
Physiological and Pathogenic Roles of Prolyl Isomerase Pin1 in Metabolic Regulations via Multiple Signal Transduction Pathway Modulations

 and
and
Abstract
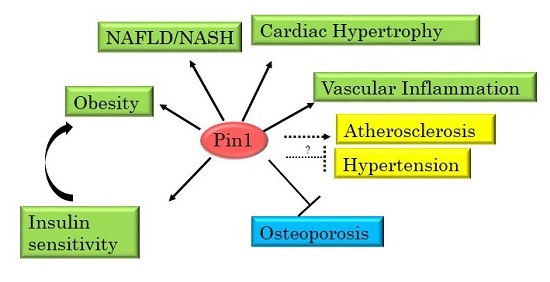
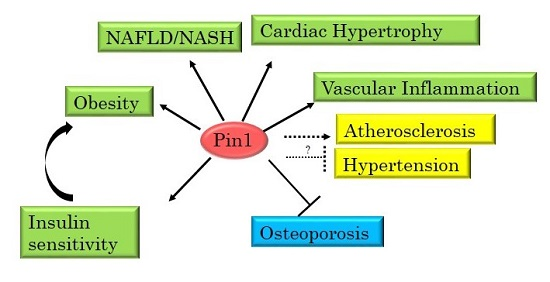
:
1. Introduction
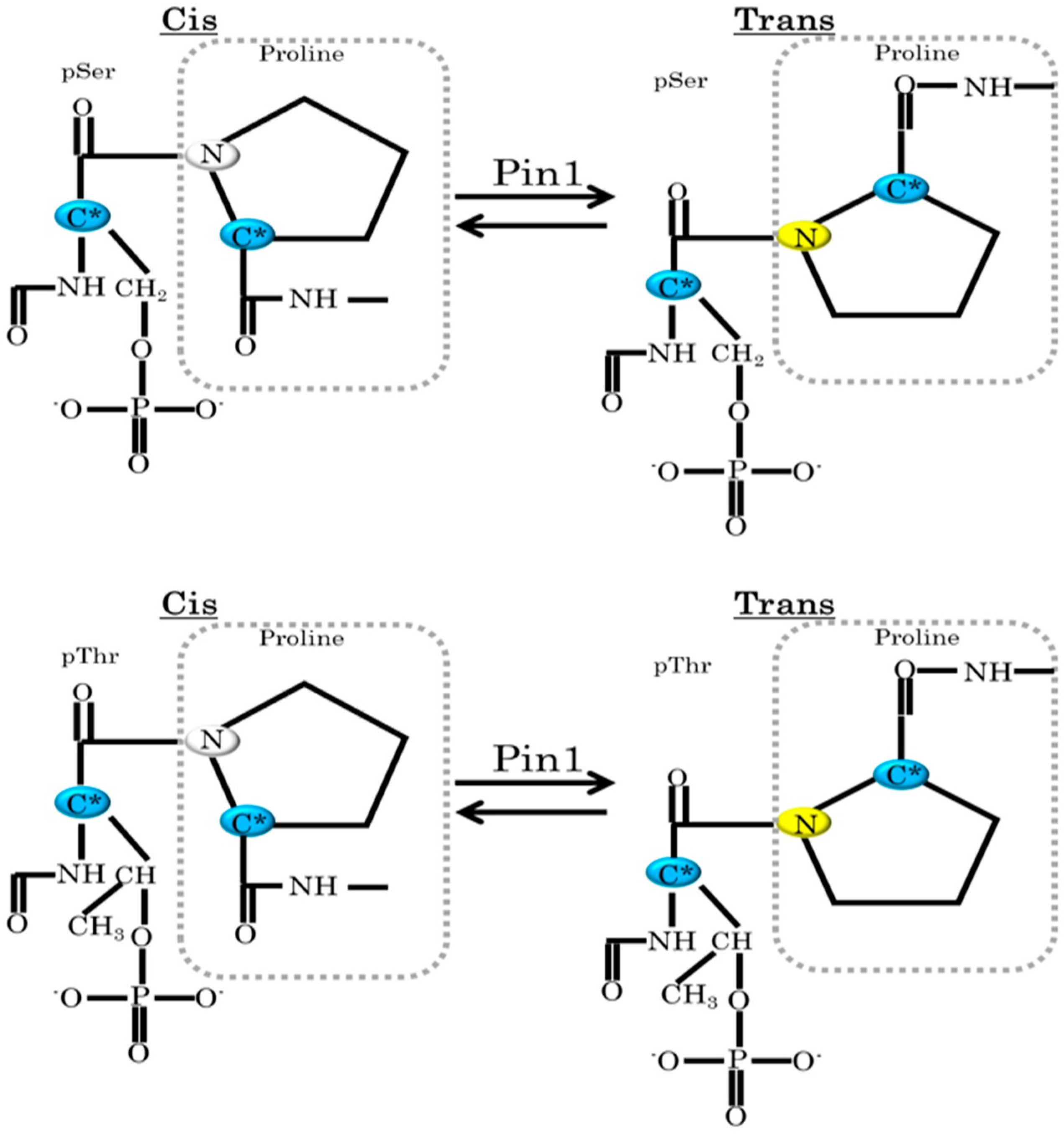
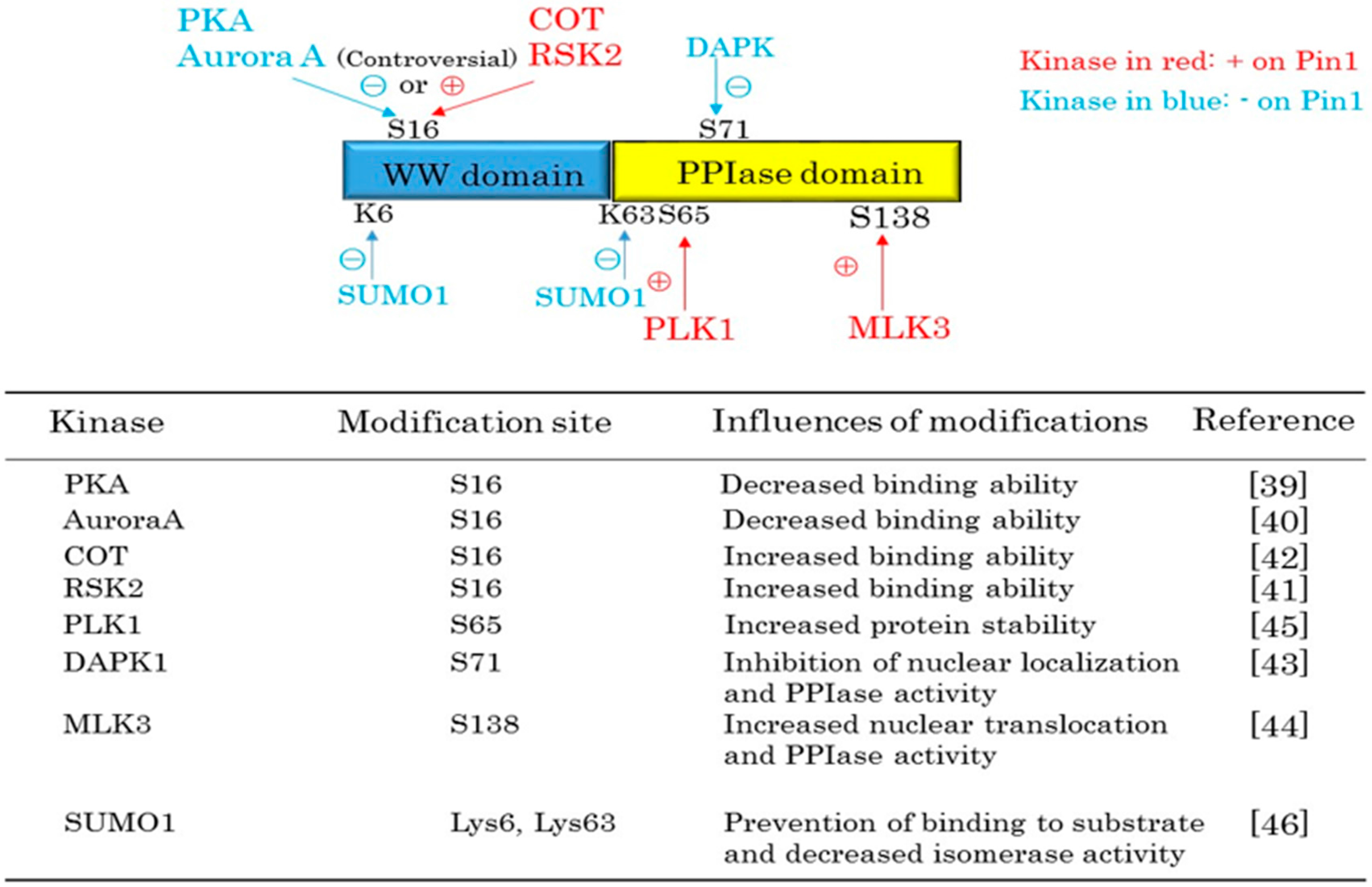
2. Pin1 Functions Are Regulated by Protein Kinases and SUMOylation
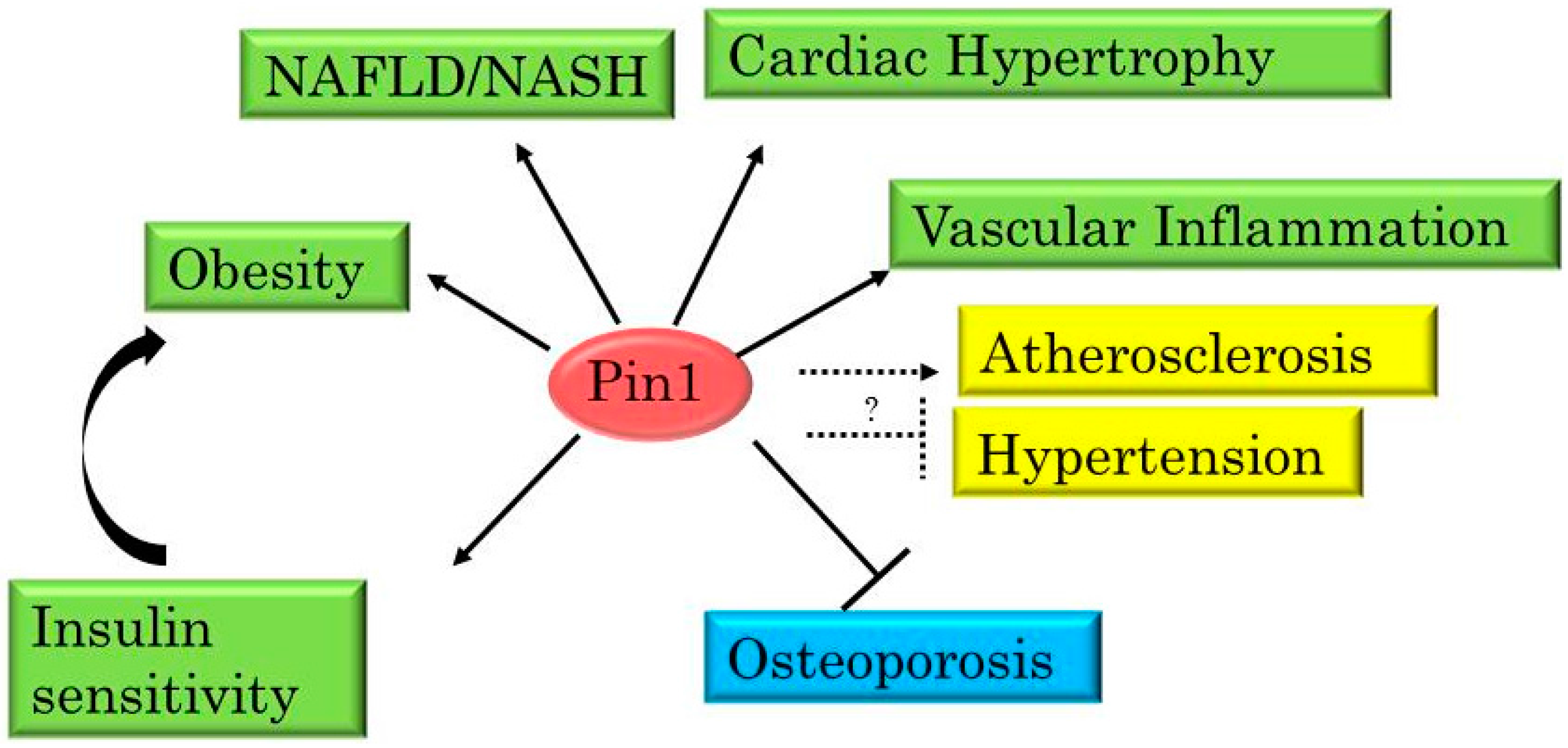
3. Pin1 Is Highly Involved in the Development of Metabolic Syndrome
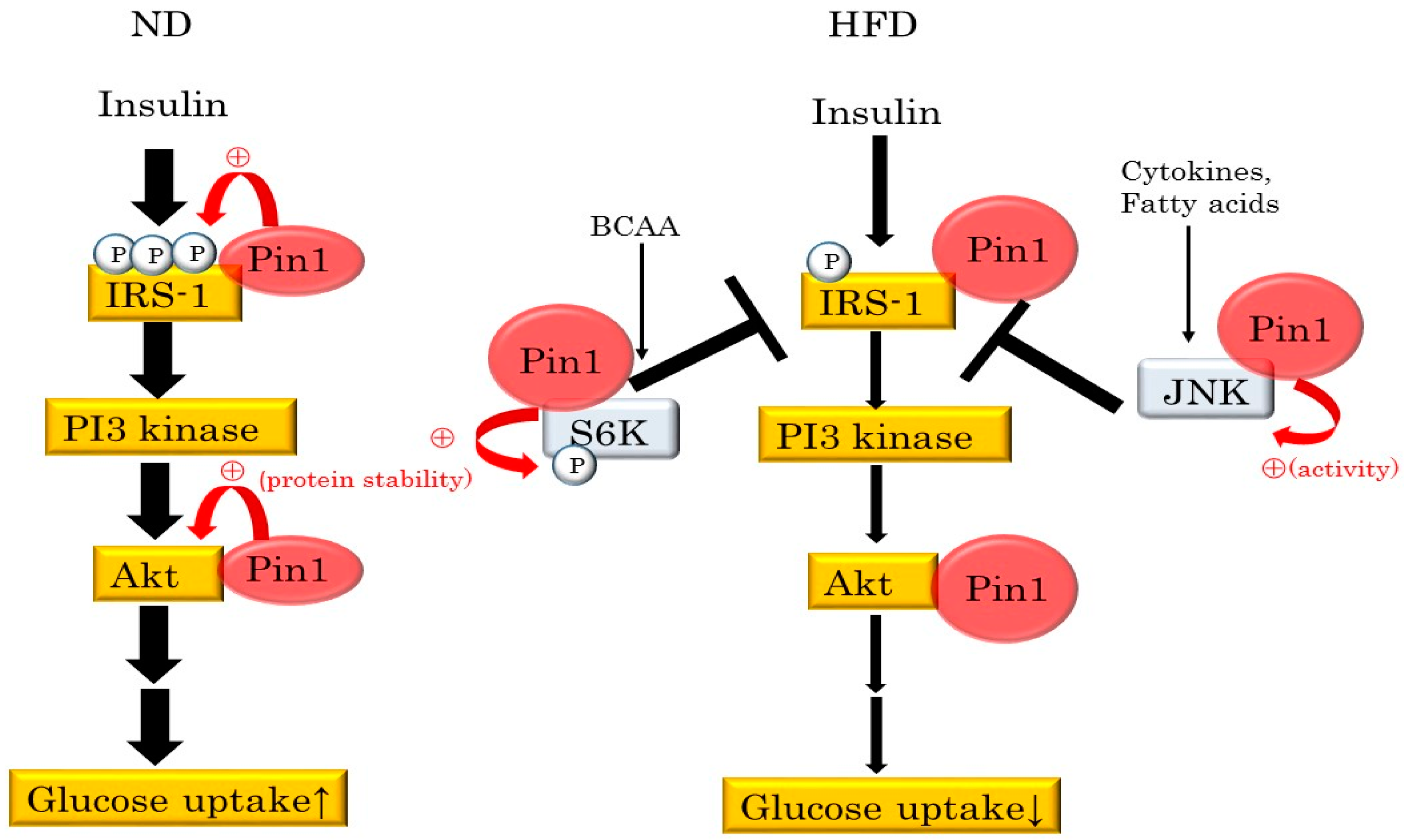
4. Pin1 Controls Insulin Signaling
5. Pin1 Suppresses Gluconeogenesis
6. Pin1 Is Essential for Adipogenesis and Is Involved in Obesity
7. Pin1 Plays a Critical Role in the Development of NASH
8. The Role of Pin1 in Hypertension
9. The Roles of Pin1 in Atherosclerosis and Cardiac Dysfunction
10. The Role of Pin1 in Bone Formation and Osteoporosis
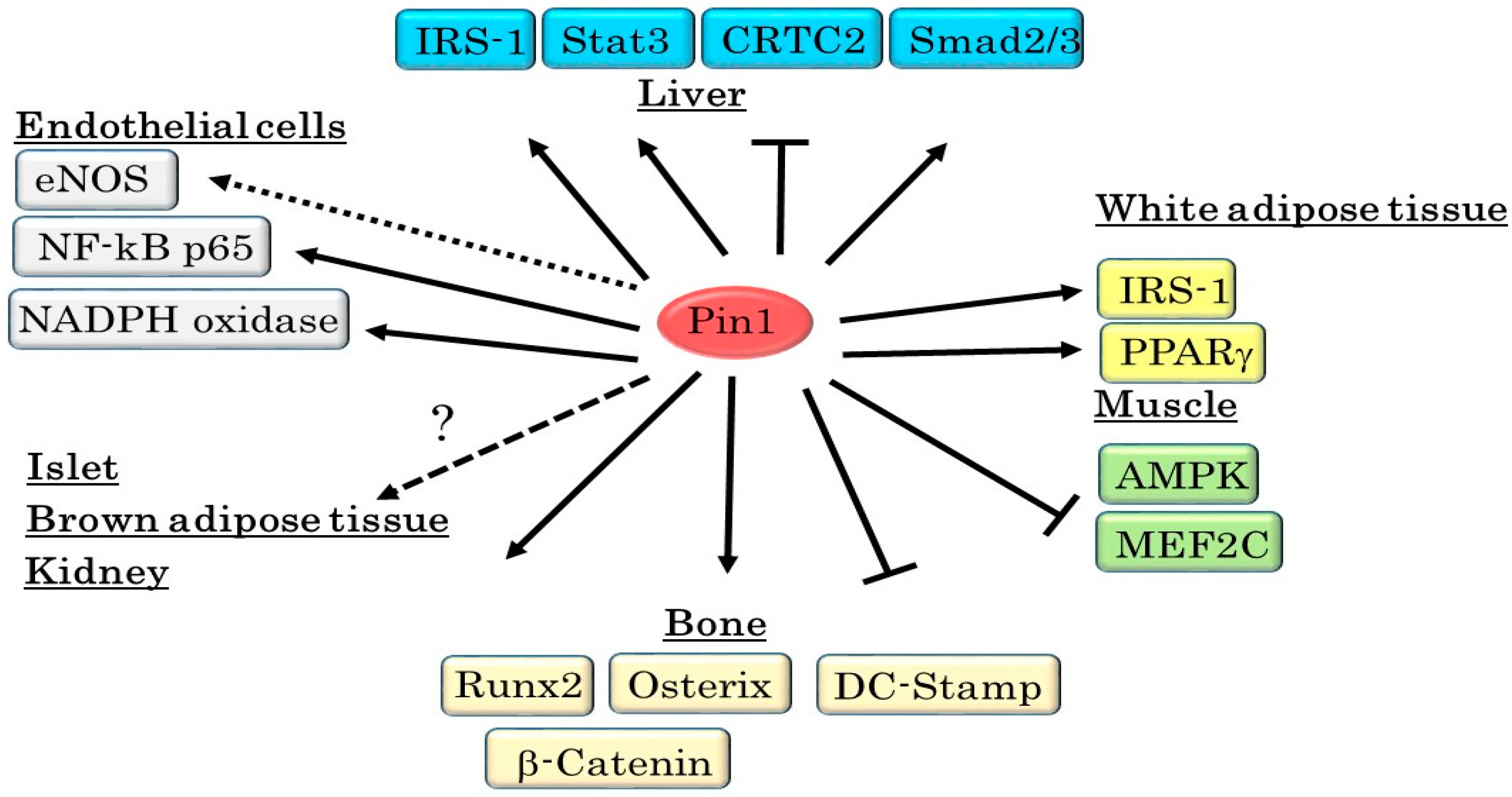
11. Conclusions
Author Contributions
Conflicts of Interest
References
- Göthel, S.F.; Marahiel, M.A. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell. Mol. Life Sci. 1999, 55, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 36, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: A specific cytosolic binding protein for cyclosporin, A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Hanes, S.D.; Hunter, T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 1996, 380, 544–547. [Google Scholar] [PubMed]
- Yaron, A.; Naider, F. Proline-dependent structural and biological properties of peptides and proteins. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 31–81. [Google Scholar] [CrossRef] [PubMed]
- Dugave, C.; Demange, L. Cis-trans isomerization of organic molecules and biomolecules: Implications and applications. Chem. Rev. 2003, 103, 2475–2532. [Google Scholar] [CrossRef] [PubMed]
- Vanhoof, G.; Goossens, F.; de Meester, I.; Hendriks, D.; Scharpé, S. Proline motifs in peptides and their biological processing. FASEB J. 1995, 9, 736–744. [Google Scholar] [PubMed]
- Reimer, U.; Scherer, G.; Drewello, M.; Kruber, S.; Schutkowski, M.; Fischer, G. Side-chain effects on peptidyl-prolyl cis/trans isomerisation. J. Mol. Biol. 1998, 279, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.H.; Ning, Y.M.; Sanchez, E.R. A new first step in activation of steroid receptors—Hormone-induced switching of FKBP51 and FKBP52 immunophilins. J. Biol. Chem. 2002, 277, 4597–4600. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef]
