
Stable Isotope-Assisted Evaluation of Different Extraction Solvents for Untargeted Metabolomics of Plants

 , ,
, ,
Abstract
:1. Introduction
2. Results
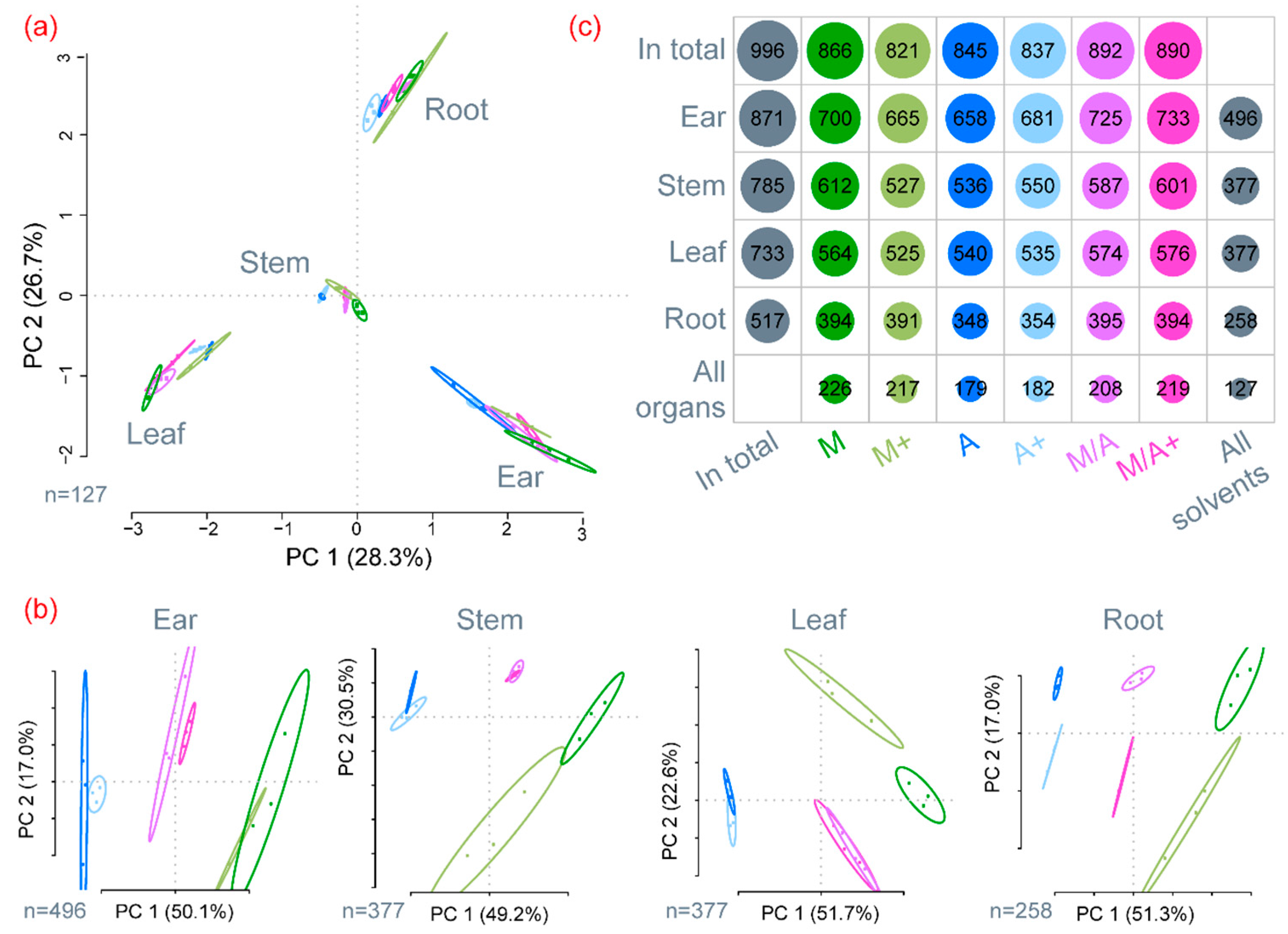
2.1. General Description of Extract Compositions
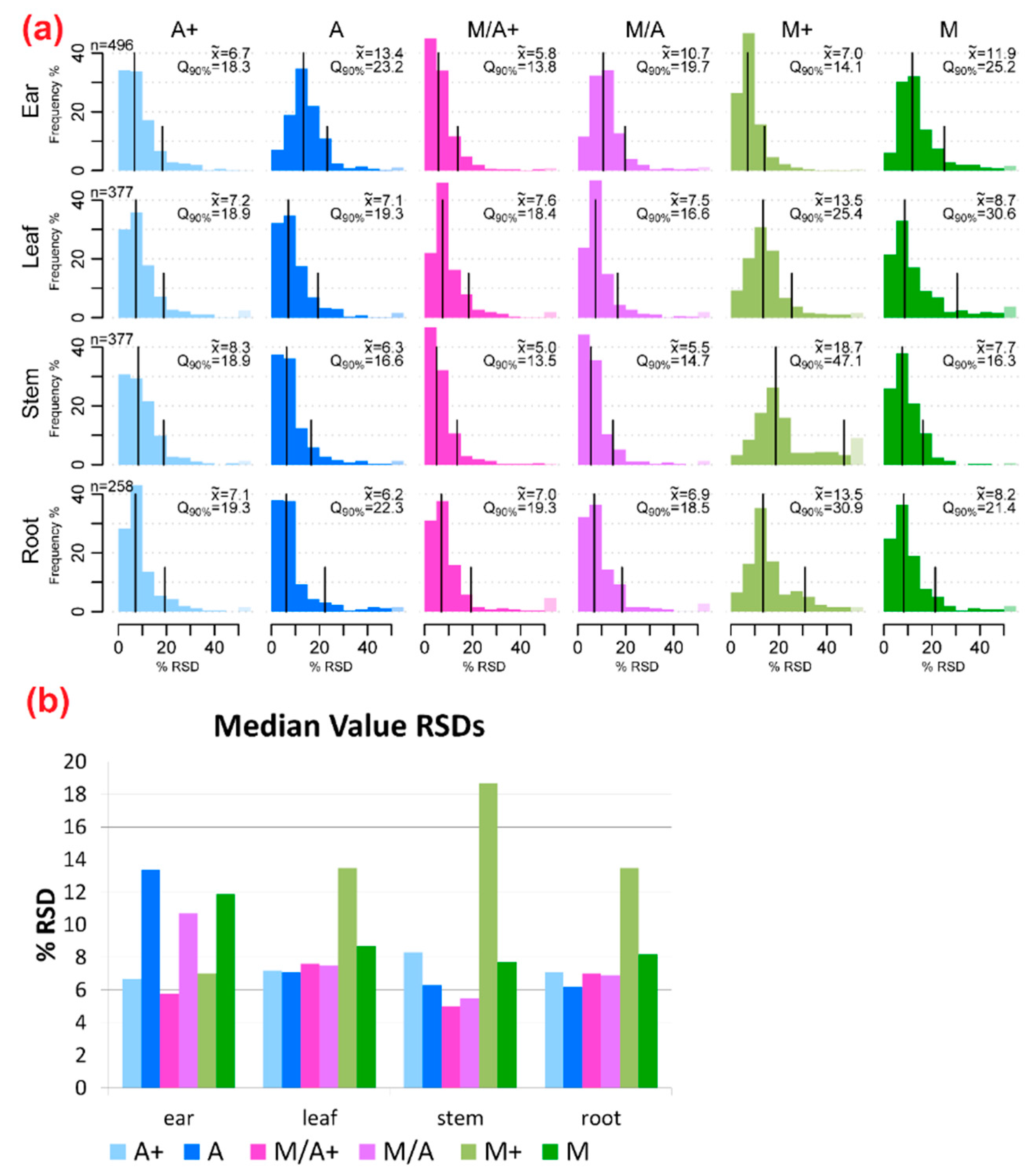
2.2. Precision of Metabolite Abundances
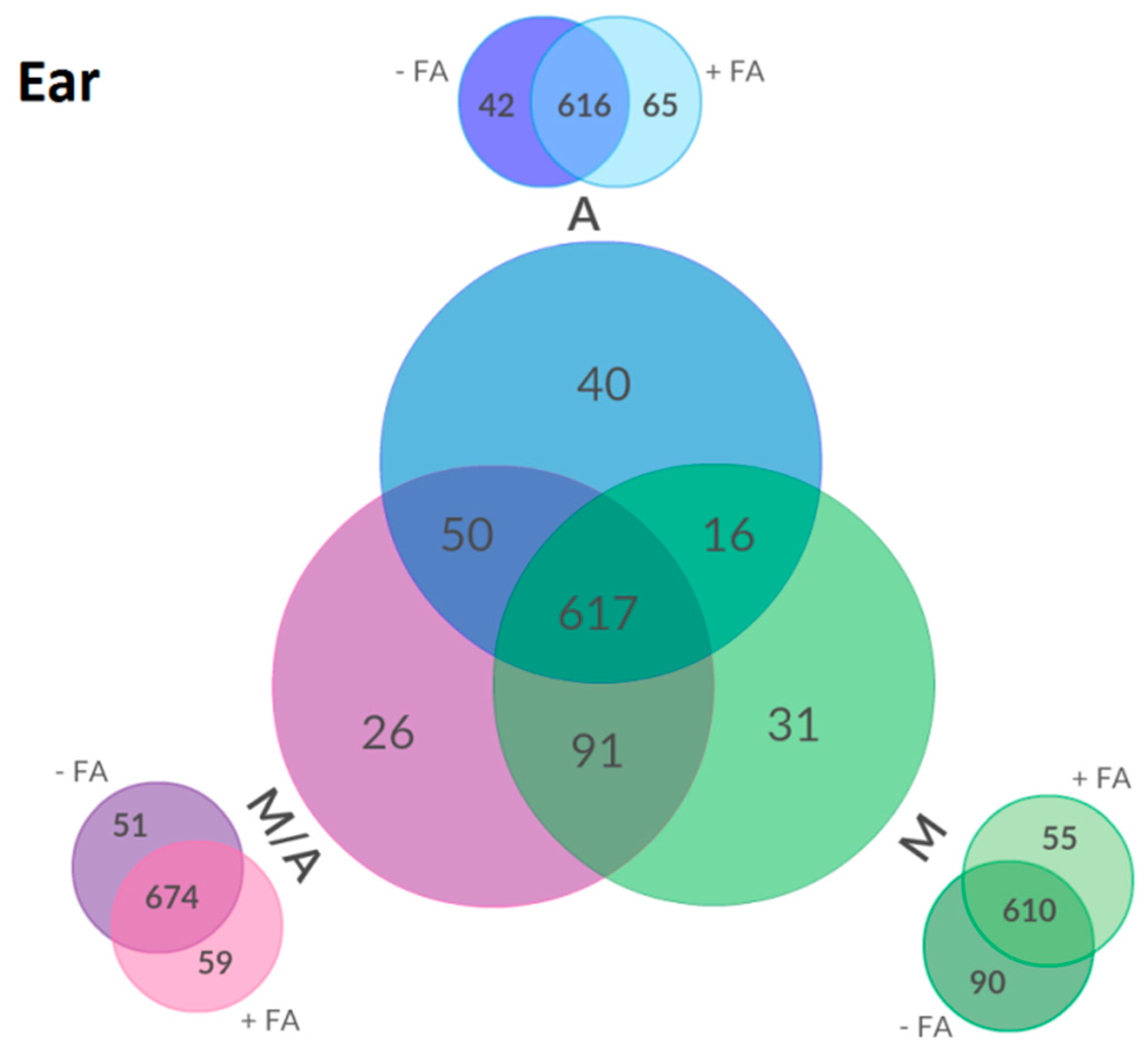
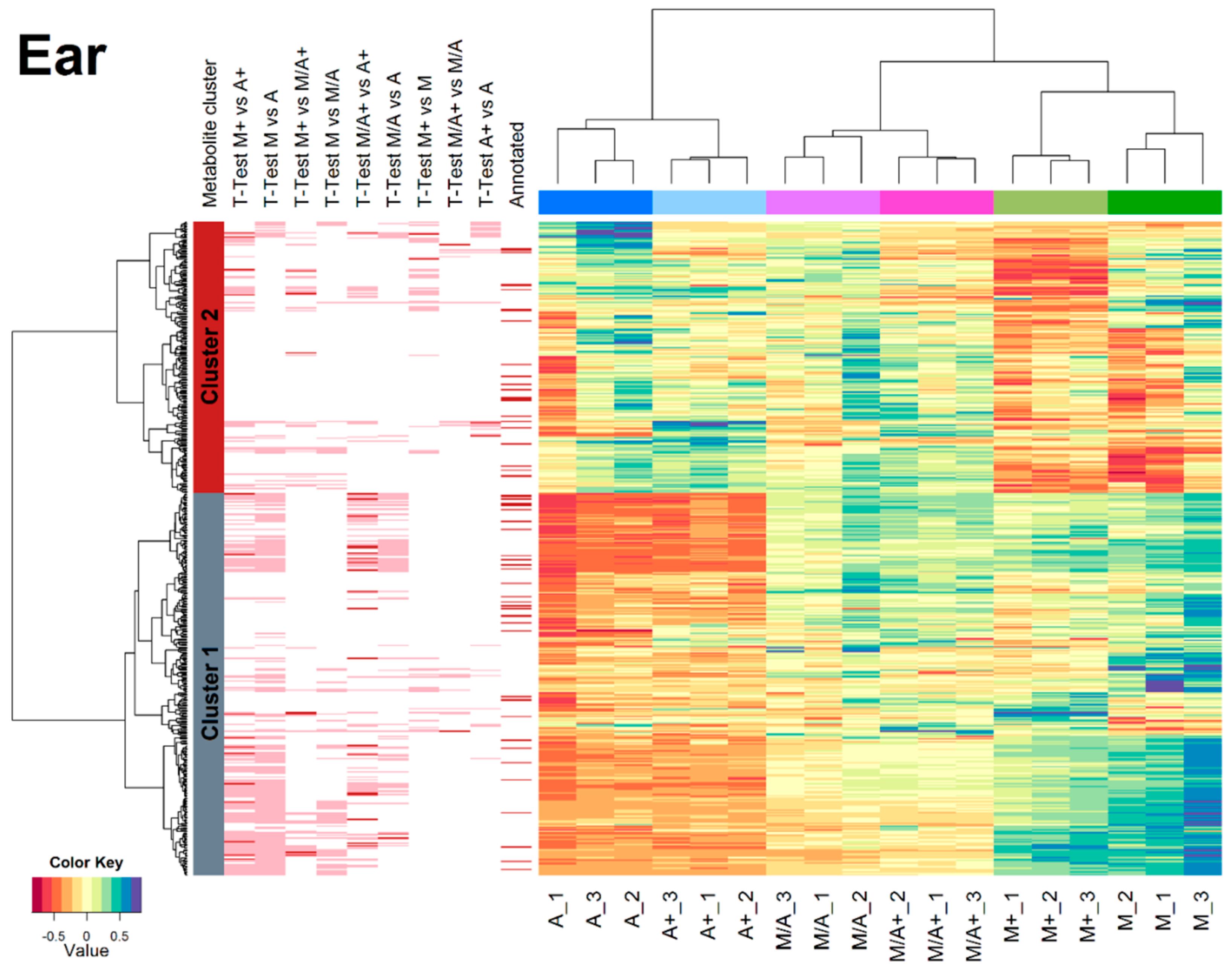
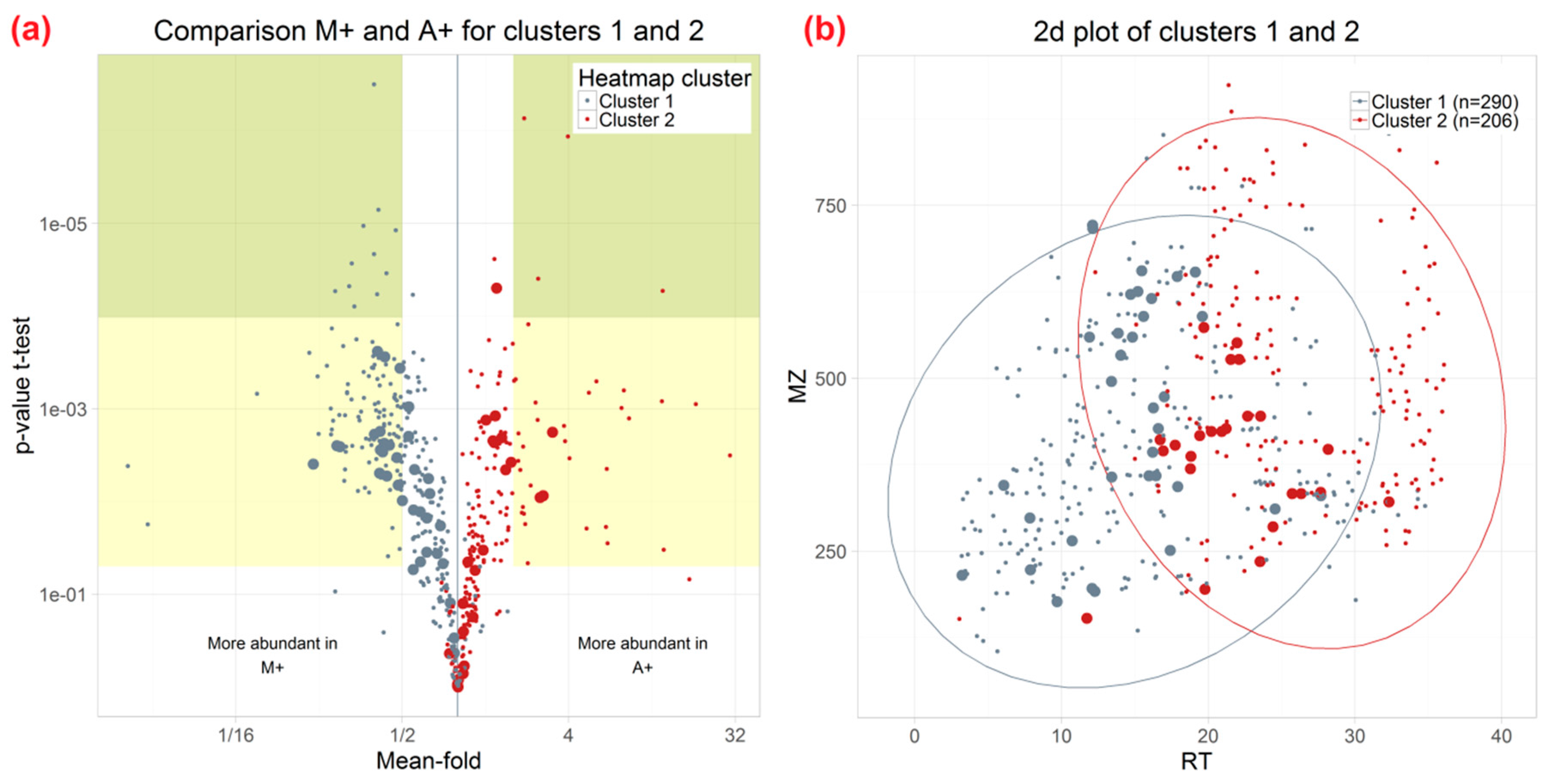
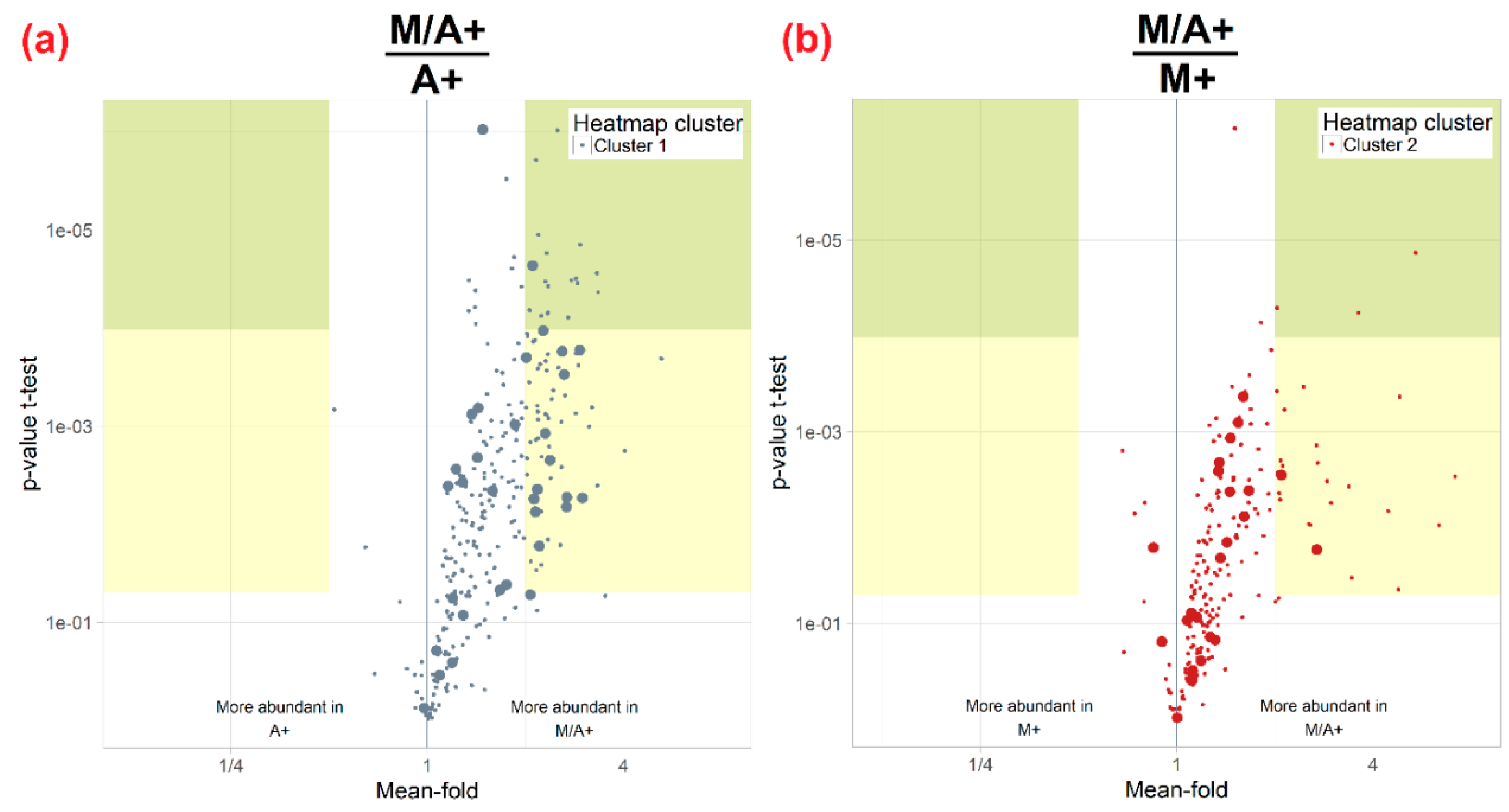
2.3. Effect of Different Extraction Mixtures on the Metabolic Composition of Wheat Ear Extracts
3. Discussion
3.1. General Discussion of Extract Composition and Precision of Metabolite Abundances
3.2. Effect of Acidification of Extraction Mixtures with 0.1% Formic Acid
3.3. Influence of Extraction Mixture on Relative Abundance of Shared Metabolites
4. Materials and Methods
4.1. Chemicals and Plant Material
4.2. Cultivation of Wheat
4.3. Sampling and Pretreatment
4.4. Extraction with Different Extraction Mixtures
4.5. LC-HRMS Analysis
4.6. Data Processing
4.6.1. LC-HRMS Data Processing
4.6.2. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| A | extraction mixture (acetonitrile 75%, water 25%) |
| A+ | extraction mixture (acetonitrile 75%, water 25%, formic acid: 0.1%) |
| ACN | acetonitril |
| CHCl3 | chloroform |
| DAD | diode array detector |
| DB | database |
| DON | deoxynivalenol |
| D3G | DON-3-glucoside |
| EIC | extracted ion chromatogram |
| ESI | electrospray ionisation |
| FA | formic acid |
| GC | gas chromatography |
| HCA | hierarchical cluster analysis |
| HDMS | high definition mass spectrometry |
| HILIC | hydrophilic interaction liquid chromatography |
| H2O | water |
| ICR | ion cyclotron resonance |
| IM | ion mobility |
| IPA | isopropanol |
| IT | ion trap |
| LC | liquid chromatography |
| LC-HRMS | liquid chromatography high resolution mass spectrometry |
