Receptors for Insulin-Like Growth Factor-2 and Androgens as Therapeutic Targets in Triple-Negative Breast Cancer

 ,
,  ,
,
Abstract
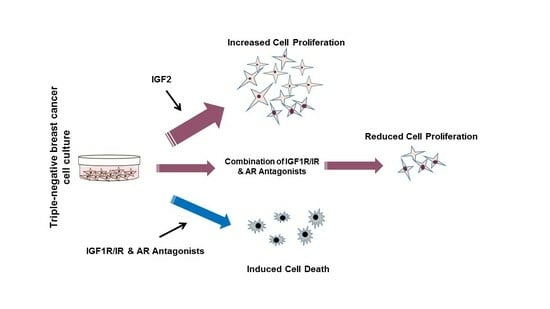
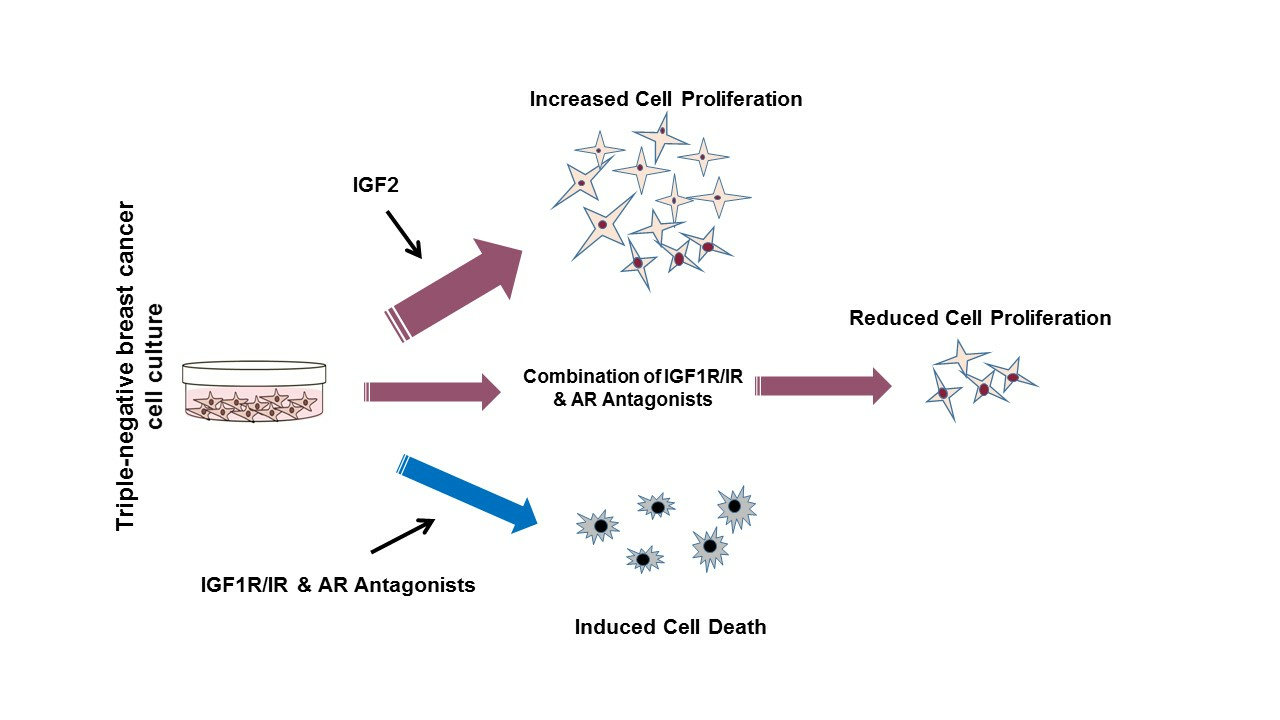
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
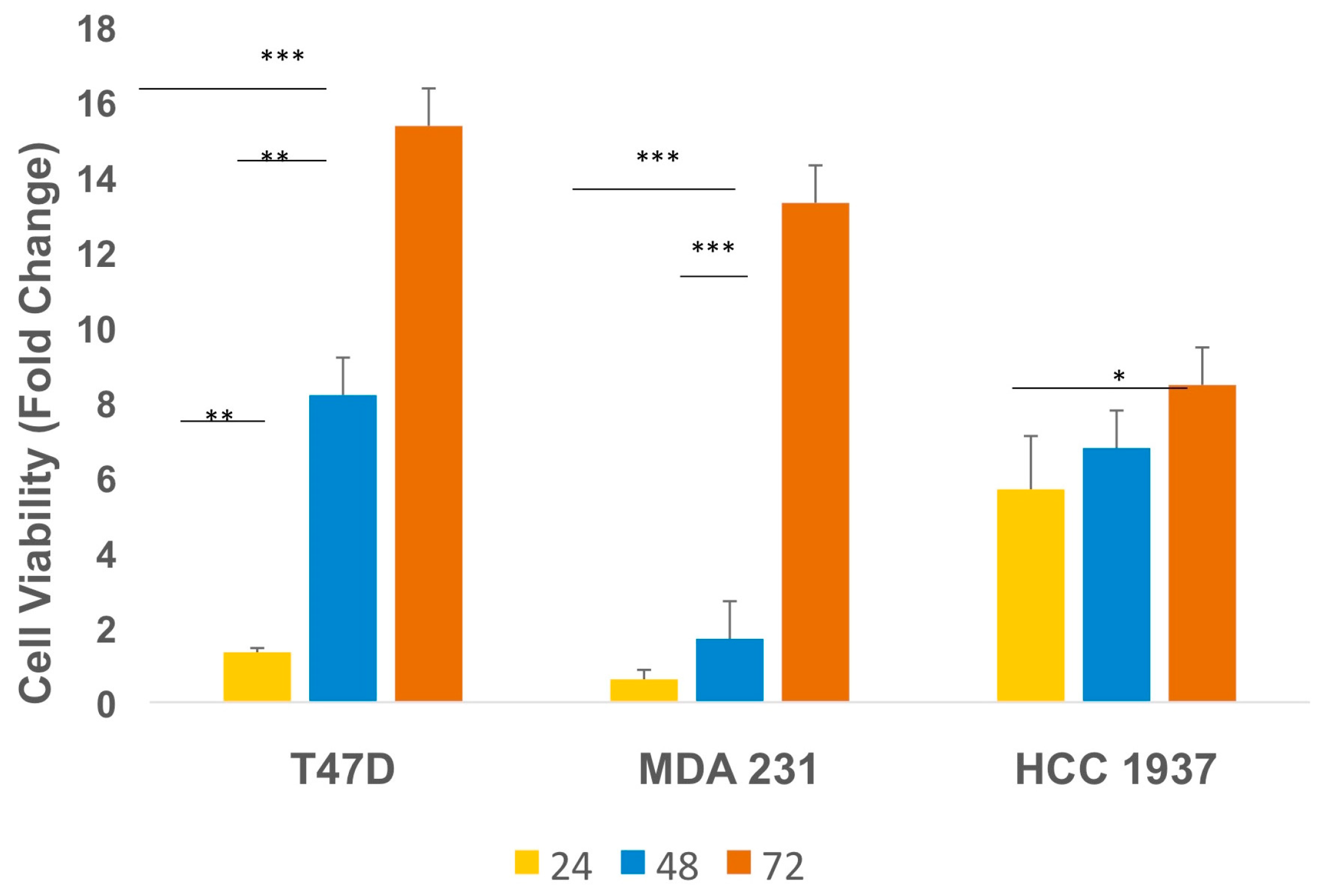
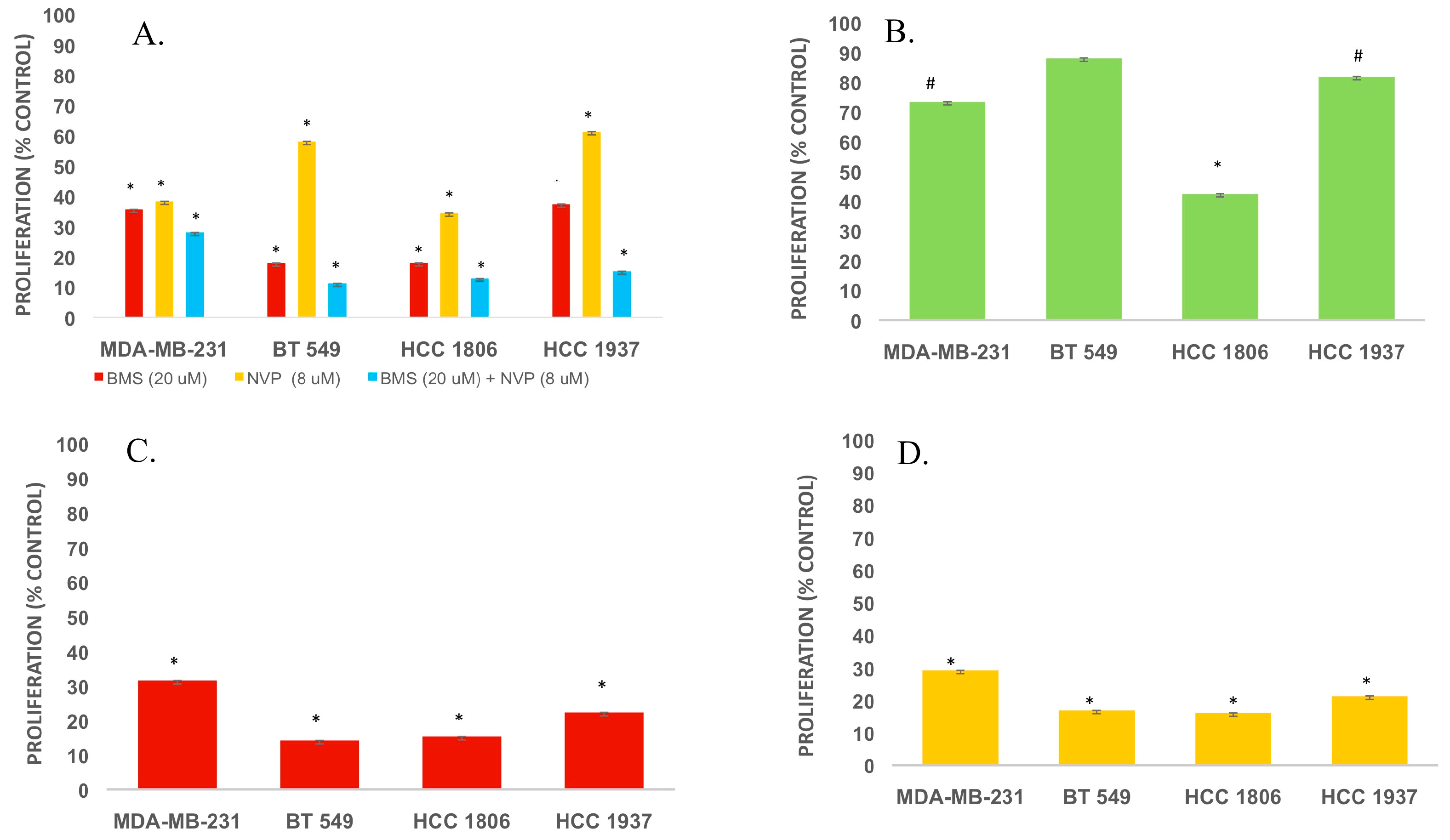
2.1. IGF2 Promotes the Viability of TNBC Cells
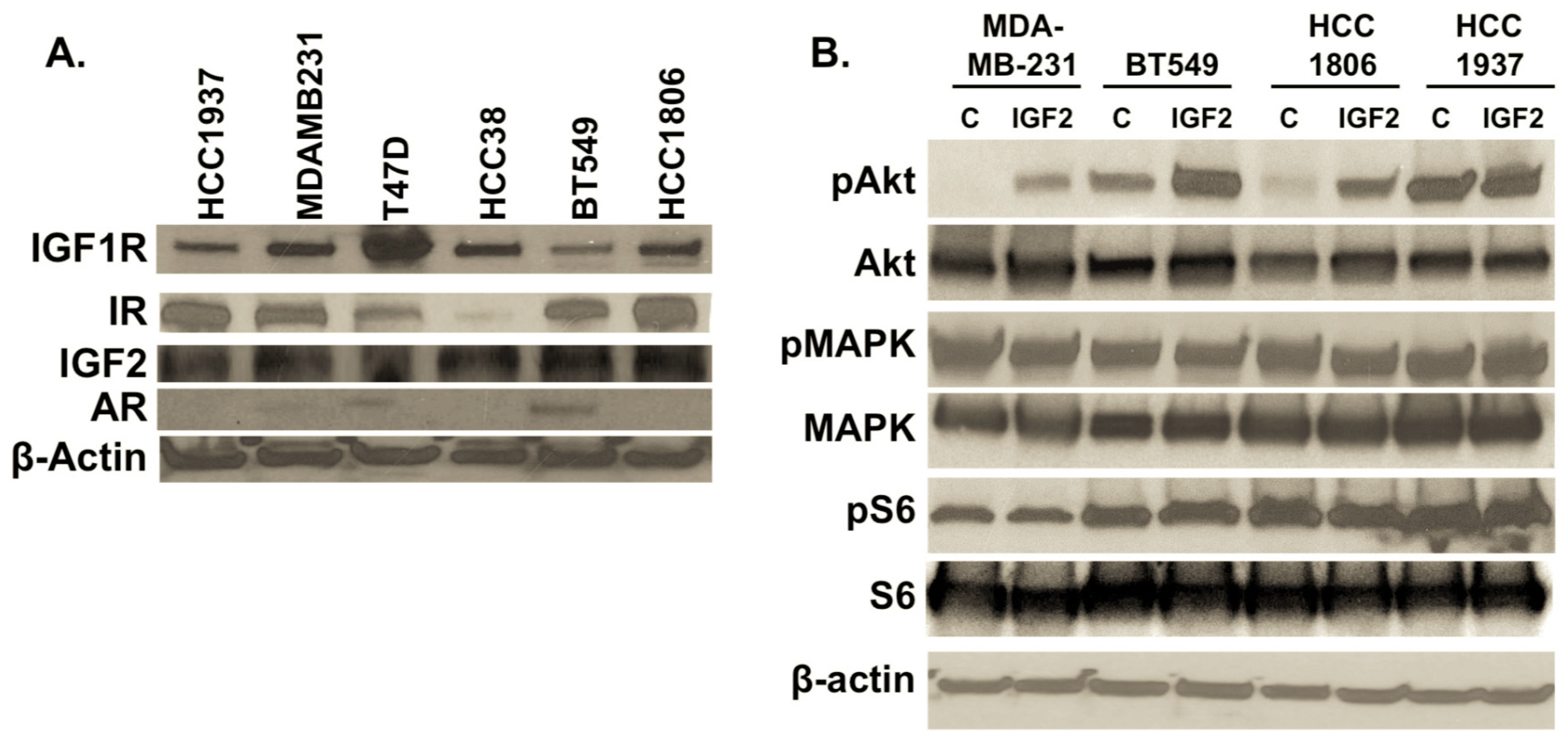
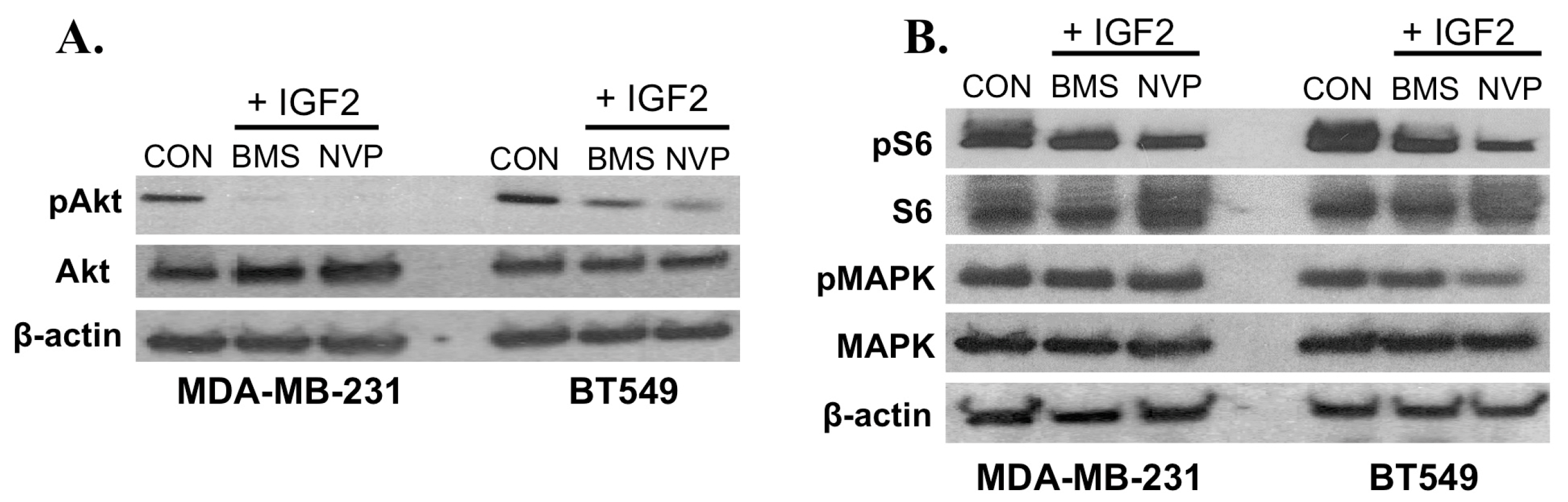
2.2. IGF2 Treatment Impacts Downstream TNBC Signaling Molecules
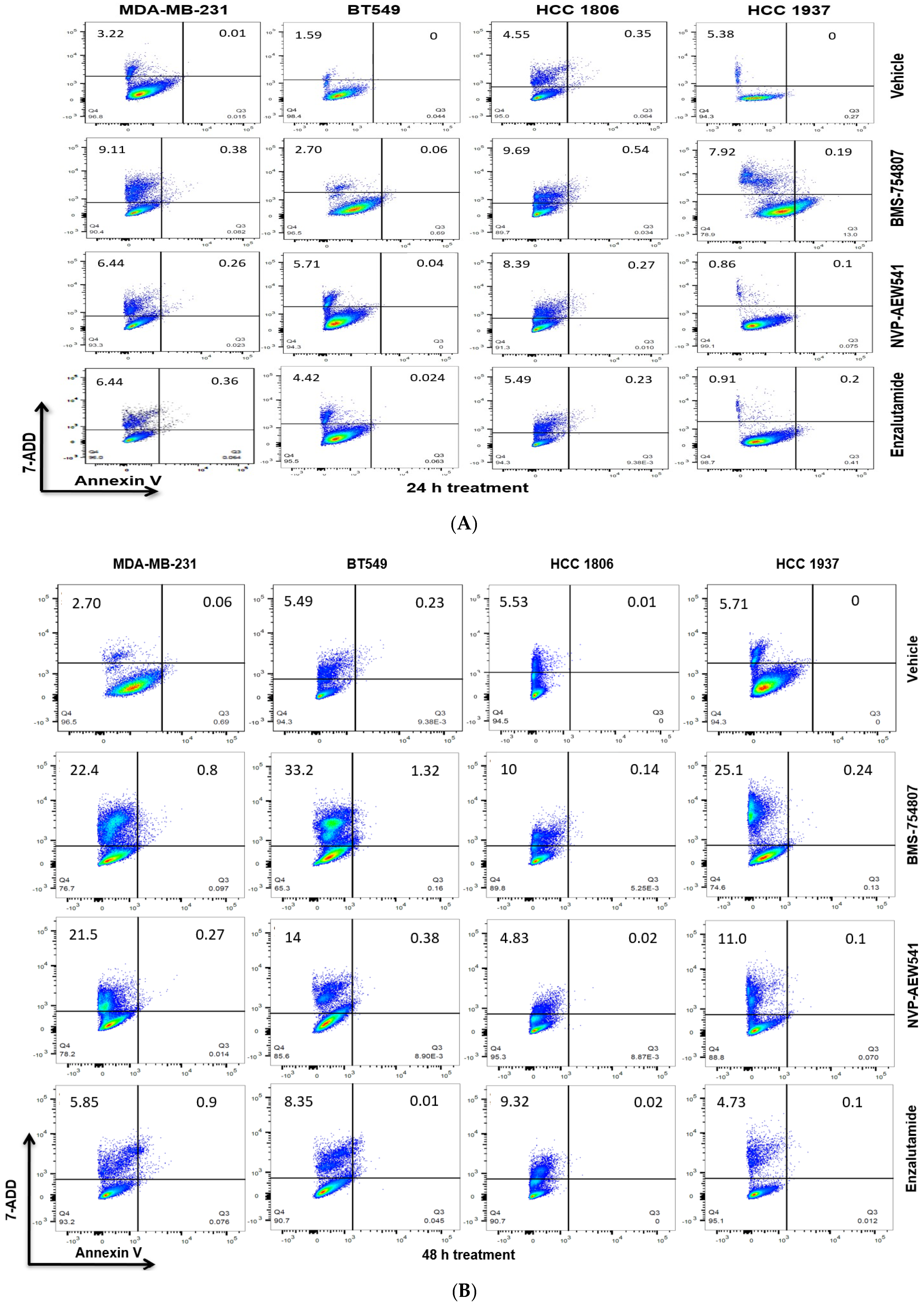
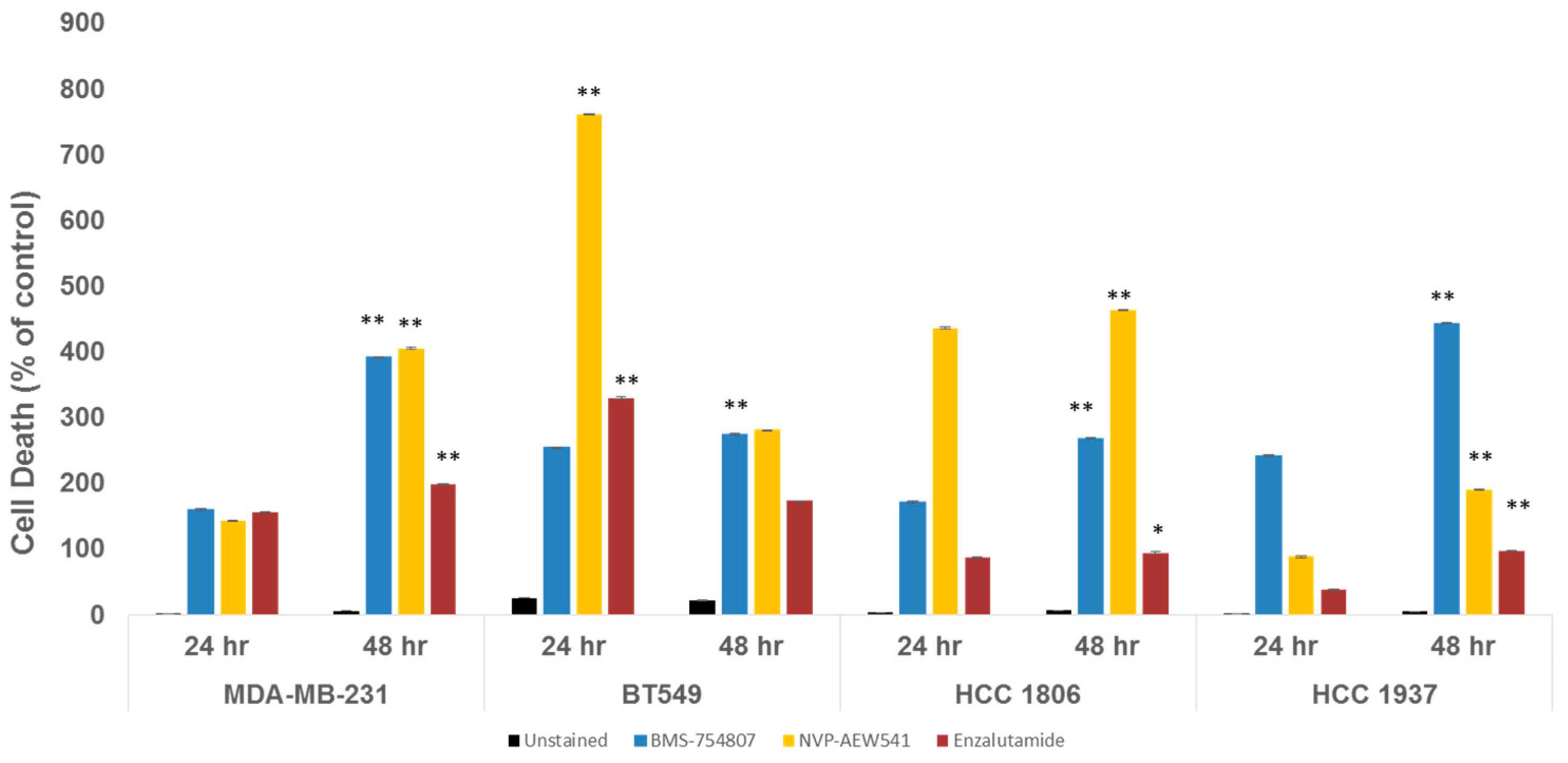
2.3. Inhibition of TNBC Proliferation and Induction of Cell Death by IGF1R/IR and AR Antagonists
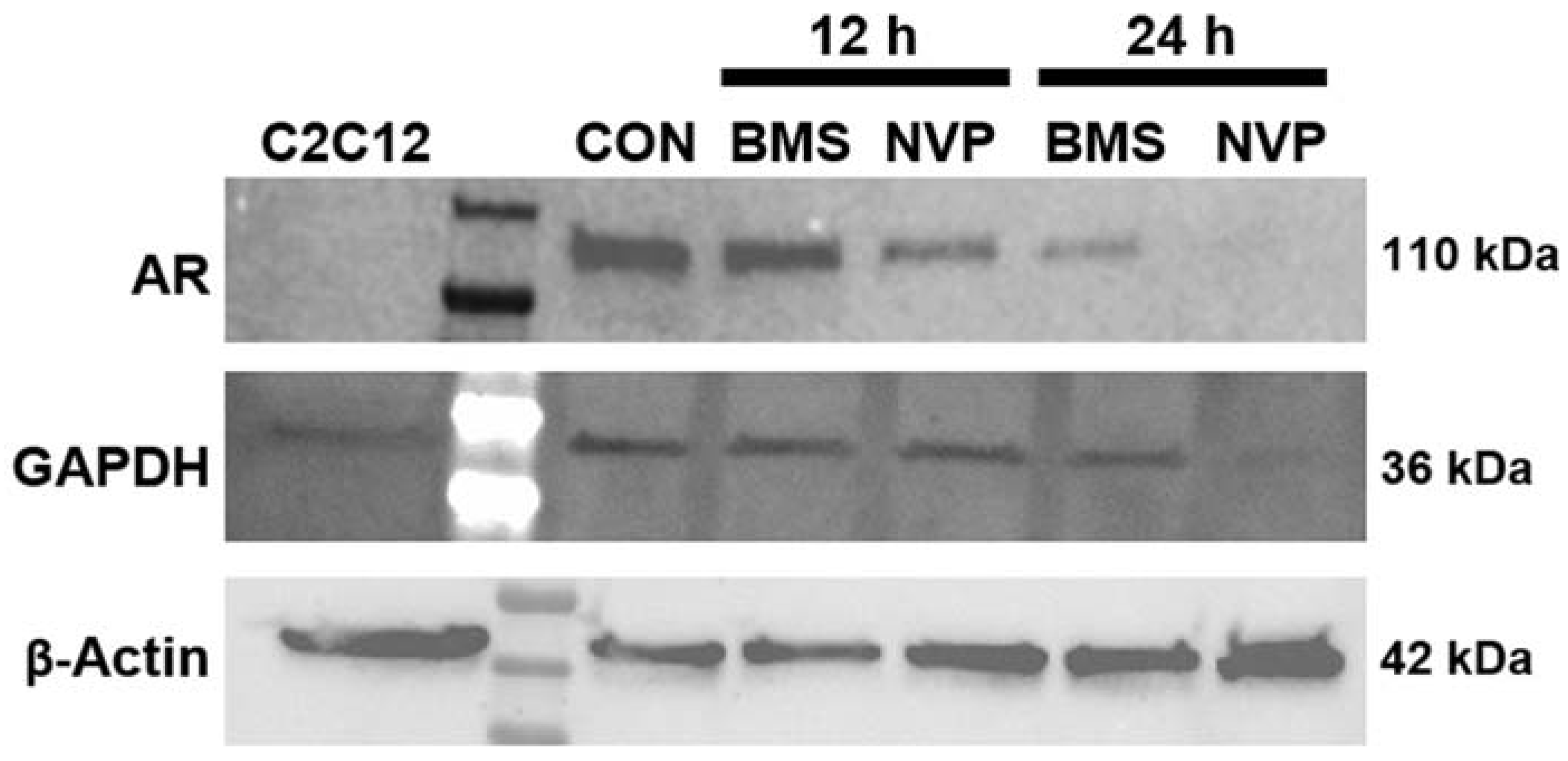
2.4. BMS-754807 and NVP-AEW541 Alter AR Expression in Mesenchyma-Subtype TNBC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability and Proliferation Assays
4.3. Assays of Cell Viability and Apoptosis/Cell Death by Flow Cytometry
4.4. Inhibition Assays
4.5. Gel Electrophoresis and Immunoblotting
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IGF1R | Insulin-like growth factor-1 receptor |
| AR | Androgen receptor |
| TNBC | Triple-negative breast cancer |
| IGF2 | Insulin-like growth factor-2 |
| BC | Breast cancer |
| PR | Progesterone receptor |
| IGF1 | Insulin-like growth factor-1 |
| IGF | Insulin-like growth factor |
| IGFBPs | IGF-binding proteins |
| ERβ | Estrogen receptor-β |
| IGF2R | IGF2 receptor |
| IR | Insulin receptor |
References
- Alter, R.; Barne, C.; Burke, A.; Gansler, T.; Gapstur, S.; Gaudet, M.; Kramer, J.; Newsman, L.A.; Niemeyer, D.; Richards, C.; et al. Breast Cancer Facts and Figures 2013–2014, 2013. American Cancer Society. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/breast-cancer-facts-and-figures/breast-cancer-facts-and-figures-2013-2014.pdf (accessed on 27 May 2017).