- Fujiyama, S.; Yanagida, M.; Hayano, T.; Miura, Y.; Isobe, T.; Fujimori, F.; Uchida, T.; Takahashi, N. Isolation and proteomic characterization of human Parvulin-associating preribosomal ribonucleoprotein complexes. J. Biol. Chem. 2002, 277, 23773–23780. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.W.; Bayer, P. Small family with key contacts: Par14 and Par17 parvulin proteins, relatives of Pin1, now emerge in biomedical research. Perspect. Med. Chem. 2008, 2, 11–20. [Google Scholar]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Dhariwala, F.A.; Rajadhyaksha, M.S. An unusual member of the Cdk family: Cdk5. Cell. Mol. Neurobiol. 2008, 28, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Himpel, S.; Tegge, W.; Frank, R.; Leder, S.; Joost, H.G.; Becker, W. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J. Biol. Chem. 2000, 275, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [PubMed]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3α-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Moretto-Zita, M.; Jin, H.; Shen, Z.; Zhao, T.; Briggs, S.P.; Xu, Y. Phosphorylation stabilizes Nanog by promoting its interaction with Pin1. Proc. Natl. Acad. Sci. USA 2010, 107, 13312–13317. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Sakoda, H.; Kushiyama, A.; Zhang, J.; Ono, H.; Fujishiro, M.; Kikuchi, T.; Fukushima, T.; Yoneda, M.; Ohno, H.; et al. Peptidyl-prolyl Cis/Trans Isomerase NIMA-interacting 1 Associates with Insulin Receptor Substrate-1 and Enhances Insulin Actions and Adipogenesis. J. Biol. Chem. 2011, 286, 20812–20822. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Shiraki, T.; Horiuchi, Y.; Waku, T.; Shigenaga, A.; Otaka, A.; Ikura, T.; Igarashi, K.; Aimoto, S.; Tate, S.; et al. Proline cis/trans-isomerase Pin1 regulates peroxisome proliferator-activated receptor γ activity through the direct binding to the activation function-1 domain. J. Biol. Chem. 2010, 285, 3126–3132. [Google Scholar] [CrossRef] [PubMed]
- Lufei, C.; Koh, T.H.; Uchida, T.; Cao, X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene 2007, 26, 7656–7664. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, F.; Takahashi, K.; Uchida, C.; Uchida, T. Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G0 arrest. Biochem. Biophys. Res. Commun. 1999, 265, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.C.; Sun, A.; Ryo, A.; Zhou, X.Z.; Yu, Z.X.; Huang, H.K.; Uchida, T.; Bronson, R.; Bing, G.; Li, X.; et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 2003, 424, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.M.; Ryo, A.; Wulf, G.G.; Lee, S.W.; Niu, T.; Petkova, V.; Lu, K.P. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001, 20, 3459–3472. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.C.; Lu, K.P. Pin 1 regulates turnover and subcellular localization of β-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A.; Zhou, X.Z.; Lu, K.P. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer's disease. Biochim. Biophys. Acta 2015, 1850, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, L.; Sun, A.; Lu, P.J.; Zhou, X.Z.; Balastik, M.; Finn, G.; Wulf, G.; Lim, J.; Li, S.H.; Li, X.; et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 2006, 440, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Tindale, L.; Cumming, R.C. Age-dependent metabolic dysregulation in cancer and Alzheimer’s disease. Biogerontology 2014, 15, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Zhou, X.Z.; Liou, Y.C.; Noel, J.P.; Lu, K.P. Critical role of WW domain phosphorylation in regulating its phosphoserine-binding activity and the Pin1 function. J. Biol. Chem. 2002, 277, 2381–2384. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Que, J.; Chen, Y.C.; Lin, J.T.; Liou, Y.C.; Liao, P.C.; Liu, Y.P.; Lee, K.H.; Lin, L.C.; Hsiao, M.; et al. Pin1 acts as a negative regulator of the G2/M transition by interacting with the Aurora-A-Bora complex. J. Cell Sci. 2013, 126, 4862–4872. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Park, S.Y.; Kim, D.J.; Lee, S.H.; Woo, K.M.; Lee, K.A.; Lee, Y.J.; Cho, Y.Y.; Shim, J.H. TPA-induced cell transformation provokes a complex formation between Pin1 and 90 kDa ribosomal protein S6 kinase 2. Mol. Cell. Biochem. 2012, 367, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Khanal, P.; Kim, J.Y.; Yun, H.J.; Lim, S.C.; Shim, J.H.; Choi, H.S. COT phosphorylates prolyl-isomerase Pin1 to promote tumorigenesis in breast cancer. Mol. Carcinog. 2015, 54, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chen, C.H.; Suizu, F.; Huang, P.; Schiene-Fischer, C.; Daum, S.; Zhang, Y.J.; Goate, A.; Chen, R.H.; Zhou, X.Z.; et al. Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol. Cell 2011, 42, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, V.; Mishra, R.; Sondarva, G.; Das, S.; Lee, T.H.; Bakowska, J.C.; Tzivion, G.; Malter, J.S.; Rana, B.; Lu, K.P.; et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc. Natl. Acad. Sci. USA 2012, 109, 8149–8154. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Yuan, J.; Saxena, K.; Martin, B.; Kappel, S.; Lindenau, C.; Kramer, A.; Naumann, S.; Daum, S.; Fischer, G.; et al. Polo-like kinase 1-mediated phosphorylation stabilizes Pin1 by inhibiting its ubiquitination in human cells. J. Biol. Chem. 2005, 280, 36575–36583. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Chang, C.C.; Lee, T.H.; Luo, M.; Huang, P.; Liao, P.H.; Wei, S.; Li, F.A.; Chen, R.H.; Zhou, X.Z.; et al. SENP1 deSUMOylates and regulates Pin1 protein activity and cellular function. Cancer Res. 2013, 73, 3951–3962. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Gores, G.J.; Hirsova, P.; Kirby, M.; Miles, L.; Jaeschke, A.; Kohli, R. Mixed lineage kinase 3 deficient mice are protected against the high fat high carbohydrate diet-induced steatohepatitis. Liver Int. 2014, 34, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Otani, Y.; Sakoda, H.; Zhang, J.; Guo, Y.; Okubo, H.; Kushiyama, A.; Fujishiro, M.; Kikuch, T.; Fukushima, T.; et al. Role of Pin1 protein in the pathogenesis of nonalcoholic steatohepatitis in a rodent model. J. Biol. Chem. 2012, 287, 44526–44535. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Zhang, J.; Zhang, L.; Xue, G.; Wang, P.; Meng, Q.; Liang, W. Essential role of Pin1 via STAT3 signalling and mitochondria-dependent pathways in restenosis in type 2 diabetes. J. Cell. Mol. Med. 2013, 17, 989–1005. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Castello, L.; Battista, R.; Capretti, G.; Chiandotto, S.; D’Amario, D.; Scavone, G.; Villano, A.; Rustighi, A.; et al. Targeting prolyl-isomerase Pin1 prevents mitochondrial oxidative stress and vascular dysfunction: Insights in patients with diabetes. Eur. Heart J. 2015, 36, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Toko, H.; Konstandin, M.H.; Doroudgar, S.; Ormachea, L.; Joyo, E.; Joyo, A.Y.; Din, S.; Gude, N.A.; Collins, B.; Völkers, M.; et al. Regulation of cardiac hypertrophic signaling by prolyl isomerase Pin1. Circ Res. 2013, 112, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Shin, S.Y.; Jue, S.S.; Kwon, I.K.; Cho, E.H.; Cho, E.S.; Park, S.H.; Kim, E.C. The role of PIN1 on odontogenic and adipogenic differentiation in human dental pulp stem cells. Stem Cells Dev. 2014, 23, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lee, S.H.; Bahn, M.; Yeo, C.Y.; Lee, K.Y. Pin1 enhances adipocyte differentiation by positively regulating the transcriptional activity of PPARγ. Mol. Cell. Endocrinol. 2016, 436, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Magli, A.; Angelelli, C.; Ganassi, M.; Baruffaldi, F.; Matafora, V.; Battini, R.; Bachi, A.; Messina, G.; Rustighi, A.; del Sal, G.; et al. Proline isomerase Pin1 represses terminal differentiation and myocyte enhancer factor 2C function in skeletal muscle cells. J. Biol. Chem. 2010, 285, 34518–34527. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Lüscher, T.F.; Cosentino, F. Pin1 inhibitor Juglone prevents diabetic vascular dysfunction. Int. J. Cardiol. 2016, 203, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, M.; Ozenirler, S.; Yücel, A.A.; Yılmaz, G. Can serum pin1 level be regarded as an indicative marker of nonalcoholic steatohepatitis and fibrotic stages? Digestion 2014, 90, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lee, J.A.; Park, S.G.; Lee, D.H.; Kim, S.J.; Kim, H.J.; Uchida, C.; Uchida, T.; Park, B.C.; Cho, S. A critical step for JNK activation: Isomerization by the prolyl isomerase Pin1. Cell Death Differ. 2012, 19, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Choi, H.K.; Shim, J.H.; Kang, K.W.; Dong, Z.; Choi, H.S. The prolyl isomerase Pin1 interacts with a ribosomal protein S6 kinase to enhance insulin-induced AP-1 activity and cellular transformation. Carcinogenesis 2009, 30, 671–681. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005, 437, 1109–1111. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Sakoda, H.; Kushiyama, A.; Ono, H.; Fujishiro, M.; Horike, N.; Yoneda, M.; Ohno, H.; Tsuchiya, Y.; Kamata, H.; et al. Pin1 associates with and induces translocation of CRTC2 to the cytosol, thereby suppressing cAMP-responsive element transcriptional Activity. J. Biol. Chem. 2010, 285, 33018–33027. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Furumai, K.; Fukuda, T.; Akiyama, H.; Takezawa, M.; Asano, T.; Fujimori, F.; Uchida, C. Prolyl isomerase Pin1 regulates mouse embryonic fibroblast differentiation into adipose cells. PLoS ONE 2012, 7, e31823. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Ogawa, W.; Asakawa, A.; Okamoto, Y.; Nishizawa, A.; Matsumoto, M.; Teshigawara, K.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006, 3, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Guo, L.; Xie, L.Q.; Zhang, Y.Y.; Liu, X.H.; Zhang, Y.; Zhu, H.; Yang, P.Y.; Lu, H.J.; Tang, Q.Q. Proteome profiling of mitotic clonal expansion during 3T3-L1 adipocyte differentiation using iTRAQ-2DLC-MS/MS. J. Proteome Res. 2014, 13, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.B.; Peroni, O.D.; Fryer, L.G.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2Cα and native bovine protein phosphatase-2Ac. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Iwashita, M.; Sakoda, H.; Ono, H.; Nagata, K.; Matsunaga, Y.; Fukushima, T.; Fujishiro, M.; Kushiyama, A.; Kamata, H.; et al. Prolyl isomerase Pin1 negatively regulates AMP-activated protein kinase (AMPK) by associating with the CBS domain in the γ subunit. J. Biol. Chem. 2015, 290, 24255–24266. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Chiang, K.N.; Lai, C.Y.; He, D.; Wang, G.; Ramkumar, R.; Uchida, T.; Ryo, A.; Lu, K.; Liu, F. Pin1 promotes transforming growth factor-β-induced migration and invasion. J. Biol. Chem. 2010, 285, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Hien, T.T.; Lim, S.C.; Jun, D.W.; Choi, H.S.; Yoon, J.H.; Cho, I.J.; Kang, K.W. Pin1 induction in the fibrotic liver and its roles in TGF-β1 expression and Smad2/3 phosphorylation. J. Hepatol. 2014, 60, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Aragón, E.; Goerner, N.; Zaromytidou, A.I.; Xi, Q.; Escobedo, A.; Massagué, J.; Macias, M.J. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011, 25, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Königsrainer, A.; Weng, H.; Dooley, S.; Ten Dijke, P. Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, E.W.; Stegeman, C.A.; Heeringa, P.; Henning, R.H.; van Goor, H. Protective role of endothelial nitric oxide synthase. J. Pathol. 2003, 199, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [PubMed]