| LTQ | linear trap quadrupole |
| MeOH | methanol |
| MS | mass spectrometry |
| M | extraction mixture (methanol 75%, water 25%) |
| M+ | extraction mixture (methanol 75%, water 25%, formic acid: 0.1%) |
| M/A | extraction mixture (methanol 37,5%, acetonitrile 37,5%, water 25%) |
| M/A+ | extraction mixture (methanol 37,5%, acetonitrile 37,5%, water 25%, formic acid: 0.1%)) |
| RP | reversed phase |
| PC | principle component |
| PCA | principle component analysis |
| PDA | photodiode array |
| QC | quality control |
| RSD | relative standard deviation |
| SQ | single quadrupole |
| (U)HPLC | (ultra) high performance liquid chromatography |
| UFLC | ultra fast liquid chromatography |
References
- Kim, H.K.; Verpoorte, R. Sample preparation for plant metabolomics. Phytochem. Anal. 2010, 21, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef] [PubMed]
- Bruce, S.J.; Tavazzi, I.; Parisod, V.; Rezzi, S.; Kochhar, S.; Guy, P.A. Investigation of human blood plasma sample preparation for performing metabolomics using ultrahigh performance liquid chromatography/mass spectrometry. Anal. Chem. 2009, 81, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Tulipani, S.; Llorach, R.; Urpi-Sarda, M.; Andres-Lacueva, C. Comparative analysis of sample preparation methods to handle the complexity of the blood fluid metabolome: When less is more. Anal. Chem. 2012, 85, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; O’Maille, G.; Smith, C.A.; Brandon, T.R.; Uritboonthai, W.; Qin, C.; Trauger, S.A.; Siuzdak, G. Solvent-dependent metabolite distribution, clustering, and protein extraction for serum profiling with mass spectrometry. Anal. Chem. 2005, 78, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.W.-M. Considerations of Sample Preparation for Metabolomics Investigation. In The Handbook of Metabolomics; Humana Press: New York City, NY, USA, 2012; pp. 7–27. [Google Scholar]
