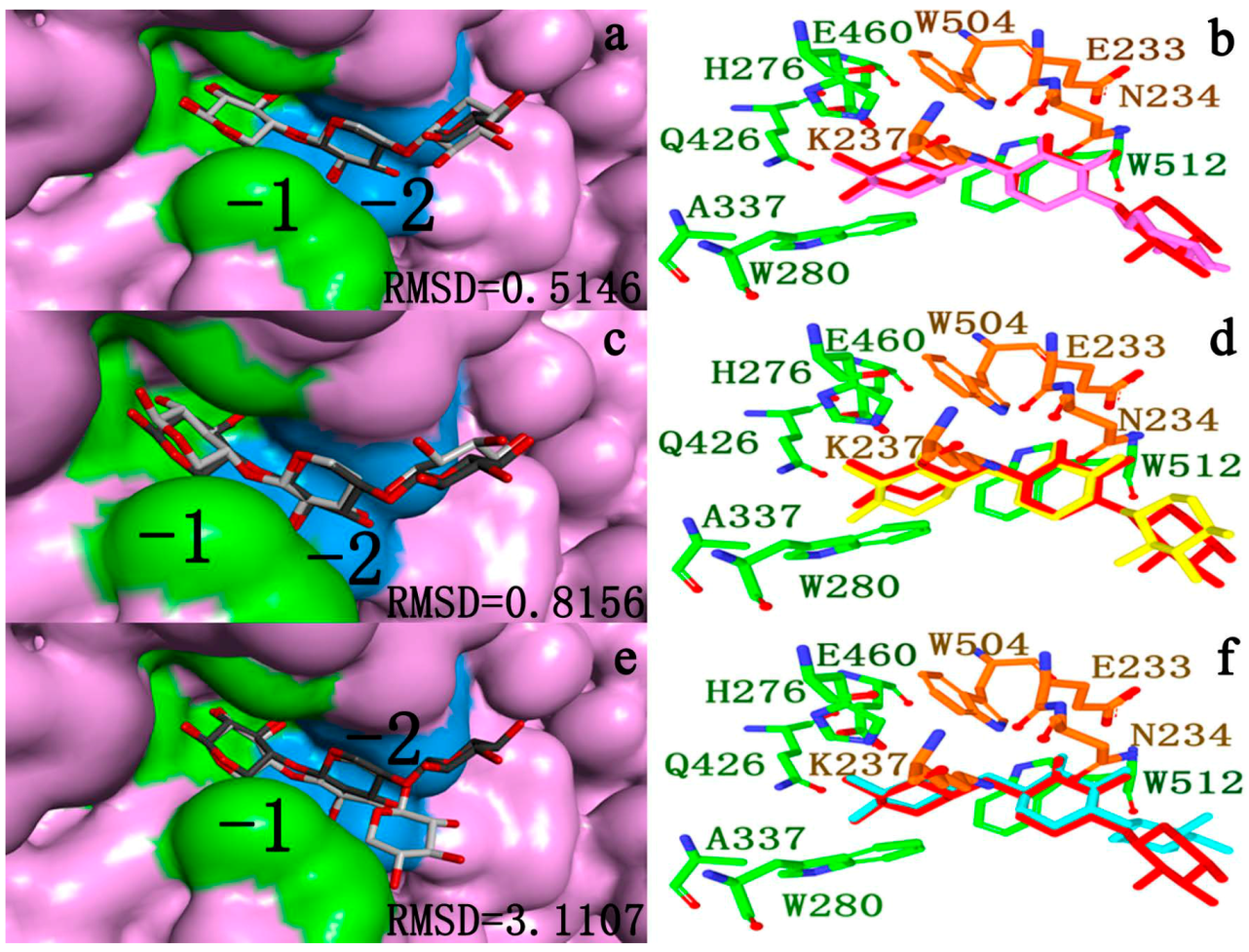
2.4.2. Docking Validation


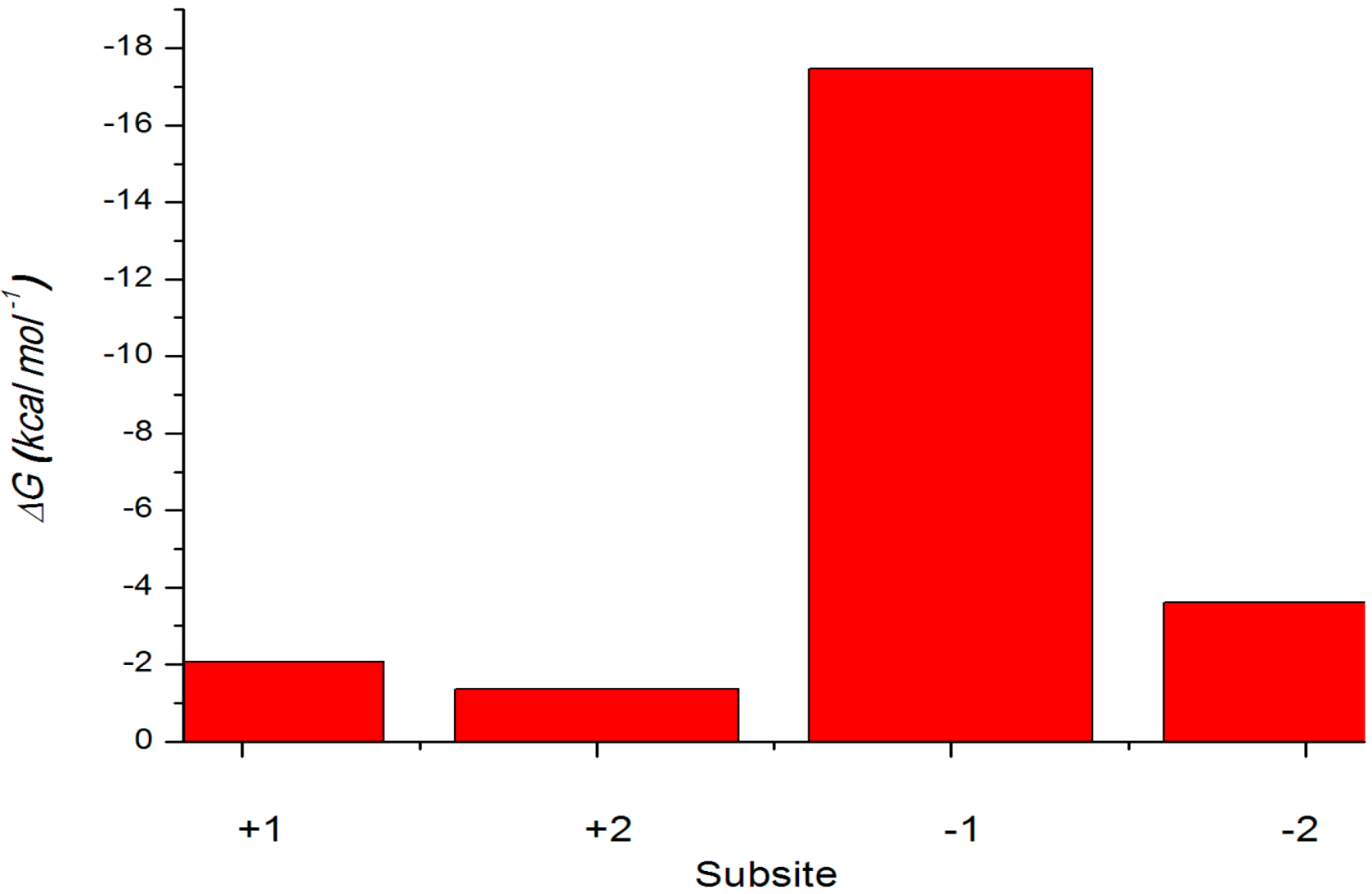
2.4.3. Energy Analyses of the Complexes
| Energy Components | X4(c)–Xyn10A | X4(sb)–Xyn10A |
|---|---|---|
| ∆Eele | −40.72 | −98.71 |
| ∆EvdW | −11.66 | −47.24 |
| ∆GPB | 53.63 | 112.02 |
| ∆Gnp | −2.92 | −7.01 |
| Nonpolar | −14.58 | −54.25 |
| Polar | 12.91 | 13.31 |
| ∆Gbind | −1.67 | −40.94 |

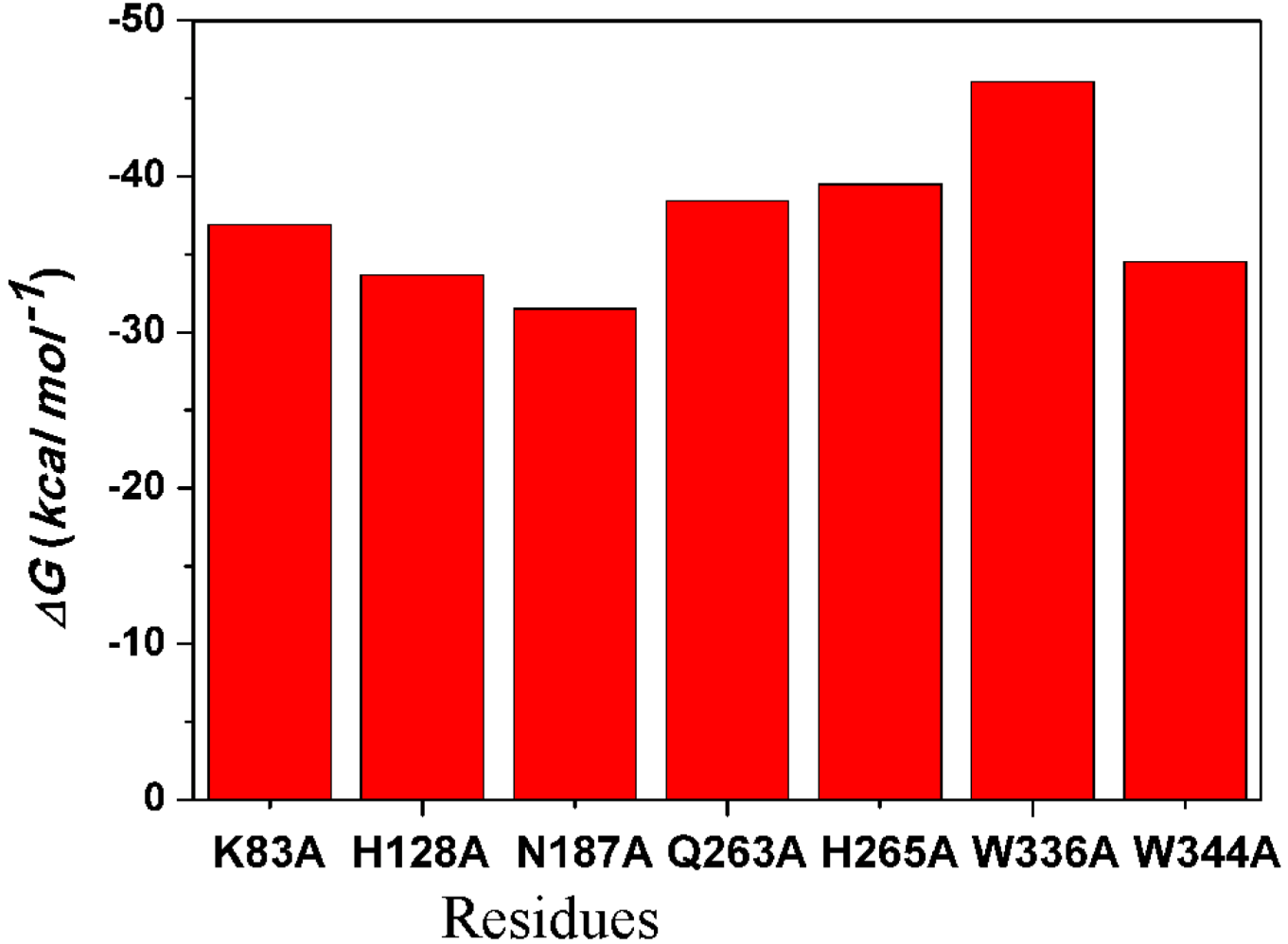
2.4.4. Computational Mutagenesis of Active Site Residues

3. Experimental Section
3.1. Homology Protein Modeling
3.2. Molecular Dynamics (MD) Simulation
3.3. Docking Study
3.4. Molecular Mechanics-Poisson–Boltzmann Surface Area (MM-PBSA) Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bryant, M.P.; Small, N.; Bouma, C.; Robinson, I.M. Characteristics of ruminal anaerobic cellulolytic cocci and Cillobacterium cellulosolvens n. sp. J. Bacteriol. 1958, 76, 529–537. [Google Scholar]
- Scott, H.W.; Dehority, B.A. Vitamin requirements of several cellulolytic rumen bacteria. J. Bacteriol. 1965, 89, 1165–1175. [Google Scholar]
- Hungate, R.E. Microorganisms in the rumen of cattle fed a constant ration. Can. J. Microbiol. 1957, 3, 289–311. [Google Scholar]
- Greve, L.C.; Labavitch, J.M.; Hungate, R.E. Alpha-l-arabinofuranosidase from Ruminococcus albus 8: Purification and possible role in hydrolysis of alfalfa cell wall. Appl. Environ. Microbiol. 1984, 47, 1135–1140. [Google Scholar]
- Greve, L.C.; Labavitch, J.M.; Stack, R.J.; Hungate, R.E. Muralytic activities of Ruminococcus albus 8. Appl. Environ. Microbiol. 1984, 47, 1141–1145. [Google Scholar]
- Hungate, R.E.; Stack, R.J. Phenylpropanoic acid: Growth factor for Ruminococcus albus. Appl. Environ. Microbiol. 1982, 44, 79–83. [Google Scholar]
- Han, X.; Gao, J.; Shang, N.; Huang, C.H.; Ko, T.P.; Chen, C.C.; Chan, H.C.; Cheng, Y.S.; Zhu, Z.; Wiegel, J.; et al. Structural and functional analyses of catalytic domain of GH10 xylanase from Thermoanaerobacterium saccharolyticum JW/SL-YS485. Porteins 2013, 81, 1256–1265. [Google Scholar]