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Bégin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Reis-Filho, J.S.; Tutt, A.N. Triple negative tumours: A critical review. Histopathology 2008, 52, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Haffty, B.G.; Yang, Q.; Reiss, M.; Kearney, T.; Higgins, S.A.; Weidhaas, J.; Harris, L.; Hait, W.; Toppmeyer, D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J. Clin. Oncol. 2006, 24, 5652–5657. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-negative breast cancer: Molecular subtypes and new targets for therapy. Am. Soc. Clin. Oncol. Educ. Book ASCO Am. Soc. Clin. Oncol. Meet. 2015, 35, e31–e39. [Google Scholar] [CrossRef] [PubMed]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Belardi, V.; Gallagher, E.J.; Novosyadlyy, R.; LeRoith, D. Insulin and IGFs in obesity-related breast cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Clemmons, D.R.; Arteaga, C.L. Regulation of breast cancer growth by insulin-like growth factors. J. Steroid Biochem. Mol. Biol. 1990, 37, 805–809. [Google Scholar] [CrossRef]
- Richardson, A.E.; Hamilton, N.; Davis, W.; Brito, C.; De Leon, D. Insulin-like growth factor-2 (IGF-2) activates estrogen receptor-alpha and -beta via the IGF-1 and the insulin receptors in breast cancer cells. Growth Factors 2011, 29, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D.; Yee, D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol. Cancer Ther. 2007, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D.; Yee, D. The IGF system and breast cancer. Endocr. Relat. Cancer 2001, 8, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Moretta, D.; Almaguel, F.; Wall, N.R.; De Leon, M.; De Leon, D. Differential effect of proIGF-II and IGF-II on resveratrol induced cell death by regulating survivin cellular localization and mitochondrial depolarization in breast cancer cells. Growth Factors 2007, 25, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Khajah, M.A.; Al Saleh, S.; Mathew, P.M.; Luqmani, Y.A. Differential effect of growth factors on invasion and proliferation of endocrine resistant breast cancer cells. PLoS ONE 2012, 7, e41847. [Google Scholar] [CrossRef] [PubMed]