- Duportet, X.; Aggio, R.B.M.A.; Carneiro, S.; Villas-Bôas, S.G. The biological interpretation of metabolomic data can be misled by the extraction method used. Metabolomics 2012, 8, 410–421. [Google Scholar] [CrossRef]
- Theodoridis, G.; Gika, H.; Franceschi, P.; Caputi, L.; Arapitsas, P.; Scholz, M.; Masuero, D.; Wehrens, R.; Vrhovsek, U. LC-MS based global metabolite profiling of grapes: solvent extraction protocol optimisation. Metabolomics 2012, 8, 175–185. [Google Scholar] [CrossRef]
- Martin, A.C.; Pawlus, A.; Jewett, E.M.; Wyse, D.L.; Angerhoferc, C.K.; Hegeman, A.D. Evaluating solvent extraction systems using metabolomics approaches. RSC Adv. 2014, 4, 26325–26334. [Google Scholar] [CrossRef]
- t'Kindt, R.; Morreel, K.; Deforce, D.; Boerian, W.; van Bocxlaer, J. Joint GC-MS and LC-MS platforms for comprehensive plant metabolomics: Repeatability and sample pre-treatment. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3572–3580. [Google Scholar] [CrossRef] [PubMed]
- Redestig, H.; Kusano, M.; Ebana, K.; Kobayashi, M.; Oikawa, A.; Okazaki, Y.; Matsuda, F.; Arita, M.; Fujita, N.; Saito, K. Exploring molecular backgrounds of quality traits in rice by predictive models based on high-coverage metabolomics. BMC Syst. Biol. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Vaclavikova, M.; Dzuman, Z.; Vaclavik, L.; Ovesna, J.; Hajslova, J. Rapid LC-MS-based metabolomics method to study the Fusarium infection of barley. J. Sep. Sci. 2014, 37, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Bueschl, C.; Kluger, B.; Lemmens, M.; Adam, G.; Wiesenberger, G.; Maschietto, V.; Marocco, A.; Strauss, J.; Bödi, S.; Thallinger, G.G.; et al. A novel stable isotope labelling assisted workflow for improved untargeted LC-HRMS based metabolomics research. Metabolomics 2014, 10, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Hennig, K.; de Vos, R.C.; Maliepaard, C.; Dekker, M.; Verkerk, R.; Bonnema, G. A