- Koutaniemi, S.; van Gool, M.P.; Juvonen, M.; Jokela, J.; Hinz, S.W.; Schols, H.A.; Tenkanen, M. Distinct roles of carbohydrate esterase family CE16 acetyl esterases and polymer-acting acetyl xylan esterases inxylan deacetylation. J. Biotechnol. 2013, 168, 684–692. [Google Scholar]
- Gong, Y.Y.; Yin, X.; Zhang, H.M.; Wu, M.C.; Tang, C.D.; Wang, J.Q.; Pang, Q.F. Cloning, expression of a feruloyl esterase from Aspergillus usamii E001 and its applicability in generating ferulic acid from wheat bran. J. Ind. Microbiol. Biotechnol. 2013, 40, 1433–1441. [Google Scholar]
- Shinozaki, A.; Kawakami, T.; Hosokawa, S.; Sakamoto, T. A novel GH43 α-l-arabinofuranosidase of Penicillium chrysogenum that preferentially degrades single-substituted arabinosyl side chains in arabinan. Enzym. Microb. Technol. 2014, 58, 80–86. [Google Scholar]
- Rogowski, A.; Baslé, A.; Farinas, C.S.; Solovyova, A.; Mortimer, J.C.; Dupree, P.; Gilbert, H.J.; Bolam, D.N. Evidence that GH115 α-glucuronidase activity, which is required to degrade plant biomass, is dependent on conformational flexibility. J. Biol. Chem. 2014, 289, 53–56. [Google Scholar]
- Ravanal, M.C.; Alegría-Arcos, M.; Gonzalez-Nilo, F.D.; Eyzaguirre, J. Penicillium purpurogenum produces two GH family 43 enzymes with β-xylosidase activity, one monofunctional and the other bifunctional: Biochemical and structural analyses explain the difference. Arch. Biochem. Biophys. 2013, 540, 117–124. [Google Scholar]
- Moon, Y.H.; Iakiviak, M.; Bauer, S.; Mackie, R.I.; Cann, I.K. Biochemical analyses of multiple endoxylanases from the rumen bacterium Ruminococcus albus 8 and their synergistic activities with accessory hemicellulose-degrading enzymes. Appl. Environ. Microbiol. 2011, 77, 5157–5169. [Google Scholar]
- Valerie, N.; Camelia, B.; Mark, N.; David, R.; Rose, R.; Antony, J.W.; Stephen, G.W. Insights into transition state stabilization of theb-1,4-glycosidase Cex by covalent intermediate accumulation in active site mutants nature structural. Biology 1998, 5, 812–818. [Google Scholar]