- Westley, B.R.; May, F.E. Role of insulin-like growth factors in steroid modulated proliferation. J. Steroid Biochem. Mol. Biol. 1994, 51, 1–9. [Google Scholar] [CrossRef]
- Kelley, K.M.; Oh, Y.; Gargosky, S.E.; Gucev, Z.; Matsumoto, T.; Hwa, V.; Ng, L.; Simpson, D.M.; Rosenfeld, R.G. Insulin-like growth factor-binding proteins (IGFBPs) and their regulatory dynamics. Int. J. Biochem. Cell Biol. 1996, 28, 619–637. [Google Scholar] [CrossRef]
- Lodhia, K.A.; Tienchaiananda, P.; Haluska, P. Understanding the Key to Targeting the IGF Axis in Cancer: A Biomarker Assessment. Front. Oncol. 2015, 5, 142. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C. IGF binding proteins in cancer: Mechanistic and clinical insights. Nat. Rev. Cancer 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.; Marquez-Garban, D.; Mah, V.; Fernando, G.; Elshimali, Y.; Garbán, H.; Elashoff, D.; Vadgama, J.; Goodglick, L.; Pietras, R. Biologic roles of estrogen receptor-beta and insulin-like growth factor-2 in triple-negative breast cancer. BioMed Res. Int. 2015, 2015, 925703. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A. IGFBP-6 five years on; not so ‘forgotten’? Growth Horm. IGF Res. 2005, 15, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin. Cancer Res. 2012, 18, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dawood, S.; Holmes, M.D.; Collins, L.C.; Schnitt, S.J.; Cole, K.; Marotti, J.D.; Hankinson, S.E.; Colditz, G.A.; Tamimi, R.M. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin. Cancer Res. 2011, 17, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Koo, J.; Park, H.S.; Kim, J.-H.; Choi, S.-Y.; Lee, J.H.; Park, B.-W.; Lee, K.S. Expression of androgen receptors in primary breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 2010, 21, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Fassan, M.; Cascione, L.; Guler, G.; Balci, S.; Irkkan, C.; Paisie, C.; Lovat, F.; Morrison, C.; Zhang, J.; et al. Androgen receptor status is a prognostic marker in non-basal triple negative breast cancers and determines novel therapeutic options. PLoS ONE 2014, 9, e88525. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.E.; Kang, S.H.; Lee, S.J.; Bae, Y.K. Androgen receptor expression predicts decreased survival in early stage triple-negative breast cancer. Ann. Surg. Oncol. 2015, 22, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Mrklic, I.; Pogorelic, Z.; Capkun, V.; Tomic, S. Expression of androgen receptors in triple negative breast carcinomas. Acta Histochem. 2013, 115, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Grogg, A.; Trippel, M.; Pfaltz, K.; Lädrach, C.; Droeser, R.A.; Cihoric, N.; Salhia, B.; Zweifel, M.; Tapia, C. Androgen receptor status is highly conserved during tumor progression of breast cancer. BMC Cancer 2015, 15, 872. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Lind, H.T.; Spoelstra, N.S.; Babbs, B.L.; Heinz, R.E.; Elias, A.; Jedlicka, P.; Jacobsen, B.M.; et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol. Cancer Ther. 2015, 14, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Palma, G.; Frasci, G.; Chirico, A.; Esposito, E.; Siani, C.; Saturnino, C.; Arra, C.; Ciliberto, G.; Giordano, A.; D’Aiuto, M. Triple negative breast cancer: Looking for the missing link between biology and treatments. Oncotarget 2015, 6, 26560–26574. [Google Scholar] [CrossRef] [PubMed]
- Rechoum, Y.; Rovito, D.; Iacopetta, D.; Barone, I.; Andò, S.; Weigel, N.L.; O’Malley, B.W.; Brown, P.H.; Fuqua, S.A.W. AR collaborates with ERalpha in aromatase inhibitor-resistant breast cancer. Breast Cancer Res. Treat. 2014, 147, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Minami, C.A.; Chung, D.U.; Chang, H.R. Management options in triple-negative breast cancer. Breast Cancer Basic Clin. Res. 2011, 5, 175–199. [Google Scholar]