metabolomics approach to identify factors influencing glucosinolate thermal degradation rates in Brassica vegetables. Food Chem. 2014, 155, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Cottet, K.; Genta-Jouve, G.; Fromentin, Y.; Oonne, G.; Duplais, C.; Laprévote, O.; Michel, S.; Lallemand, M.-C. Comparative LC-MS-based metabolite profiling of the ancient tropical rainforest tree Symphonia globulifera. Phytochemistry 2014, 108, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, M.; Zhang, A.; Ni, B.; Dong, H.; Wang, X. UPLC-Q-TOF-HDMS analysis of constituents in the root of two kinds of Aconitum using a metabolomics approach. Phytochem. Anal. 2013, 24, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Scherling, C.; Roscher, C.; Giavalisco, P.; Schulze, E.-D.; Weckwerth, W. Metabolomics unravel contrasting effects of biodiversity on the performance of individual plant species. PLoS ONE 2010, 5, e12569. [Google Scholar] [CrossRef] [PubMed]
- Glauser, G.; Veyrat, N.; Rochat, B.; Wolfender, J.L.; Turlings, T.C. Ultra-high pressure liquid chromatography-mass spectrometry for plant metabolomics: A systematic comparison of high-resolution quadrupole-time-of-flight and single stage Orbitrap mass spectrometers. J. Chromatogr. A 2013, 1292, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Doerfler, H.; Sun, X.; Wang, L.; Engelmeier, D.; Lyon, D.; Weckwerth, W. mzGroupAnalyzer-predicting pathways and novel chemical structures from untargeted high-throughput metabolomics data. PLoS ONE 2014, 9, e96188. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Ellis, D.I. Metabolomics: Current analytical platforms and methodologies. TrAC Trends Anal. Chem. 2005, 24, 285–294. [Google Scholar]