- Gebler, J.; Gilkes, N.R.; Claeyssens, M.; Wilson, D.B.; Béguin, P.; Wakarchuk, W.W.; Kilburn, D.G.; Miller, R.C., Jr.; Warren, R.A.; Withers, S.G. Stereoselective hydrolysis catalyzed by related β-1,4-xylanases and β-1,4-glucanases. J. Biol. Chem. 1992, 267, 12559–12561. [Google Scholar]
- Sinnott, M.L. Catalytic mechanisms of enzymic glycosyl transfer. Chem. Rev. 1990, 90, 1171–1202. [Google Scholar]
- McCarter, J.D.; Withers, S.G. Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 1994, 4, 885–892. [Google Scholar]
- Dean, J.F.D.; Gamble, H.R.; Anderson, J.D. The ethylene biosynthesis-inducing xylanase: Its induction in Trichoderma viride and certain plant pathogens. Phytopathology 1989, 79, 1071–1078. [Google Scholar]
- Shabir, N.; Benedita, A.P.; José, A.M.P.; Harry, J.G.; Maria, J.R. Putting an N-terminal end to the Clostridium thermocellum xylanase Xyn10B story: Crystal structure of the CBM22–1–GH10 modules complexed with xylohexaose. J. Struct. Biol. 2010, 172, 353–362. [Google Scholar]
- Dean, J.F.D.; Anderson, J.D. Purification and physical characterization of the enzyme produced by Trichoderma viride. Plant Physiol. 1991, 95, 316–323. [Google Scholar]
- Wu, S.C.; Kauffmann, S.; Darvill, A.G.; Albersheim, P. Purification, cloning and characterization of two xylanases from Magnaporthe grisea, the rice blast fungus. Mol. Plant Microb. Interact. 1995, 8, 506–514. [Google Scholar]
- Giesbert, S.; Lepping, H.B.; Tenberge, K.B.; Tudzynski, P. The Xylanolytic system of Claviceps purpurea: Cytological evidence for secretion of xylanases in infected rye tissue and molecular characterization of two xylanase genes. Phytopathology 1998, 88, 1020–1030. [Google Scholar]
- Rouau, X. Investigations into the effects of an enzyme preparation for baking on wheat dough pentosans. J. Cereal Sci. 1993, 18, 145–157. [Google Scholar]
