Glucocorticoids Improve Myogenic Differentiation In Vitro by Suppressing the Synthesis of Versican, a Transitional Matrix Protein Overexpressed in Dystrophic Skeletal Muscles

 ,
,
Abstract
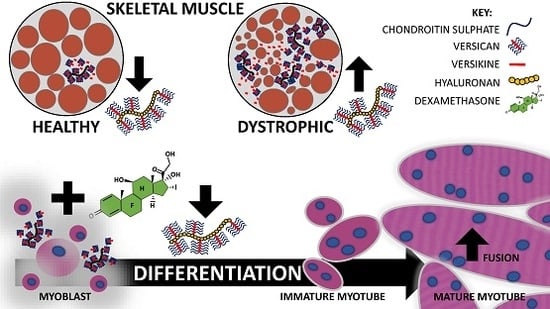
:
1. Introduction
2. Results
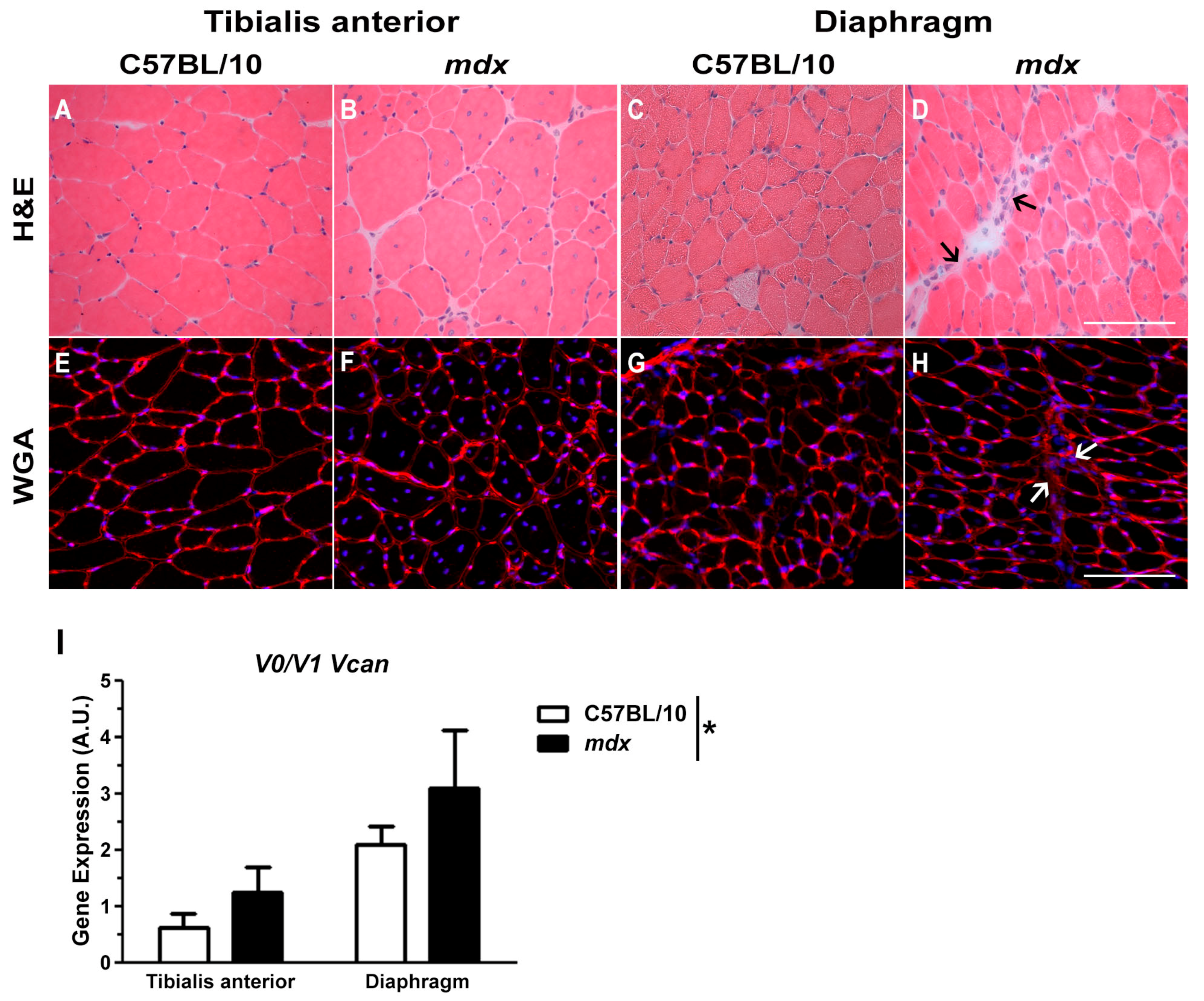
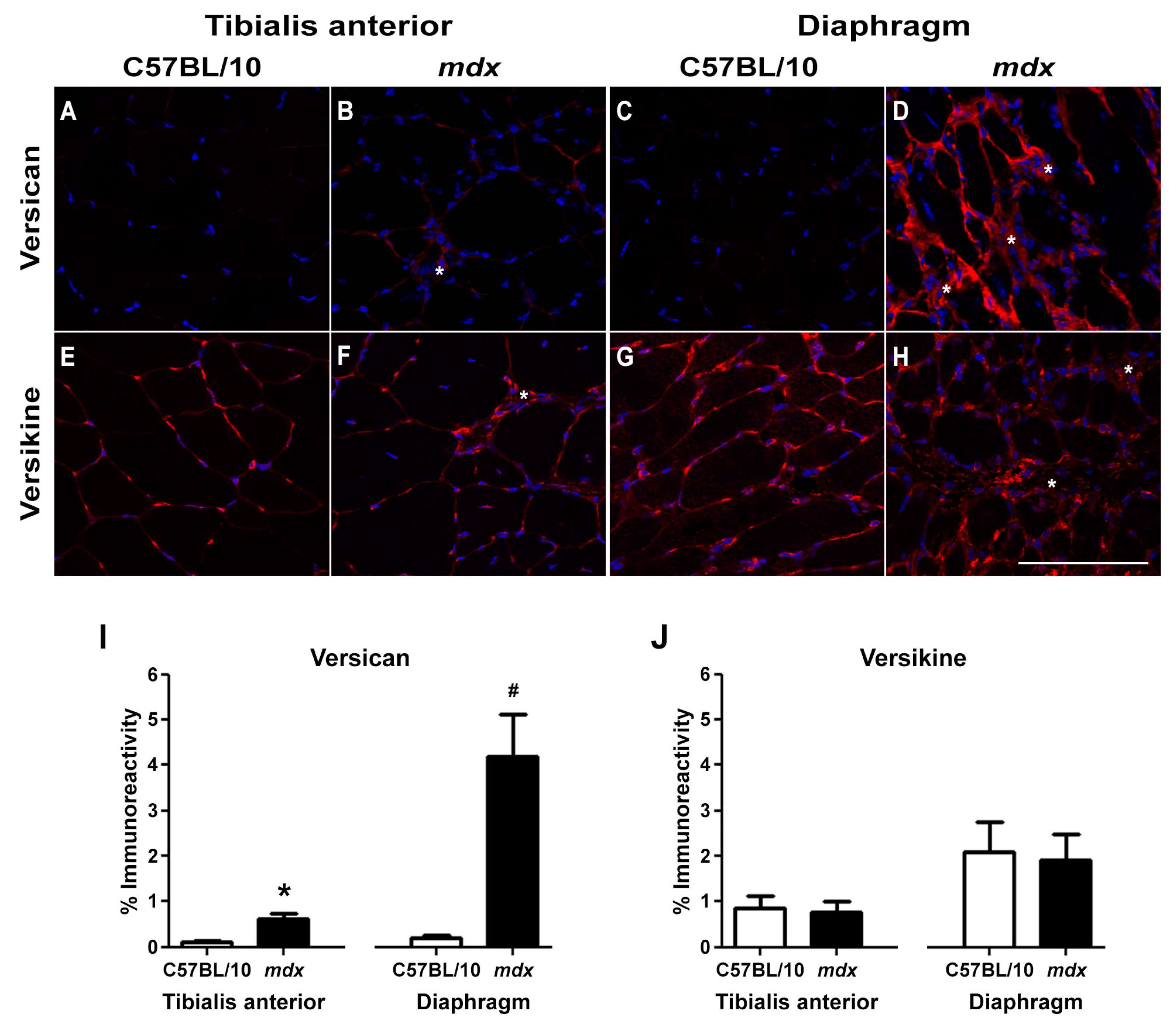
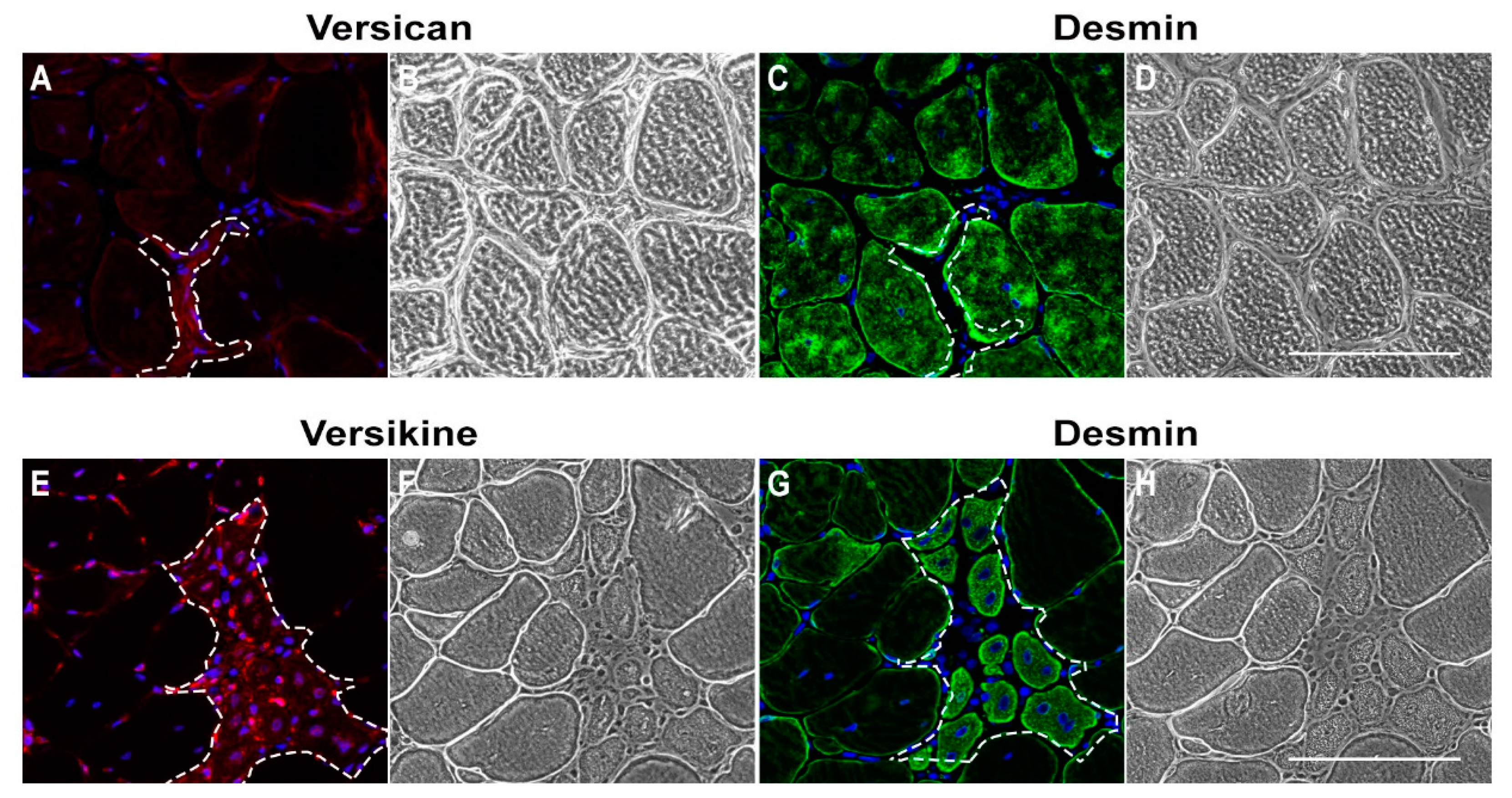
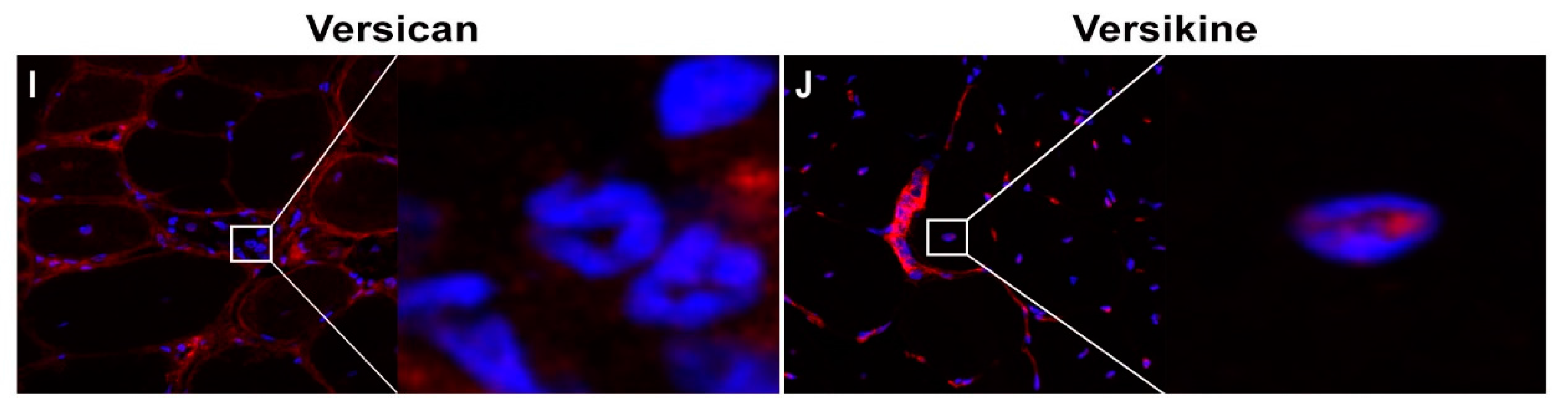
2.1. Versican Expression Is Increased in Dystrophic Muscles and Correlate with the Severity of Pathology
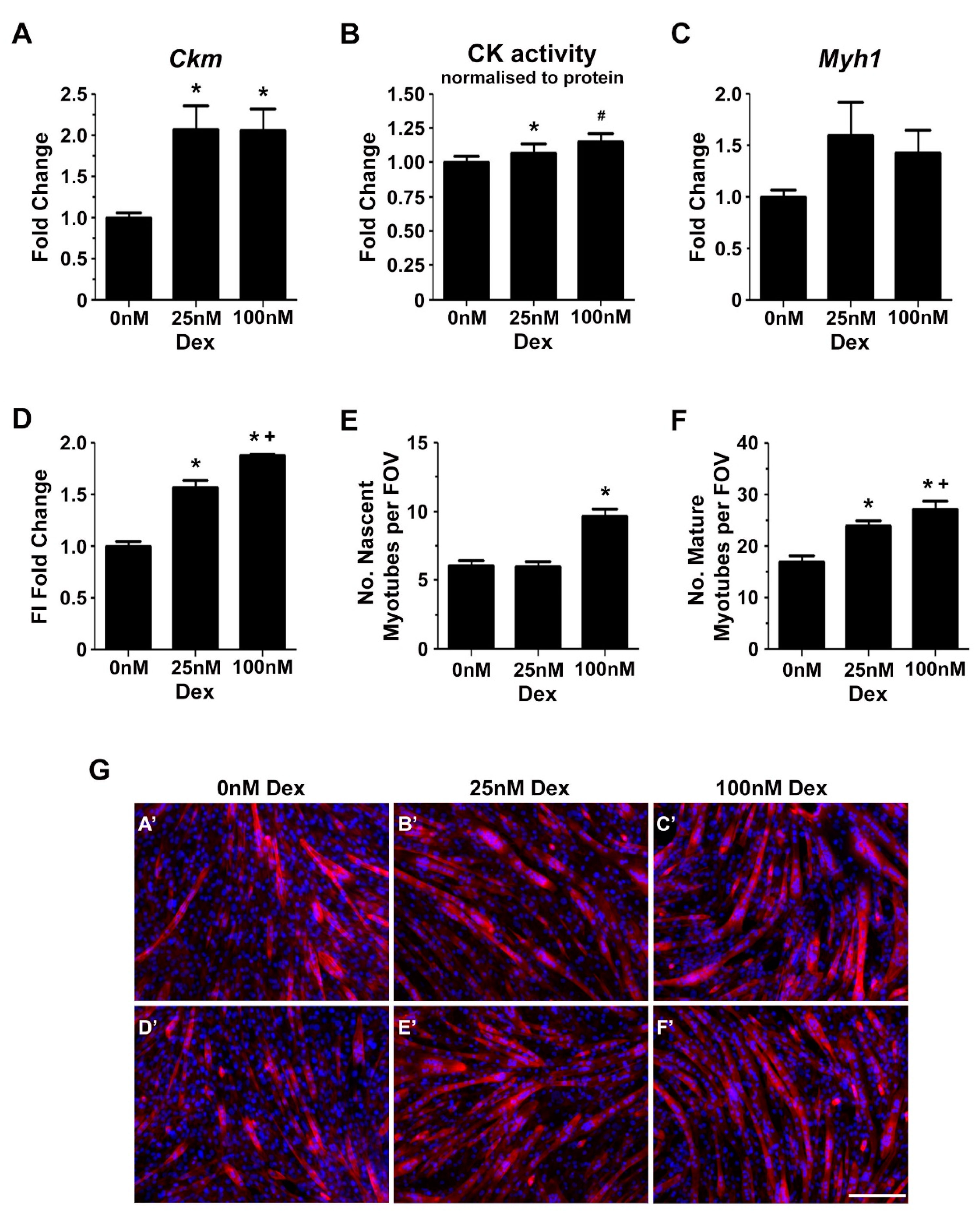
2.2. Glucocorticoids Enhance Myoblast Fusion and Myotube Formation
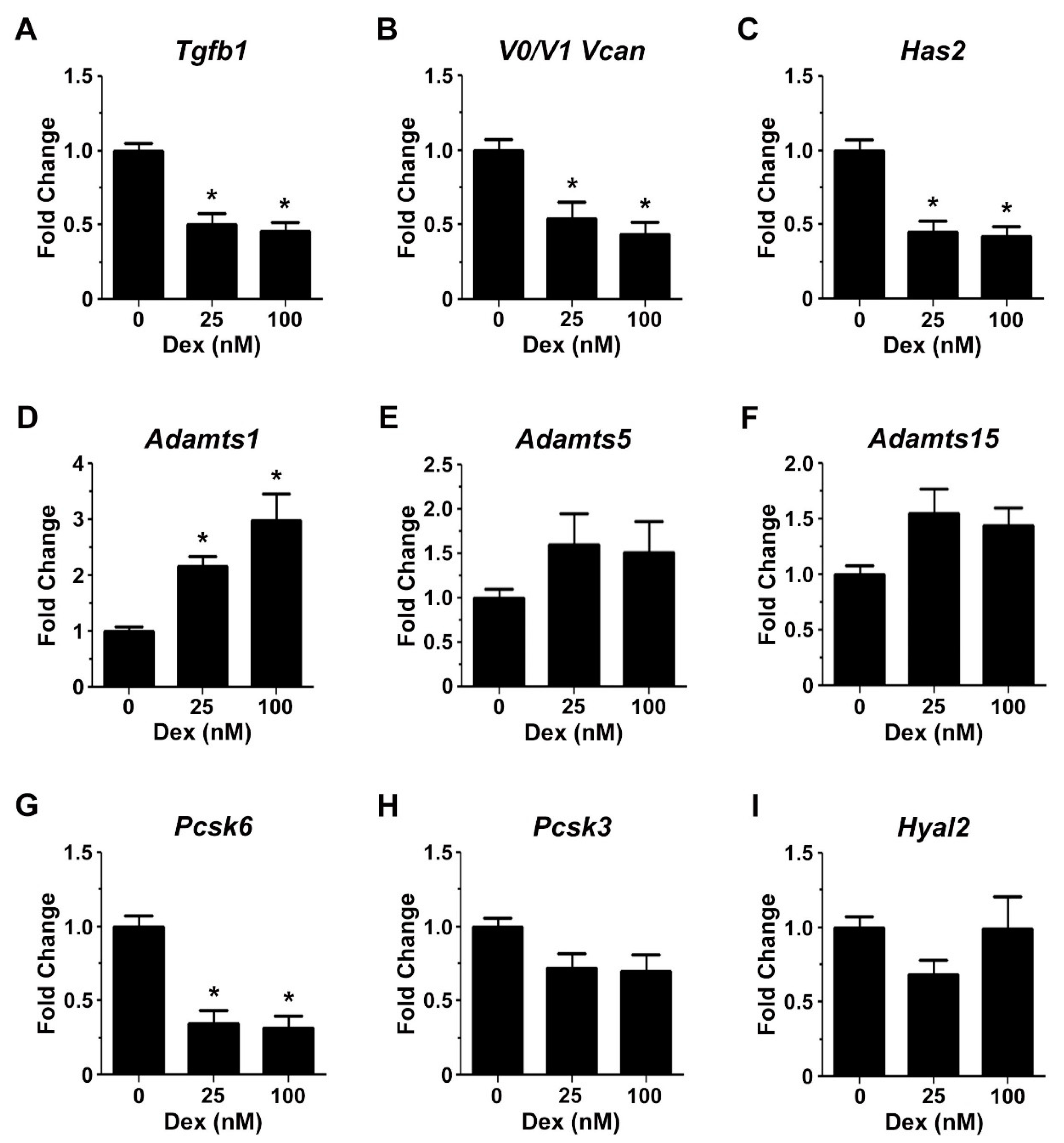
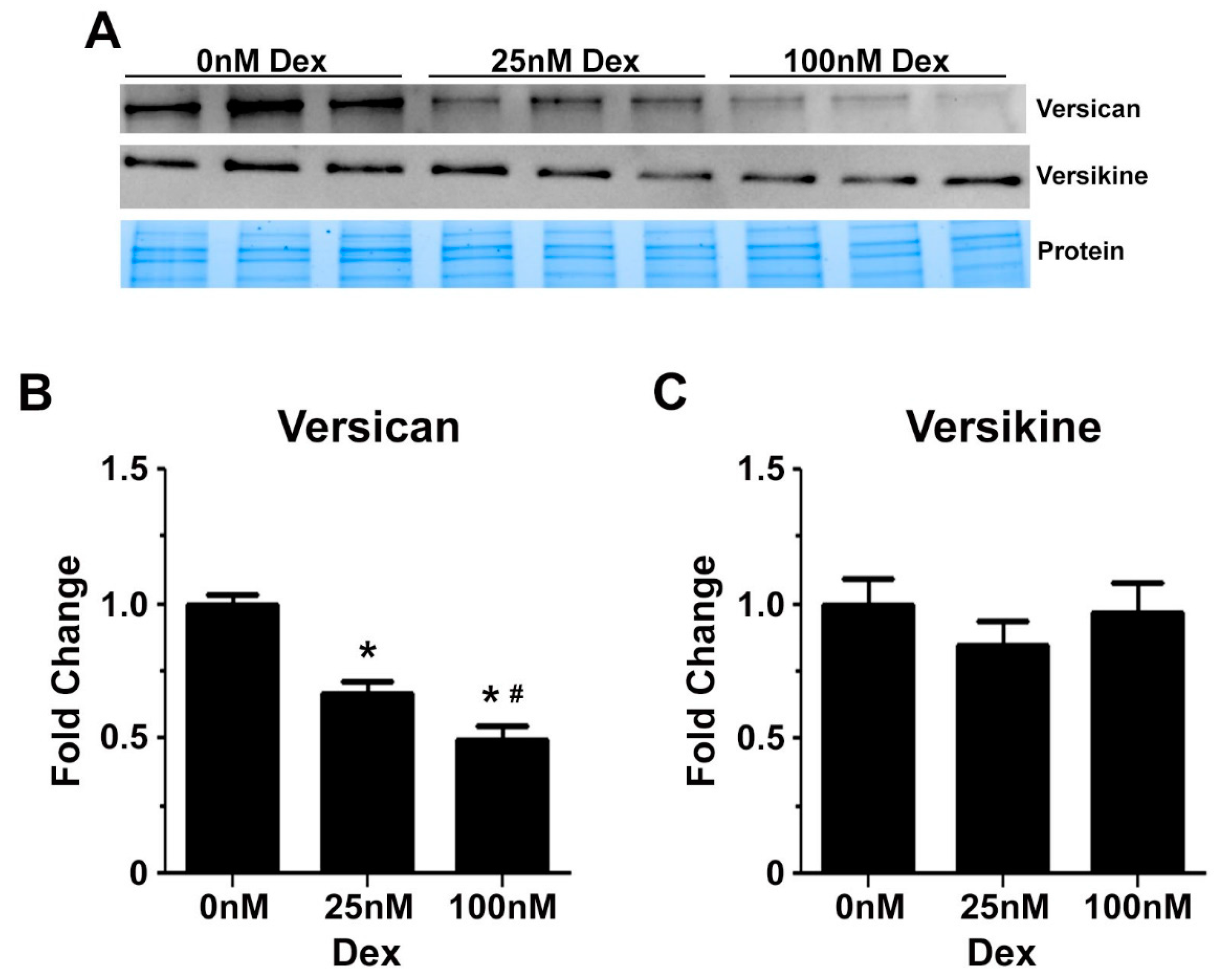