- Huang, X.; Chen, Y., Jr.; Cho, K.; Nikolskiy, L.; Crawford, P.A.; Patti, G.J. X13CMS: Global Tracking of Isotopic Labels in Untargeted Metabolomics. Anal. Chem. 2014, 86, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Chokkathukalam, A.; Jankevics, A.; Creek, D.J.; Achcar, F.; Barrett, M.P.; Breitling, R. mzMatch–ISO: An R tool for the annotation and relative quantification of isotope-labelled mass spectrometry data. Bioinformatics 2013, 29, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Leeming, M.G.; Isaac, A.P.; Pope, B.J.; Cranswick, N.; Wright, C.E.; Ziogas, J.; O'Hair, R.A.J.; Donald, W.A. High-resolution twin-ion metabolite extraction (HiTIME) mass spectrometry: Nontargeted detection of unknown drug metabolites by isotope labeling, liquid chromatography mass spectrometry, and automated high-performance computing. Anal. Chem. 2015, 87, 4104–4109. [Google Scholar] [CrossRef] [PubMed]
- Kessler, N.; Walter, F.; Persicke, M.; Albaum, S.P.; Kalinowski, J.; Goesmann, A.; Niehaus, K.; Nattkemper, T.W. Allocator: An interactive web platform for the analysis of metabolomic LC-ESI-MS datasets, enabling semi-automated, user-revised compound annotation and mass isotopomer ratio analysis. PLoS ONE 2014, 9, e113909. [Google Scholar] [CrossRef] [PubMed]
- Bueschl, C.; Kluger, B.; Berthiller, F.; Lirk, G.; Winkler, S.; Krska, R.; Schuhmacher, R. MetExtract: A new software tool for the automated comprehensive extraction of metabolite-derived LC/MS signals in metabolomics research. Bioinformatics 2012, 28, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Suwa, K. Search extension transforms Wiki into a relational system: A case for flavonoid metabolite database. BioData Min. 2008, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Kopka, J.; Dörmann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Mumper, R.J. Plant Phenolics: Extraction, Analysis and Their Antioxidant and Anticancer Properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef] [PubMed]
- Verpoorte, R.; Choi, Y.H.; Mustafa, N.R.; Kim, H.K. Metabolomics: back to basics. Phytochem. Rev. 2008, 7, 525–537. [Google Scholar] [CrossRef]