- Khosravi-Shahi, P.; Cabezon-Gutierrez, L.; Custodio-Cabello, S. Metastatic triple negative breast cancer: Optimizing treatment options, new and emerging targeted therapies. Asia. Pac. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, N.; Azuma, M.; Ikarashi, M.; Yamamoto, M.; Sato, M.; Watanabe, K.; Yamashiro, K.; Takahashi, M. The therapeutic candidate for immune checkpoint inhibitors elucidated by the status of tumor-infiltrating lymphocytes (TILs) and programmed death ligand 1 (PD-L1) expression in triple negative breast cancer (TNBC). Breast Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Li, Y.; Song, W.; Xu, Y.; Yang, F.; Zhang, W.; Yin, Y.; Guan, X. Antiproliferative Effect of Androgen Receptor Inhibition in Mesenchymal Stem-Like Triple-Negative Breast Cancer. Cell. Physiol. Biochem. 2016, 38, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Lopez, M.D.; Montero, J.C.; Morales, J.C.; Prat, A.; Pandiella, A.; Ocana, A. Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer 2014, 14, 302. [Google Scholar] [CrossRef] [PubMed]
- Iams, W.T.; Lovly, C.M. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin. Cancer Res. 2015, 21, 4270–4277. [Google Scholar] [CrossRef] [PubMed]
- Pietras, R.J.; Marquez-Garban, D.C. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Gariboldi, M.B.; Taiana, E.; Bonzi, M.C.; Craparotta, I.; Pagin, M.; Monti, E. Co-targeting the IGF system and HIF-1 inhibits migration and invasion by (triple-negative) breast cancer cells. Br. J. Cancer 2014, 110, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, B.C.; Creighton, C.J.; Tsimelzon, A.; Chan, B.T.-Y.; Hilsenbeck, S.G.; Wang, T.; Carboni, J.M.; Gottardis, M.M.; Huang, F.; Chang, J.C.; et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin. Cancer Res. 2011, 17, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Ma, J.; Shao, N.; Shi, Y.; Liu, R.; Li, W.; Lin, Y.; Wang, S. Co-Targeting IGF-1R and Autophagy Enhances the Effects of Cell Growth Suppression and Apoptosis Induced by the IGF-1R Inhibitor NVP-AEW541 in Triple-Negative Breast Cancer Cells. PLoS ONE 2017, 12, e0169229. [Google Scholar] [CrossRef] [PubMed]