2.3. Glucocorticoids Regulate the Expression of Genes Associated with Transitional Matrix Synthesis and Processing during Myogenic Differentiation
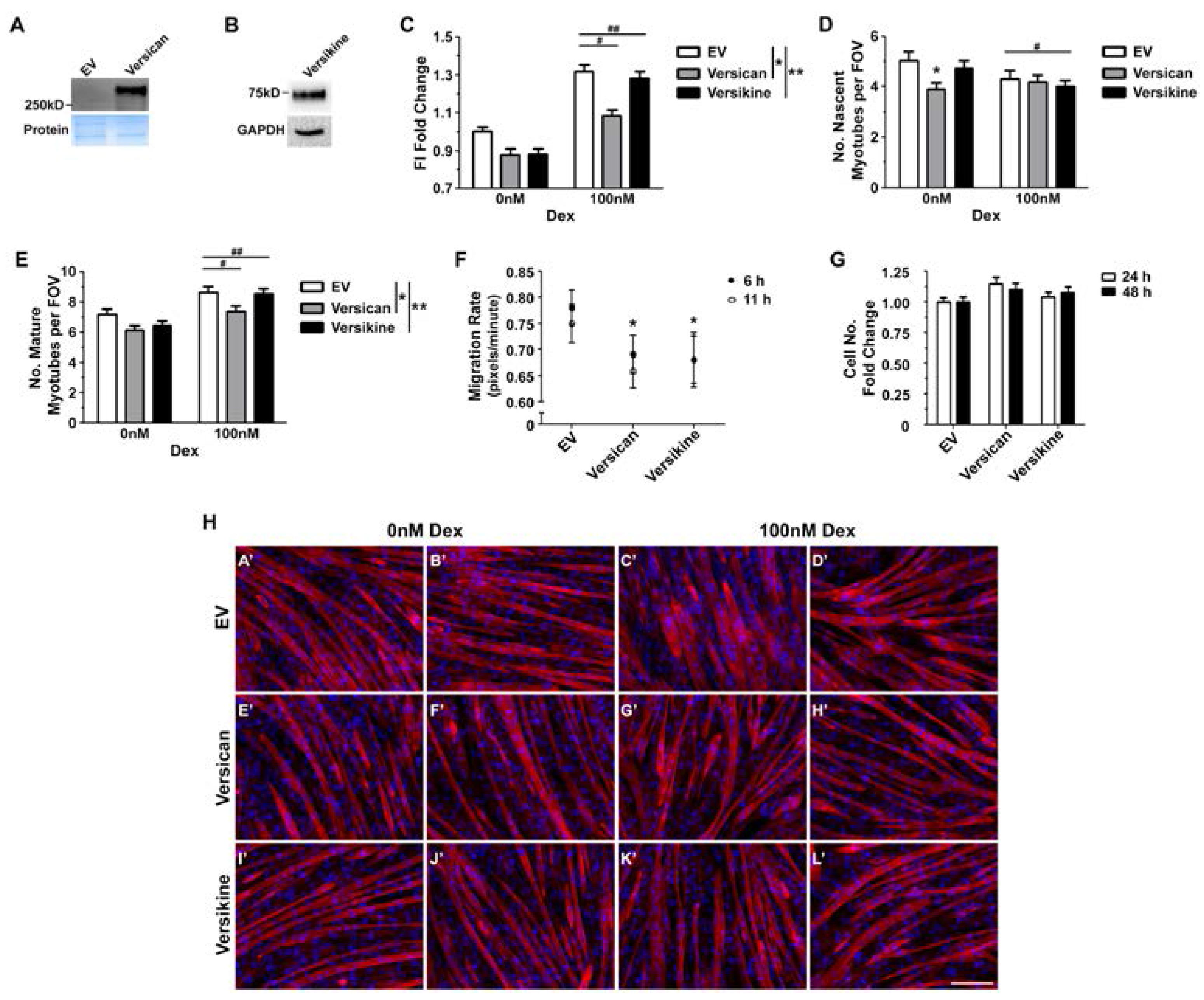
2.4. Glucocorticoids Rescue Myotube Formation in Differentiating Myoblasts Treated with Exogenous Versican and Versikine
3. Discussion
4. Materials and Methods
4.1. Mouse Models
4.2. Skeletal Muscle Immunohistochemistry and Histology
4.3. Cell Culture and Expression Constructs
4.4. Glucocorticoid Treatment of Differentiating C2C12 Cells
4.5. RNA Extraction, Reverse Transcription, and Quantitative RT-PCR
4.6. Versican or Versikine Treatment and C2C12 Myoblast Differentiation