- Mushtaq, M.Y.; Choi, Y.H.; Verpoorte, R.; Wilson, E.G. Extraction for metabolomics: Access to the metabolome. Phytochem. Anal. 2014, 25, 291–306. [Google Scholar] [CrossRef] [PubMed]
- De Vos, R.C.; Moco, S.; Lommen, A.; Keurentjes, J.J.; Bino, R.J.; Hall, R.D. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2007, 2, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, S.; Schumppb, O.; Bohnia, N.; Bujarda, A.; Azzollinia, A.; Monodc, M.; Gindrob, K.; Wolfendera, J.-C. Detection of metabolite induction in fungal co-cultures on solid media by high-throughput differential ultra-high pressure liquid chromatography–time-of-flight mass spectrometry fingerprinting. J. Chromatogr. A 2013, 1292, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.R.; Arnon, D.I. The Water culture method for growing plants without soil. Circ. CA Agric. Exp. Stat. 1950, 347, 32. [Google Scholar]
- Kluger, B.; Bueschl, C.; Lemmens, M.; Berthiller, F.; Häubl, G.; Jaunecker, G.; Adam, G.; Krska, R.; Schuhmacher, R. Stable isotopic labelling-assisted untargeted metabolic profiling reveals novel conjugates of the mycotoxin deoxynivalenol in wheat. Anal. Bioanal. Chem. 2013, 405, 5031–5036. [Google Scholar] [CrossRef] [PubMed]
- Simader, A.M.; Kluger, B.; Neumann, N.K.N.; Bueschl, C.; Lemmens, M.; Lirk, G.; Krska, R.; Schuhmacher, R. QCScreen: A software tool for data quality control in LC-HRMS based metabolomics. BMC Bioinf. 2015, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrioli, P.G.; Eng, J.K.; Hubley, R.; Vogelzang, M.; Deutsch, E.W.; Raught, B.; Pratt, B.; Nilsson, E.; Angeletti, R.H.; Apweiler, R.; et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004, 22, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. Avaliable online: http://cran.fiocruz.br/web/packages/dplR/vignettes/timeseries-dplR.pdf (accessed on 15 March 2016).
