UltraPse: A Universal and Extensible Software Platform for Representing Biological Sequences


Abstract
:
1. Introduction
2. Results and Discussion
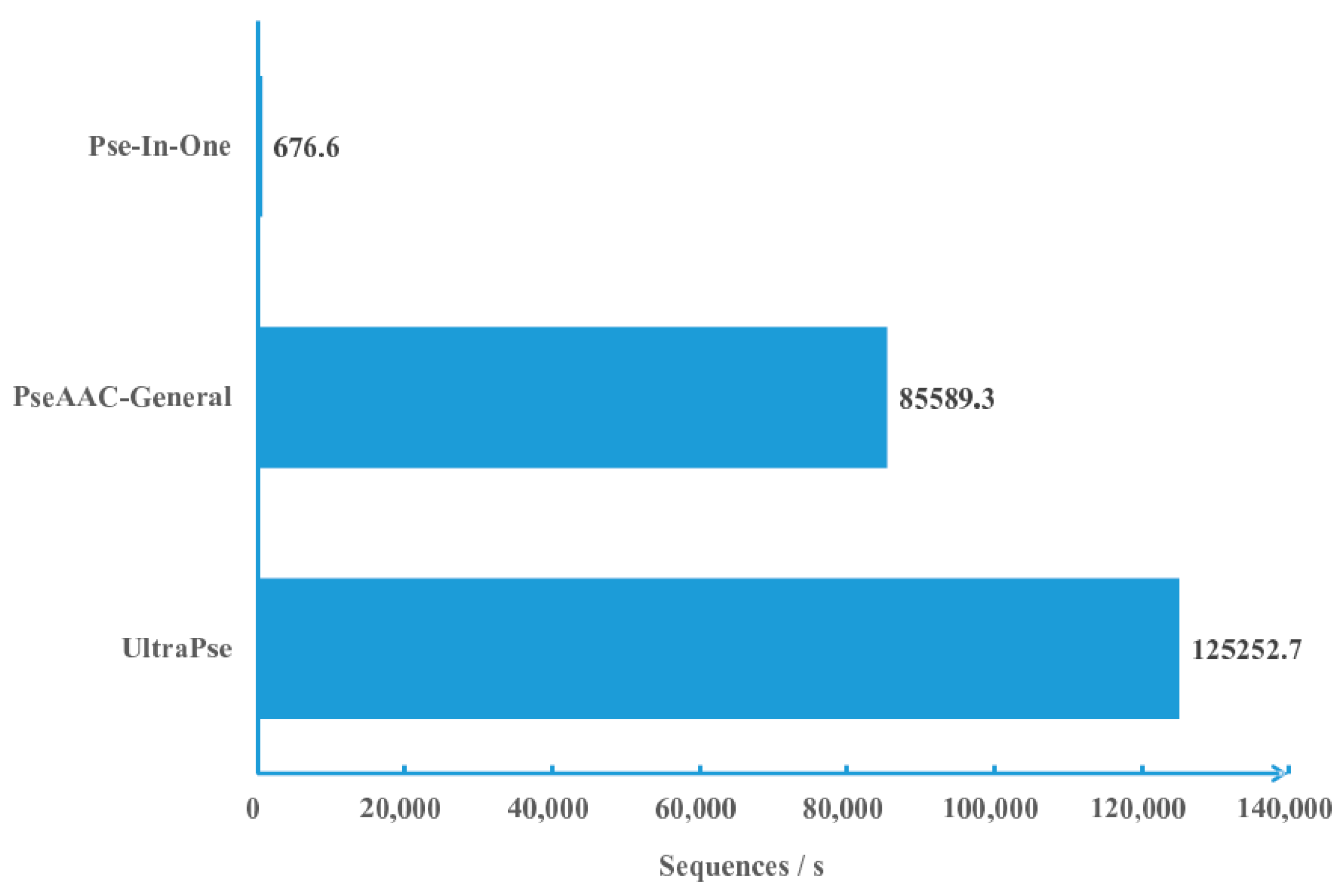
2.1. Computational Efficiency Analysis
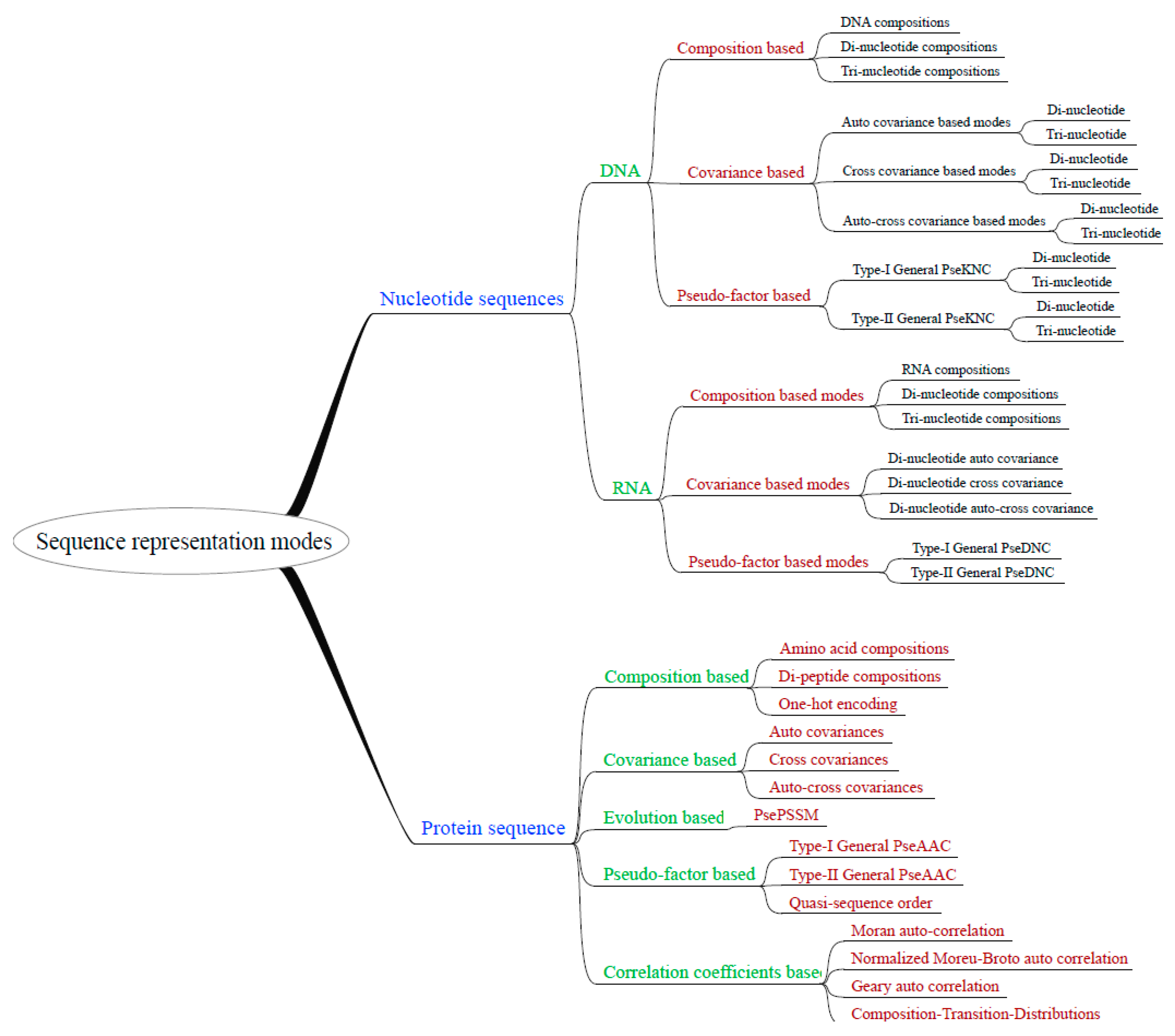
2.2. Flexibility and Extensibility
2.3. Compatibility and Robustness
2.4. Technical Detail Comparison
2.5. Future Works in Plan
2.6. Availability
3 Methods
3.1. Efficiency Comparison Protocols
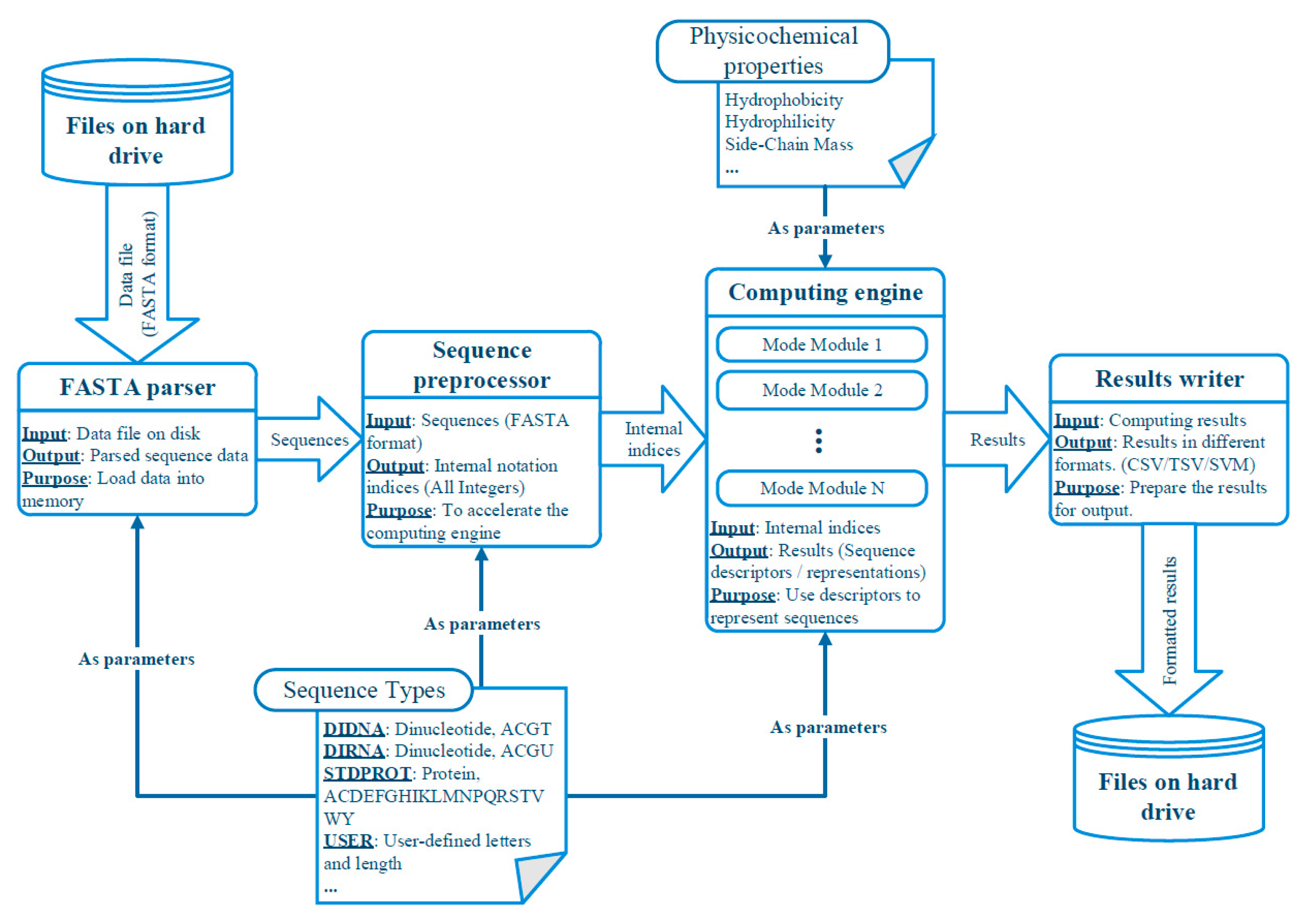
3.2. Abstracted Software Design
3.3. Implementation Technology
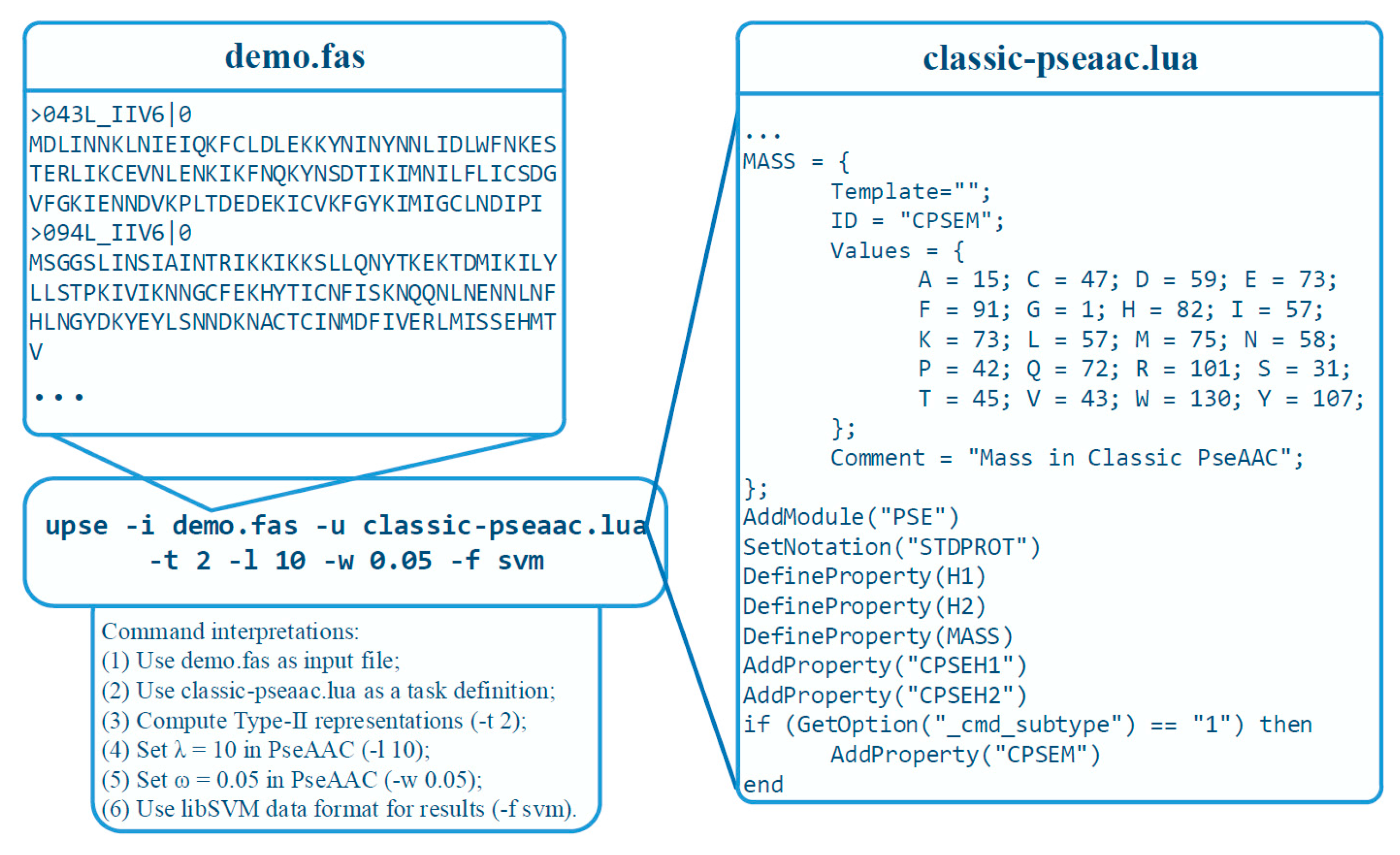
3.4. A Practical Example
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AmPseAAC | Amphiphilic pseudo amino acid composition |
| BEM | Binary extension module |
| BSO | Binary shared objects |
| GPL | General public license |
| GUI | Graphical user interfaces |
| PseAAC | Pseudo-amino acid composition |
| PseDNC | Pseudo-dinucleotide composition |
| PseKNC | Pseudo-k nucleotide composition |
| TDF | Task definition file |
References
- Jiao, Y.-S.; Du, P.-F. Predicting Golgi-resident protein types using pseudo amino acid compositions: Approaches with positional specific physicochemical properties. J. Theor. Biol. 2016, 391, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.-S.; Du, P.-F. Predicting protein submitochondrial locations by incorporating the positional-specific physicochemical properties into Chou’s general pseudo-amino acid compositions. J. Theor. Biol. 2017, 416, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Brahnam, S.; Lumini, A. High performance set of PseAAC and sequence based descriptors for protein classification. J. Theor. Biol. 2010, 266, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Brahnam, S.; Lumini, A. Prediction of protein structure classes by incorporating different protein descriptors into general Chou’s pseudo amino acid composition. J. Theor. Biol. 2014, 360, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, S.; Xiao, W.; Li, Y.; Hu, W.; Huang, L.; Zheng, X.; Zhou, S.; Yang, H. Protein submitochondrial localization from integrated sequence representation and SVM-based backward feature extraction. Mol. Biosyst. 