- Morrow, V.A.; Foufelle, F.; Connell, J.M.; Petrie, J.R.; Gould, G.W.; Salt, I.P. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J. Biol. Chem. 2003, 278, 31629–31639. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Seo, J.; Park, J.H.; Jo, C.; Choi, Y.J.; Soh, J.W.; Jo, I. Cyclin-dependent kinase 5 phosphorylates endothelial nitric oxide synthase at serine 116. Hypertension 2010, 55, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Bernier, S.G.; Haldar, S.; Michel, T. Bradykinin-regulated interactions of the mitogen-activated protein kinase pathway with the endothelial nitric-oxide synthase. J. Biol. Chem. 2000, 275, 30707–30715. [Google Scholar] [CrossRef] [PubMed]
- Chiasson, V.L.; Munshi, N.; Chatterjee, P.; Young, K.J.; Mitchell, B.M. Pin1 deficiency causes endothelial dysfunction and hypertension. Hypertension 2011, 58, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.A.; Jue, S.S.; Bae, W.J.; Heo, S.H.; Shin, S.I.; Kwon, I.K.; Lee, S.C.; Kim, E.C. PIN1 inhibition suppresses osteoclast differentiation and inflammatory responses. J. Dent. Res. 2015, 94, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Torres, C.M.; Qian, J.; Chen, F.; Mintz, J.D.; Stepp, D.W.; Fulton, D.; Venema, R.C. Pin1 prolyl isomerase regulates endothelial nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Kennard, S.; Ruan, L.; Buffett, R.J.; Fulton, D.; Venema, R.C. TNFα reduces eNOS activity in endothelial cells through serine 116 phosphorylation and Pin1 binding: Confirmation of a direct, inhibitory interaction of Pin1 with eNOS. Vasc. Pharmacol. 2016, 81, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Koinuma, D.; Miyazawa, K.; Uchida, T.; Saitoh, M.; Kawabata, M.; Hanai, J.; Akiyama, H.; Abe, M.; Miyazono, K.; et al. Pin1 down-regulates transforming growth factor-β (TGF-β) signaling by inducing degradation of Smad proteins. J. Biol. Chem. 2009, 284, 6109–6115. [Google Scholar] [CrossRef] [PubMed]
- Fila, C.; Metz, C.; van der Sluijs, P. Juglone inactivates cysteine-rich proteins required for progression through mitosis. J. Biol. Chem. 2008, 283, 21714–21724. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Yu, X.F.; Qu, S.C.; Qu, X.R.; Jiang, Y.F.; Sui, D.Y. Juglone, from Juglans mandshruica Maxim, inhibits growth and induces apoptosis in human leukemia cell HL-60 through a reactive oxygen species-dependent mechanism. Food Chem. Toxicol. 2012, 50, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Jha, B.K.; Jung, H.J.; Seo, I.; Suh, S.I.; Suh, M.H.; Baek, W.K. Juglone induces cell death of Acanthamoeba through increased production of reactive oxygen species. Exp. Parasitol. 2015, 159, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Liu, G.J.; Li, Z.Y.; Wang, X.H. Pin1 in cardiovascular dysfunction: A potential double-edge role. Int. J. Cardiol. 