- van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Šidák, Z. Rectangular Confidence Regions for the Means of Multivariate Normal Distributions. J. Am. Stat. Assoc. 1967, 62, 626–633. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant (Organ) | Purpose of Study | Fresh or Dried/Extraction Solvent 1 | Clean up 1 | Instrument | Reference |
|---|---|---|---|---|---|
| Rice (kernels) | To explore the molecular background of quality traits in rice by predictive models based on high-coverage metabolomics | Fresh/MeOH:H2O (5:95) | Dilution in 0.1% acetic acid solution, filtration, evaporation and dissolving in H2O | LC-QTOF-MS, IT-MS | [11] |
| Barley | To study the Fusarium infection of barley | Fresh/MeOH:H2O (50:50) (untargeted) and ACN:H2O (84:16) (targeted for mycotoxins DON and D3G) | Filter (0.22 µm) | UHPLC–QTOF-MS | [12] |
| Wheat, maize (ears at flowering stage, kernels) | Development of a novel stable isotope labelling-assisted workflow for improved untargeted LC–HRMS | Fresh/MeOH:H2O (75:25) + 0.1% FA | Dilution to final ratio MeOH:H2O 1:1 + 0.1% FA | HPLC-ESI-Orbitrap | [13] |
| Brassica vegetables (leaves) | Identification of factors influencing glucosinolate thermal degradation rates | Freeze-dried/MeOH:H20 (75:25) + 0.1% FA | Filter (0.2 µm) | HPLC-QTOF | [14] |
| Vitis vinifera (grapes) | Solvent extraction protocol optimisation | Fresh/MeOH:H2O:CHCl3 (17 different mixtures) | SPE for aqueous fraction | RP- and HILIC-UPLC-TOF | [8] |
| Symphonia globulifera (leaves, roots, latex, bark, seeds, pericarps and flowers) | Comparative LC-MS-based metabolite profiling | Dried/MeOH | After evaporation ethyl acetate and H2O, dried, reconstituted in MeOH | UHPLC G2-HDMS | [15] |
| Aconitum (root) | Analysis of constituents in the root | Fresh/MeOH:H2O (75/25) | Filter (0.22 µm) | UHPLC–QTOF–HDMS | [16] |
| Medicago x varia; Knautia arvensis; Lotus corniculatus; Bellis perennis, Leontodon autumnalis (leaves) | Contrasting effects of biodiversity on the performance of individual plant species | Fresh/MeOH | Drying, redissolved in 50% MeOH | UHPLC-FT-ICR-MS | [17] |
| Rhus typhina L., Lythrum salicaria L., Monarda Fistulosa L. (aerial tissue; berries, leaves, stem, flowers, buds) | Evaluation of solvent extraction systems | Dried/eight solvent systems (hexane, dichlormethane, ethyl acetate, methanol, isopropanol, water aqueous ethanol (70%), dichloromethane-methanol (50:50); and additional extract partitioning | For some samples extract partitioning with hexane and dichloromethane | UPLC-ESI-SQ-MS | [9] |
| Arabidopsis thaliana (leaves) | Joint GC- and LC-MS platforms and evaluation of repeatability and sample pre-treatment | Fresh/MeOH:H2O (80:20) and CHCl3:MeOH:H2O (20:60:20) with various protocols | Various protocols | LC-QTOF-MS | [10] |
| Arabidopsis thaliana (leaves) | A systematic comparison of high-resolution quadrupole-time-of-flight and single-stage Orbitrap mass spectrometers | Fresh/IPA:FA (99.5:0.5) | Evaporation, resuspended in MeOH:H2O, SPE (C18) MeOH:H2O 80:20 | UHPLC-QTOF, UHPLC-Exactive Orbitrap | [18] |
| Arabidopsis thaliana (rosette leaves) | Prediction of pathways and novel chemical structures | Fresh/MeOH:H2O (80:20) | Dried, redissolved in MeOH:H2O (50:50), filter (STAGE tip), remove lipids with chloroform | LC-HRMS Orbitrap | [19] |
| ID | Accurate Mass m/z of Most Abundant Feature | RT (min) | Cluster | Name or Substance Class | Assigned Molecular Formula | Number of db Hits | p-Value M+ vs. A+ | Fold Change A+/M+ |
|---|---|---|---|---|---|---|---|---|
| 1 | 215.1394 | 3.22 | 1 | d-Desthiobiotin | C10H18N2O3 | 1 | 0.0025 | 0.2 |