- Courtin, C.M.; Roelants, A.; Delcour, J.A. Fractionation–reconstitution experiments provide insight into the role of endoxylanases in bread-making. J. Agric. Food Chem. 1999, 47, 1870–1877. [Google Scholar]
- Courtin, C.M.; Delcour, J.A. Arabinoxylans and endoxylanases in wheat flour bread-making. J. Cereal Sci. 2002, 35, 225–243. [Google Scholar]
- Bedford, M.R. Factors influencing the use of enzymes in cerealbaseddiets. In Recent Advances in Enzymes in Grain Processing, Laboratory of Food Chemistry; Courtin, C.M., Veraverbeke, W.S., Delcour, J.A., Eds.; K.U. Leuven: Leuven, Belgium, 2003; pp. 371–380. [Google Scholar]
- Borght, A.V.D.; Goesaert, H.; Veraverbeke, W.S.; Delcour, J.A. Fractionation of wheat and wheat flour into starch and gluten: Overview of the main processes and the factors involved. J. Cereal Sci. 2005, 41, 221–237. [Google Scholar]
- Flatman, R.; McLauchlan, W.R.; Juge, N.; Furniss, C.; Berrin, J.G.; Hughes, R.K.; Manzanares, P.; Ladbury, J.E.; Brien, R.O.; Williamson, G. Interactions defining the specificity between fungal xylanases and the xylanase-inhibiting protein XIP-I from wheat. Biochem. J. 2002, 365, 773–781. [Google Scholar]
- Vanswemarliere, E.; Bourgois, T.M.; Rombouts, S.; van Camoenhout, S.; Volckaert, G.; Strelkov, S.V.; Delcour, J.A.; Rabijns, A.; Courtin, C.M. Crystallographic analysis shows substrate binding at the −3 to +1 active-site subsites and at the surface of glycoside hydrolase family 11 endo-1,4-β-xylanases. Biochem. J. 2008, 410, 71–79. [Google Scholar]
- Ducros, V.; Charnock, S.J.; Derewenda, U.; Derewenda, Z.S.; Dauteri, Z.; Dupont, C.; Shareck, F.; Morosoli, R.; Kluepfel, D.; Davies, G.J. Substrate specificity in glycoside hydrolase family 10. J. Biol. Chem. 2000, 275, 23020–23026. [Google Scholar]
- Koshland, D.E. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 12, W252–W258. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar]