2016, 212, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Mechanisms of disease—Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Tun-Kyi, A.; Finn, G.; Greenwood, A.; Nowak, M.; Lee, T.H.; Asara, J.M.; Tsokos, G.C.; Fitzgerald, K.; Israel, E.; Li, X.; et al. Essential role for the prolyl isomerase Pin1 in toll-like receptor signaling and type I interferon-mediated immunity. Nat. Immunol. 2011, 12, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Makni-Maalej, K.; Boussetta, T.; Hurtado-Nedelec, M.; Belambri, S.A.; Gougerot-Pocidalo, M.A.; El-Benna, J. The TLR7/8 agonist CL097 primes N-formyl-methionyl-leucyl-phenylalanine-stimulated NADPH oxidase activation in human neutrophils: Critical role of p47phox phosphorylation and the proline isomerase Pin1. J. Immunol. 2012, 189, 4657–4665. [Google Scholar] [CrossRef] [PubMed]
- Boussetta, T.; Gougerot-Pocidalo, M.A.; Hayem, G.; Ciappelloni, S.; Raad, H.; Arabi Derkawi, R.; Bournier, O.; Kroviarski, Y.; Zhou, X.Z.; Malter, J.S.; et al. The prolyl isomerase Pin1 acts as a novel molecular switch for TNF-α-induced priming of the NADPH oxidase in human neutrophils. Blood 2010, 116, 5795–5802. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Yoon, W.J.; Ryoo, H.M. Pin1, the master orchestrator of bone cell differentiation. J. Cell. Physiol. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.J.; Hu, J.; Ali, A.; Pastor, J.; Shiizaki, K.; Blank, R.D.; Kuro-o, M.; Malter, J.S. Pin1 null mice exhibit low bone mass and attenuation of BMP signaling. PLoS ONE 2013, 8, e63565. [Google Scholar] [CrossRef] [PubMed]
- Yoon, W.J.; Islam, R.; Cho, Y.D.; Ryu, K.M.; Shin, H.R.; Woo, K.M.; Baek, J.H.; Ryoo, H.M. Pin1 plays a critical role as a molecular switch in canonical BMP signaling. J. Cell. Physiol. 2015, 230, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Yoon, W.J.; Islam, R.; Cho, Y.D.; Woo, K.M.; Baek, J.H.; Uchida, T.; Komori, T.; van Wijnen, A.; Stein, J.L.; Lian, J.B.; et al. Pin1-mediated Runx2 modification is critical for skeletal development. J. Cell. Physiol. 2013, 228, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Yoon, W.J.; Cho, Y.D.; Kim, W.J.; Bae, H.S.; Islam, R.; Woo, K.M.; Baek, J.H.; Bae, S.C.; Ryoo, H.M. Prolyl isomerase Pin1-mediated conformational change and subnuclear focal accumulation of Runx2 are crucial for fibroblast growth factor 2 (FGF2)-induced osteoblast differentiation. J. Biol. Chem. 2014, 289, 8828–8838. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, Y.H.; Kim, Y.J.; Choi, H.S.; Yeo, C.Y.; Lee, K.Y. Prolyl isomerase Pin1 enhances osteoblast differentiation through Runx2 regulation. FEBS Lett. 2013, 587, 3640–3647. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jeong, H.M.; Han, Y.; Cheong, H.; Kang, B.Y.; Lee, K.Y. Prolyl isomerase Pin1 regulates the osteogenic activity of Osterix. Mol. Cell. Endocrinol. 2015, 400, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.R.; Islam, R.; Yoon, W.J.; Lee, T.; Cho, Y.D.; Bae, H.S.; Kim, B.S.; Woo, K.M.; Baek, J.H.; Ryoo, H.M. Pin1-mediated Modification Prolongs the Nuclear Retention of β-Catenin in Wnt3a-induced Osteoblast Differentiation. J. Biol. Chem. 2016, 291, 5555–5565. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Bae, H.S.; Yoon, W.J.; Woo, K.M.; Baek, J.H.; Kim, H.H.; Uchida, T.; Ryoo, H.M. Pin1 regulates osteoclast fusion through suppression of the master regulator of cell fusion DC-STAMP. J. Cell. Physiol. 2014, 229, 2166–2174. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Functional Changes Mediated By Pin1 | Reference |
|---|---|---|
| IRS-1 | Enhances Tyrosine Phosphorylation | [26] |
| CRTC2 | Inhibits Nuclear Translocation | [64] |
| CREB | Suppresses Transcriptional Activity | [65] |
| Stat3 | Upregulates Transcriptional Activity | [28] |
| AMPK | Decreases Phosphorylation of the α Subunit | [77] |
| PPARγ | Enhances Transcriptional Activity | [53] |
| Smad | Enhances Protein Stability and Phosphorylation | [79,80] |
| NF-κB p65 | Increases Protein Stability | [101] |
| eNOS | Increases or Decreases eNOS Activity | [90,91,92,93] |
| Runx2 | Enhances Transcriptional Activity | [111,112,113] |
| Osterix | Increases Protein Stability | [114] |
| β-Catenin | Increases Protein Stability | [115] |
| DC-STAMP | Decreases Protein Stability | [116] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakatsu, Y.; Matsunaga, Y.; Yamamotoya, T.; Ueda, K.; Inoue, Y.; Mori, K.; Sakoda, H.; Fujishiro, M.; Ono, H.; Kushiyama, A.; et al. Physiological and Pathogenic Roles of Prolyl Isomerase Pin1 in Metabolic Regulations via Multiple Signal Transduction Pathway Modulations. Int. J. Mol. Sci. 2016, 17, 1495. https://doi.org/10.3390/ijms17091495
Nakatsu Y, Matsunaga Y, Yamamotoya T, Ueda K, Inoue Y, Mori K, Sakoda H, Fujishiro M, Ono H, Kushiyama A, et al. Physiological and Pathogenic Roles of Prolyl Isomerase Pin1 in Metabolic Regulations via Multiple Signal Transduction Pathway Modulations. International Journal of Molecular Sciences. 2016; 17(9):1495. https://doi.org/10.3390/ijms17091495
Chicago/Turabian StyleNakatsu, Yusuke, Yasuka Matsunaga, Takeshi Yamamotoya, Koji Ueda, Yuki Inoue, Keiichi Mori, Hideyuki Sakoda, Midori Fujishiro, Hiraku Ono, Akifumi Kushiyama, and et al. 2016. "Physiological and Pathogenic Roles of Prolyl Isomerase Pin1 in Metabolic Regulations via Multiple Signal Transduction Pathway Modulations" International Journal of Molecular Sciences 17, no. 9: 1495. https://doi.org/10.3390/ijms17091495
APA StyleNakatsu, Y., Matsunaga, Y., Yamamotoya, T., Ueda, K., Inoue, Y., Mori, K., Sakoda, H., Fujishiro, M., Ono, H., Kushiyama, A., & Asano, T. (2016). Physiological and Pathogenic Roles of Prolyl Isomerase Pin1 in Metabolic Regulations via Multiple Signal Transduction Pathway Modulations. International Journal of Molecular Sciences, 17(9), 1495. https://doi.org/10.3390/ijms17091495