2014, 11, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Chen, W.; Yuan, L.-F.; Li, Z.-Q.; Ding, H. Using Over-Represented Tetrapeptides to Predict Protein Submitochondria Locations. Acta Biotheor 2013, 61, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.-C.; Peng, Y.; Liu, L.; Chen, W.; Yang, L.; Fan, G.-L. Predicting peroxidase subcellular location by hybridizing different descriptors of Chou’ pseudo amino acid patterns. Anal. Biochem. 2014, 458, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A.; Gupta, D.; Garg, A. Identifying Bacterial Virulent Proteins by Fusing a Set of Classifiers Based on Variants of Chou’s Pseudo Amino Acid Composition and on Evolutionary Information. IEEE-ACM Trans. Comput. Biol. Bioinform. 2012, 9, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Pseudo Amino Acid Composition and its Applications in Bioinformatics, Proteomics and System Biology. Curr. Proteom. 2009, 6, 262–274. [Google Scholar] [CrossRef]
- Qiu, W.-R.; Sun, B.-Q.; Xiao, X.; Xu, Z.-C.; Chou, K.-C. iHyd-PseCp: Identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget 2016, 7, 44310–44321. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, Y.-X.; Ding, J.; Wu, L.-Y.; Deng, N.-Y. Phogly–PseAAC: Prediction of lysine phosphoglycerylation in proteins incorporating with position-specific propensity. J. Theor. Biol. 2015, 379, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhang, L.; Liu, Z.; Xiao, X.; Chou, K.-C. pSumo-CD: Predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics 2016, 32, 3133–3141. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Waris, M.; Hayat, M. Prediction of Protein Submitochondrial Locations by Incorporating Dipeptide Composition into Chou’s General Pseudo Amino Acid Composition. J. Membr. Biol. 2016, 249, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.-M.; Chen, W.; Lin, H.; Chou, K.-C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013, 442, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-Z.; Fang, J.-A.; Xiao, X.; Chou, K.