- Botti, G.; Collina, F.; Scognamiglio, G.; Rao, F.; Peluso, V.; De Cecio, R.; Piezzo, M.; Landi, G.; De Laurentiis, M.; Cantile, M.; et al. Programmed Death Ligand 1 (PD-L1) Tumor Expression Is Associated with a Better Prognosis and Diabetic Disease in Triple Negative Breast Cancer Patients. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Hirshfield, K.M.; Ganesan, S. Triple-negative breast cancer: Molecular subtypes and targeted therapy. Curr. Opin. Obstet. Gynecol. 2014, 26, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Thike, A.A.; Yong-Zheng Chong, L.; Cheok, P.Y.; Li, H.H.; Wai-Cheong Yip, G.; Huat Bay, B.; Tse, G.M.; Iqbal, J.; Tan, P.H. Loss of androgen receptor expression predicts early recurrence in triple-negative and basal-like breast cancer. Mod. Pathol. 2014, 27, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Shi, Y.X.; Li, Z.M.; Jiang, W.Q. Expression and clinical significance of androgen receptor in triple negative breast cancer. Chin. J. Cancer 2010, 29, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Xu, S.; Zhang, Q.; Zhao, W. The expression and clinical significance of the androgen receptor and E-cadherin in triple-negative breast cancer. Med. Oncol. 2012, 29, 526–533. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Peng, R.; Yuan, Z.; Wang, S.; Peng, J.; Lin, G.; Jiang, X.; Qin, T. Prognostic value of androgen receptor expression in operable triple-negative breast cancer: A retrospective analysis based on a tissue microarray. Med. Oncol. 2012, 29, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Christenson, J.L.; Elias, A.; Richer, J.K. Androgen Receptor Biology in Triple Negative Breast Cancer: A Case for Classification as AR+ or Quadruple Negative Disease. Horm. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Traina, T.A. Androgen receptor-positive, triple-negative breast cancer. Cancer 2017, 123, 1686–1688. [Google Scholar] [CrossRef] [PubMed]