| 12 | 298.0975 | 7.84 | 1 | 5′-Deoxy-5′-(methylthio)adenosine * | C11H15N5O3S | 1 | 0.0039 | 0.2 |
| 13 | 223.1080 | 7.88 | 1 | 5-Methoxy-3-indoleacetic acid or DL-Indole-3-lactic acid | C11H11NO3 | 2 | 0.0026 | 0.2 |
| 17 | 177.0547 | 9.69 | 1 | Chlorogenic acid * | C16H18O9 | 1 | 0.0053 | 0.4 |
| 29 | 559.1792 | 11.9 | 1 | Tetrahydroxyprenylflavanone-hexoside, e.g., Phellavin | C26H32O12 | 4 | 0.0004 | 0.5 |
| 31 | 196.0607 | 12.08 | 1 | 2-Carboxy-2,3-dihydro-5,6-dihydroxyindole or Dopaquinone | C9H9NO4 | 2 | 0.0025 | 0.4 |
| 32 | 721.2328 | 12.11 | 1 | Dihydrophelloside | C32H42O17 | 1 | 0.0028 | 0.4 |
| 45 | 565.1559 | 13.86 | 1 | Schaftoside * | C26H28O14 | 85 | 0.0019 | 0.4 |
| 46 | 533.1636 | 14.03 | 1 | Plumerubroside | C24H30O12 | 1 | 0.0017 | 0.4 |
| 48 | 621.2158 | 14.71 | 1 | Flavonoid-dihexoside-hydroxycinnamicacid ester, e.g., Petunoside | C37H38O19 | 9 | 0.0003 | 0.4 |
| 49 | 559.1790 | 14.83 | 1 | Tetrahydroxyprenylflavanone-hexoside, e.g., Phellavin | C26H32O12 | 4 | 0.0029 | 0.4 |
| 55 | 655.1873 | 15.46 | 1 | Dimethoxy-tetrahydroxyflavon-dihexoside, e.g., Limocitrin 3-rutinoside | C29H34O17 | 13 | 0.0024 | 0.4 |
| 60 | 359.1317 | 15.96 | 1 | Dihydroxyflavan-hexoside, e.g., Koaburanin | C21H24O8 | 3 | 0.0002 | 0.4 |
| 61 | 615.2264 | 16.14 | 1 | Flavonoid-trihexoside, e.g., Tricin 7-rutinoside-4′-glucoside | C35H44O21 | 2 | 0.0066 | 0.5 |
| 75 | 647.1590 | 17.88 | 1 | Methoxy-trihydroxyflavanol-dihexoside or dimethoxy,trihydroxyflavon-di-C-hexoside, e.g., 6-C-Arabinopyranosyl-8-C-glucopyranosyltricin | C28H32O16 | 75 | 0.0033 | 0.5 |
| 101 | 333.2042 | 26.34 | 2 | 9-Hydroperoxy-10,12,15-octadecatrienoate or isomer | C18H30O4 | 1 | 0.0018 | 3 |
| 103 | 335.2199 | 27.65 | 2 | octadecanoic acid derivatives, e.g., (9Z,11E)-(13S)-13-Hydroperoxyoctadeca-9,11-dienoic acid | C18H32O4 | 4 | 0.0086 | 3.4 |
| 108 | 321.2403 | 32.31 | 2 | cis-9,10-Epoxystearic acid | C18H34O3 | 1 | 0.0090 | 3.6 |
| Extraction Mixture | Organic Solvent (vol) | Water (vol) | Formic Acid (vol) |
|---|---|---|---|
| M | Methanol (75%) | 25% | - |
| A | Acetonitrile (75%) | 25% | - |
| M/A | Methanol/acetonitrile (37.5%/37.5%) | 25% | - |
| M+ | Methanol (75%) | 25% | 0.1% |
| A+ | Acetonitrile (75%) | 25% | 0.1% |
| M/A+ | Methanol/acetonitrile (37.5%/37.5%) | 25% | 0.1% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doppler, M.; Kluger, B.; Bueschl, C.; Schneider, C.; Krska, R.; Delcambre, S.; Hiller, K.; Lemmens, M.; Schuhmacher, R. Stable Isotope-Assisted Evaluation of Different Extraction Solvents for Untargeted Metabolomics of Plants. Int. J. Mol. Sci. 2016, 17, 1017. https://doi.org/10.3390/ijms17071017
Doppler M, Kluger B, Bueschl C, Schneider C, Krska R, Delcambre S, Hiller K, Lemmens M, Schuhmacher R. Stable Isotope-Assisted Evaluation of Different Extraction Solvents for Untargeted Metabolomics of Plants. International Journal of Molecular Sciences. 2016; 17(7):1017. https://doi.org/10.3390/ijms17071017
Chicago/Turabian StyleDoppler, Maria, Bernhard Kluger, Christoph Bueschl, Christina Schneider, Rudolf Krska, Sylvie Delcambre, Karsten Hiller, Marc Lemmens, and Rainer Schuhmacher. 2016. "Stable Isotope-Assisted Evaluation of Different Extraction Solvents for Untargeted Metabolomics of Plants" International Journal of Molecular Sciences 17, no. 7: 1017. https://doi.org/10.3390/ijms17071017
APA StyleDoppler, M., Kluger, B., Bueschl, C., Schneider, C., Krska, R., Delcambre, S., Hiller, K., Lemmens, M., & Schuhmacher, R. (2016). Stable Isotope-Assisted Evaluation of Different Extraction Solvents for Untargeted Metabolomics of Plants. International Journal of Molecular Sciences, 17(7), 1017. https://doi.org/10.3390/ijms17071017