-C. iLoc-Animal: A multi-label learning classifier for predicting subcellular localization of animal proteins. Mol. Biosyst. 2013, 9, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Mohabatkar, H.; Mohammad Beigi, M.; Esmaeili, A. Prediction of GABAA receptor proteins using the concept of Chou’s pseudo-amino acid composition and support vector machine. J. Theor. Biol. 2011, 281, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.-S.; Du, P.-F. Prediction of Golgi-resident protein types using general form of Chou’s pseudo-amino acid compositions: Approaches with minimal redundancy maximal relevance feature selection. J. Theor. Biol. 2016, 402, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Wang, L. Predicting human protein subcellular locations by the ensemble of multiple predictors via protein-protein interaction network with edge clustering coefficients. PLoS ONE 2014, 9, e86879. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Some remarks on predicting multi-label attributes in molecular biosystems. Mol. Biosyst. 2013, 9, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Impacts of bioinformatics to medicinal chemistry. Med. Chem. 2015, 11, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, H.; Chou, K.-C. Pseudo nucleotide composition or PseKNC: An effective formulation for analyzing genomic sequences. Mol. Biosyst. 2015, 11, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.-M.; Lin, H.; Chou, K.-C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic. Acids Res. 2013, 41, e68. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.-M.; Lin, H.; Chou, K.-C. iSS-PseDNC: Identifying splicing sites using pseudo dinucleotide composition. Biomed. Res. Int. 2014, 2014, 623149. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.-M.; Deng, E.-Z.; Lin, H.; Chou, K.-C. iTIS-PseTNC: A sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal. Biochem. 2014, 462, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-H.; Deng, E.-Z.; Xu, L.-Q.; Ding, H.; Lin, H.; Chen, W.; Chou, K.-C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 2014, 30, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Deng, E.-Z.; Ding, H.; Chen, W.; Chou, K.-C. iPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014, 42, 12961–12972. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J.; Chen, W.; Feng, P.; Ding, H.; Lin, H.; Chou, K.-C. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015, 490, 26–33. [Google Scholar] [CrossRef]