- Schwartzberg, L.S.; Yardley, D.A.; Elias, A.D.; Patel, M.; LoRusso, P.; Burris, H.A.; Gucalp, A.; Peterson, A.C.; Blaney, M.E.; Steinberg, J.L.; et al. A Phase I/Ib Study of Enzalutamide Alone and in Combination with Endocrine Therapies in Women with Advanced Breast Cancer. Clin. Cancer Res. 2017, 23, 4046–4054. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Traina, T.A. Targeting the androgen receptor in triple-negative breast cancer. Curr. Probl. Cancer 2016, 40, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Parsons, K.; Mittal, D.; Lopez, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends. Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef] [PubMed]
- King, E.R.; Wong, K.K. Insulin-like growth factor: Current concepts and new developments in cancer therapy. Recent Anticancer Drug Discov. 2012, 7, 14–30. [Google Scholar] [CrossRef]
- Ma, C.X.; Suman, V.J.; Goetz, M.; Haluska, P.; Moynihan, T.; Nanda, R.; Olopade, O.; Pluard, T.; Guo, Z.; Chen, H.X.; et al. A phase I trial of the IGF-1R antibody Cixutumumab in combination with temsirolimus in patients with metastatic breast cancer. Breast Cancer Res. Treat. 2013, 139, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Pegram, M.; Konecny, G.; O’Callaghan, C.; Beryt, M.; Pietras, R.; Slamon, D.J. Rational combinations of trastuzumb with chemotherapeutic drugs used in the treatment of breast cancer. J. Natl. Cancer Inst. 2004, 96, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Jovanovic, B.; Sheng, Q.; Seitz, R.S.; Lawrence, K.D.; Morris, S.W.; Thomas, L.R.; Hout, D.R.; Schweitzer, B.L.; Guo, Y.; Pietenpol, J.A.; et al. Comparison of triple-negative breast cancer molecular subtyping using RNA from matched fresh-frozen versus formalin-fixed paraffin-embedded tissue. BMC Cancer 2017, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Sweet-Cordero, E.A. Two is better than one: Combining IGF1R and MEK blockade as a promising novel treatment strategy against KRAS-mutant lung cancer. Cancer Discov. 2013, 3, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar] [PubMed]