- Solomon, V.; Teplitsky, A.; Shulami, S.; Zolotnitsky, G.; Shoham, Y.; Shoham, G. Structure–specificity relationships of an intracellular xylanase from Geobacillus stearothermophilus. Acta Crystallogr. Sect. D 2007, 63, 845–859. [Google Scholar]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003, 31, 3320–3323. [Google Scholar]
- Heightman, T.D.; Vasella, A.T. ATRecent insights into inhibition, structure, and mechanism of configuration-retaining glycosidases. Angew. Chem. Int. Ed. 1999, 38, 750–770. [Google Scholar]
- Davies, G.J.; Wilson, K.S.; Henrissat, B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997, 321, 557–559. [Google Scholar]
- Wakarchuk, W.W.; Campbell, R.L.; Sung, W.L.; Davoodi, J.; Yaguchi, M. Mutational and crystallographic analyses of the active site residues of the Bacillus circulans xylanase. Protein Sci. 1994, 3, 467–475. [Google Scholar]
- Jiaheng, Z.; Jean’ne M, S. 3,3'-Dinitroamino-4,4'-azoxyfurazan and its derivatives: An assembly of diverse N–O building blocks for high-performance energetic materials. J. Am. Chem. Soc. 2014, 136, 4437–4445. [Google Scholar] [CrossRef]
- Zechel, D.L.; Withers, S.G. Glycosidase mechanisms: Anatomy of a finely tuned catalyst. Acc. Chem. Res. 2000, 33, 11–18. [Google Scholar]
- Mukherjee, S.; Balius, T.E.; Rizzo, R.C. Docking validation resources: Protein family and ligand flexibility experiments. J. Chem. Inf. Model. 2010, 50, 1986–2000. [Google Scholar]
- Ertan-Bolelli, T.; Musdal, Y.; Bolelli, K.; Yilmaz, S.; Aksoy, Y.; Yildiz, I.; Aki-Yalcin, E.; Yalcin, I. Synthesis and biological evaluation of 2-substituted-5-(4-nitrophenylsulfonamido)benzoxazoles as Human GST P1–1 Inhibitors, and description of the binding site features. Chem. Med. Chem. 2014, 9, 984–992. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar]
- Grimsley, J.K.; Calamini, B.; Wild, J.R.; Mesecar, A.D. Structural and mutational studies of organophosphorus hydrolase reveal a cryptic and functional allosteric-binding site. Arch. Biochem. Biophys. 2005, 442, 169–179. [Google Scholar]