- Liu, B.; Fang, L.; Liu, F.; Wang, X.; Chou, K.-C. iMiRNA-PseDPC: MicroRNA precursor identification with a pseudo distance-pair composition approach. J. Biomol. Struct. Dyn. 2016, 34, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, H.; Ye, J.; Lin, H.; Chou, K.-C. iRNA-PseU: Identifying RNA pseudouridine sites. Mol. Ther. Nucleic Acids 2016, 5, e332. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Long, R.; Chou, K.-C. iDHS-EL: Identifying DNase I hypersensitive sites by fusing three different modes of pseudo nucleotide composition into an ensemble learning framework. Bioinformatics 2016, 32, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, F.; Huang, D.-S.; Chou, K.-C. iPromoter-2L: A two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Hayat, M. “iSS-Hyb-mRMR”: Identification of splicing sites using hybrid space of pseudo trinucleotide and pseudo tetranucleotide composition. Comput. Methods Programs Biomed. 2016, 128, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.; Iqbal, M.; Ahmad, S.; Hayat, M. iTIS-PseKNC: Identification of Translation Initiation Site in human genes using pseudo k-tuple nucleotides composition. Comput. Biol. Med. 2015, 66, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, J.-W.; Liu, Z.; Ren, M.-W.; Shen, H.-B.; Yu, D.-J. Improving N(6)-methyladenosine site prediction with heuristic selection of nucleotide physical-chemical properties. Anal. Biochem. 2016, 508, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, Y.-Z.; Zhang, F.-Z.; Hua, H.-L.; Ye, Y.-N.; Labena, A.A.; Lin, H.; Chen, W.; Guo, F.-B. Combining pseudo dinucleotide composition with the Z curve method to improve the accuracy of predicting DNA elements: A case study in recombination spots. Mol. Biosyst. 2016, 12, 2893–2900. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, Y.; Huang, D. Recombination Hotspot/Coldspot Identification Combining Three Different Pseudocomponents via an Ensemble Learning Approach. Biomed. Res. Int. 2016, 2016, 8527435. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-R.; Jiang, S.-Y.; Xu, Z.-C.; Xiao, X.; Chou, K.-C. iRNAm5C-PseDNC: Identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget 2017, 8, 41178–41188. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-C.; Wang, P.; Qiu, W.-R.; Xiao, X. iSS-PC: Identifying Splicing Sites via Physical-Chemical Properties Using Deep Sparse Auto-Encoder. Sci. Rep. 2017, 7, 8222. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.R.; Lin, H.H.; Han, L.Y.; Jiang, L.; Chen, X.; Chen, Y.Z. PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006, 34, W32–W37. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-B.; Chou, K.-C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.-S.; Xu, Q.-S.; Liang, Y.-Z. Propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Wang, X.; Xu, C.; Gao, Y. PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou’s pseudo-amino acid compositions. Anal. Biochem. 2012, 425, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Gu, S.; Jiao, Y. PseAAC-General: Fast building various modes of general form of Chou’s pseudo-amino acid composition for large-scale protein datasets. Int. J. Mol. Sci. 2014, 15, 3495–3506. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Cao, D.-S.; Zhu, M.-F.; Xu, Q.-S. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lei, T.-Y.; Jin, D.-C.; Lin, H.; Chou, K.-C. PseKNC: A flexible web server for generating pseudo K-tuple nucleotide composition. Anal. Biochem. 2014, 456, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, X.; Brooker, J.; Lin, H.; Zhang, L.; Chou, K.-C. PseKNC-General: A cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics 2015, 31, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Fang, L.; Wang, X.; Chou, K.-C. repDNA: A Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics 2015, 31, 1307–1309. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Fang, L.; Wang, X.; Chou, K.-C. repRNA: A web server for generating various feature vectors of RNA sequences. Mol. Genet. Genom. 