- Tominaga, K.; Shimamura, T.; Kimura, N.; Murayama, T.; Matsubara, D.; Kanauchi, H.; Niida, A.; Shimizu, S.; Nishioka, K.; Tsuji, E.; et al. Addiction to the IGF2-ID1-IGF2 circuit for maintenance of the breast cancer stem-like cells. Oncogene 2017, 36, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; Christenson, J.L.; Gordon, M.A.; Greene, L.I.; Rogers, T.J.; Butterfield, K.; Babbs, B.; Spoelstra, N.S.; D’Amato, N.C.; Elias, A.; et al. Androgen Receptor Supports an Anchorage-Independent, Cancer Stem Cell-like Population in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3455–3466. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, T.B.; Tomblin, J.K. Insulin/Insulin-like growth factors in cancer: New roles for the aryl hydrocarbon receptor, tumor resistance mechanisms, and new blocking strategies. Front. Endocrinol. 2015, 6, 12. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamilton, N.; Austin, D.; Márquez-Garbán, D.; Sanchez, R.; Chau, B.; Foos, K.; Wu, Y.; Vadgama, J.; Pietras, R. Receptors for Insulin-Like Growth Factor-2 and Androgens as Therapeutic Targets in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2017, 18, 2305. https://doi.org/10.3390/ijms18112305
Hamilton N, Austin D, Márquez-Garbán D, Sanchez R, Chau B, Foos K, Wu Y, Vadgama J, Pietras R. Receptors for Insulin-Like Growth Factor-2 and Androgens as Therapeutic Targets in Triple-Negative Breast Cancer. International Journal of Molecular Sciences. 2017; 18(11):2305. https://doi.org/10.3390/ijms18112305
Chicago/Turabian StyleHamilton, Nalo, David Austin, Diana Márquez-Garbán, Rudy Sanchez, Brittney Chau, Kay Foos, Yanyuan Wu, Jaydutt Vadgama, and Richard Pietras. 2017. "Receptors for Insulin-Like Growth Factor-2 and Androgens as Therapeutic Targets in Triple-Negative Breast Cancer" International Journal of Molecular Sciences 18, no. 11: 2305. https://doi.org/10.3390/ijms18112305
APA StyleHamilton, N., Austin, D., Márquez-Garbán, D., Sanchez, R., Chau, B., Foos, K., Wu, Y., Vadgama, J., & Pietras, R. (2017). Receptors for Insulin-Like Growth Factor-2 and Androgens as Therapeutic Targets in Triple-Negative Breast Cancer. International Journal of Molecular Sciences, 18(11), 2305. https://doi.org/10.3390/ijms18112305