- Kiefer, F.; Arnold, K.; Künzli, M.; Bordoli, L.; Schwede, T. The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 2009, 37, D387–D392. [Google Scholar]
- Case, D.A.; Cheatham, T.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Computat. Chem. 2005, 26, 1668–1688. [Google Scholar]
- Langevin, P. Sur la théorie du mouvement brownien. C.R. Acad. Sci. 1908, 146, 530–533. (In French) [Google Scholar]
- Berendsen, H.J.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An W log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Gilson, M.K.; Sharp, K.A.; Honig, B.H. Calculating the electrostatic potential of molecules in solution: Method and error assessment. J. Comput. Chem. 1988, 9, 327–335. [Google Scholar]
- Fu, G.; Liu, H.; Doerksen, R.J. Molecular modeling to provideinsight into the substrate binding and catalytic mechanism of human biliverdin-IXα reductase. J. Phys. Chem. B 2012, 116, 9580–9594. [Google Scholar]
- Kuhn, B.; Kollman, P.A. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, D.; Yu, L.; Jin, H.; Guan, S.; Han, W. Molecular Modeling and MM-PBSA Free Energy Analysis of Endo-1,4-β-Xylanase from Ruminococcus albus 8. Int. J. Mol. Sci. 2014, 15, 17284-17303. https://doi.org/10.3390/ijms151017284
Zhan D, Yu L, Jin H, Guan S, Han W. Molecular Modeling and MM-PBSA Free Energy Analysis of Endo-1,4-β-Xylanase from Ruminococcus albus 8. International Journal of Molecular Sciences. 2014; 15(10):17284-17303. https://doi.org/10.3390/ijms151017284
Chicago/Turabian StyleZhan, Dongling, Lei Yu, Hanyong Jin, Shanshan Guan, and Weiwei Han. 2014. "Molecular Modeling and MM-PBSA Free Energy Analysis of Endo-1,4-β-Xylanase from Ruminococcus albus 8" International Journal of Molecular Sciences 15, no. 10: 17284-17303. https://doi.org/10.3390/ijms151017284
APA StyleZhan, D., Yu, L., Jin, H., Guan, S., & Han, W. (2014). Molecular Modeling and MM-PBSA Free Energy Analysis of Endo-1,4-β-Xylanase from Ruminococcus albus 8. International Journal of Molecular Sciences, 15(10), 17284-17303. https://doi.org/10.3390/ijms151017284