2016, 291, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.-C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Du, P.; Xu, N. Identifying human kinase-specific protein phosphorylation sites by integrating heterogeneous information from various sources. PLoS ONE 2010, 5, e15411. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Y.; Tang, J.; Du, P.-F. Predicting protein lysine phosphoglycerylation sites by hybridizing many sequence based features. Mol. Biosyst. 2017, 13, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.-C.; Tang, J.; Du, P.-F. Predicting S-sulfenylation Sites Using Physicochemical Properties Differences. Lett. Org. Chem. 2017, 14, 665–672. [Google Scholar] [CrossRef]
- Steinbiss, S.; Gremme, G.; Schärfer, C.; Mader, M.; Kurtz, S. AnnotationSketch: A genome annotation drawing library. Bioinformatics 2009, 25, 533–534. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.-C. iCar-PseCp: Identify carbonylation sites in proteins by Monte Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget 2016, 7, 34558–34570. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-R.; Xiao, X.; Lin, W.-Z.; Chou, K.-C. iMethyl-PseAAC: Identification of protein methylation sites via a pseudo amino acid composition approach. Biomed. Res. Int. 2014, 2014, 947416. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Lan, X.; Xu, R.; Zhou, J.; Wang, X.; Chou, K.-C. iDNA-Prot|dis: Identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS ONE 2014, 9, e106691. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wen, X.; Wen, L.-S.; Wu, L.-Y.; Deng, N.-Y.; Chou, K.-C. iNitro-Tyr: Prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS ONE 2014, 9, e105018. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software Functions | Sequence Types | Extensibility |
|---|---|---|
| UltraPse | DNA, RNA, Protein, User-defined types | Users can define their own sequence types, representation modes and physicochemical properties |
| PseAAC-General [46] | Protein | Users can define their own representation modes |
| PseAAC-Builder [45] | Protein | No extensibility |
| Pse-In-One [52] | DNA, RNA, Protein | Users can define their own physicochemical properties |
| PseKNC [48] | DNA, RNA | Users can define their own physicochemical properties |
| PseKNC-General [49] | DNA, RNA | Users can define their own physicochemical properties |
| Software | Output Formats | Input Formats | Data Fault Tolerant a |
|---|---|---|---|
| UltraPse | SVM b, TSV c, CSV d | Multi-line FASTA (Automatic ID recognition for UniProt, GenBank, EMBL, DDBJ and RefSeq) | User-controllable behavior on data faults |
| PseAAC-General [46] | SVM, TSV, CSV | Single-line FASTA (With restrictions on comment line) e | Automatically ignore and report data faults |
| PseAAC-Builder [45] | SVM, TSV, CSV | Single-line FASTA (With restrictions on comment line) | Automatically ignore and report data faults |
| Pse-In-One [52] | SVM, TSV, CSV | Mutlti-line FASTA | Abort processing on data faults |
| PseKNC [48] | SVM, TSV, CSV | Mutlti-line FASTA | Abort processing on data faults |
| PseKNC-General [49] | SVM, TSV, CSV | Mutlti-line FASTA | Abort processing on data faults |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, P.-F.; Zhao, W.; Miao, Y.-Y.; Wei, L.-Y.; Wang, L. UltraPse: A Universal and Extensible Software Platform for Representing Biological Sequences. Int. J. Mol. Sci. 2017, 18, 2400. https://doi.org/10.3390/ijms18112400
Du P-F, Zhao W, Miao Y-Y, Wei L-Y, Wang L. UltraPse: A Universal and Extensible Software Platform for Representing Biological Sequences. International Journal of Molecular Sciences. 2017; 18(11):2400. https://doi.org/10.3390/ijms18112400
Chicago/Turabian StyleDu, Pu-Feng, Wei Zhao, Yang-Yang Miao, Le-Yi Wei, and Likun Wang. 2017. "UltraPse: A Universal and Extensible Software Platform for Representing Biological Sequences" International Journal of Molecular Sciences 18, no. 11: 2400. https://doi.org/10.3390/ijms18112400
APA StyleDu, P. -F., Zhao, W., Miao, Y. -Y., Wei, L. -Y., & Wang, L. (2017). UltraPse: A Universal and Extensible Software Platform for Representing Biological Sequences. International Journal of Molecular Sciences, 18(11), 2400. https://doi.org/10.3390/ijms18112400