4.7. Versican or Versikine Treatment and C2C12 Myoblast Migration and Proliferation
4.8. Western Blot
4.9. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADAMTS | A disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats |
| CSPG | Chondroitin sulphate proteoglycan |
| CK | Creatine kinase |
| Ckm | Creatine kinase muscle |
| Dex | Dexamethasone |
| DMEM | Dulbecco’s modified eagle medium |
| DMD | Duchenne muscular dystrophy |
| ECM | Extracellular matrix |
| EV | Empty vector |
| FI | Fusion index |
| FOV | Field of view |
| GAG | Glycosaminoglycan |
| GLM | General linear model |
| GSK-3β | Glycogen synthase kinase-3β |
| Has | Hyaluronan synthase |
| HS | Horse serum |
| Hyal | Hyaluronidase |
| Myh1 | Myosin heavy chain 1 |
| OCT | Optimum cutting temperature |
| TA | Tibialis anterior |
| TGF-β | Transforming growth factor-β |
| Vcan | Versican |
References
- Beytia, M.; Vry, J.; Kirschner, J. Drug treatment of duchenne muscular dystrophy: Available evidence and perspectives. Acta Myol. 2012, 31, 4–8. [Google Scholar]
- Gumerson, J.D.; Michele, D.E. The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011, 2011, 210797. [Google Scholar] [CrossRef] [PubMed]
- Duance, V.C.; Stephens, H.R.; Dunn, M.; Bailey, A.J.; Dubowitz, V. A role for collagen in the pathogenesis of muscular dystrophy? Nature 1980, 284, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lu, H. Targeting fibrosis in duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Carvajal Monroy, P.L.; Grefte, S.; Kuijpers-Jagtman, A.M.; Helmich, M.P.; Wagener, F.A.; von den Hoff, J.W. Fibrosis impairs the formation of new myofibers in the soft palate after injury. Wound Repair Regen. 2015, 23, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Calve, S.; Simon, H.G. Biochemical and mechanical environment cooperatively regulate skeletal muscle regeneration. FASEB J. 2012, 26, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Calve, S.; Odelberg, S.J.; Simon, H.G. A transitional extracellular matrix instructs cell behavior during muscle regeneration. Dev. Biol. 2010, 344, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Stupka, N.; Kintakas, C.; White, J.D.; Fraser, F.W.; Hanciu, M.; Aramaki-Hattori, N.; Martin, S.; Coles, C.; Collier, F.; Ward, A.C.; et al. Versican processing by a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats proteinases-5 and -15 facilitates myoblast fusion. J. Biol. Chem. 2013, 288, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’Amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Marotta, M.; Ruiz-Roig, C.; Sarria, Y.; Peiro, J.L.; Nunez, F.; Ceron, J.; Munell, F.; Roig-Quilis, M. Muscle genome-wide expression profiling during disease evolution in mdx mice. Physiol. Genom. 2009, 37, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, P.; Torchiana, E.; Confalonieri, P.; Brugnoni, R.; Barresi, R.; Mora, M.; Cornelio, F.; Morandi, L.; Mantegazza, R. Expression of transforming growth factor-β 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J. Clin. Investig. 1995, 96, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Ishitobi, M.; Haginoya, K.; Zhao, Y.; Ohnuma, A.; Minato, J.; Yanagisawa, T.; Tanabu, M.; Kikuchi, M.; Iinuma, K. Elevated plasma levels of transforming growth factor β1 in patients with muscular dystrophy. Neuroreport 2000, 11, 4033–4035. [Google Scholar] [CrossRef] [PubMed]
- Negroni, E.; Henault, E.; Chevalier, F.; Gilbert-Sirieix, M.; Van Kuppevelt, T.H.; Papy-Garcia, D.; Uzan, G.; Albanese, P. Glycosaminoglycan modifications in duchenne muscular dystrophy: Specific remodeling of chondroitin sulfate/dermatan sulfate. J. Neuropathol. Exp. Neurol. 2014, 73, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Stephens, H.R.; Duance, V.C.; Dunn, M.J.; Bailey, A.J.; Dubowitz, V. Collagen types in neuromuscular diseases. J. Neurol. Sci. 1982, 53, 45–62. [Google Scholar] [CrossRef]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar] [PubMed]
- Wight, T.N. Provisional matrix: A role for versican and hyaluronan. Matrix Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nandadasa, S.; Foulcer, S.; Apte, S.S. The multiple, complex roles of versican and its proteolytic turnover by ADAMTS proteases during embryogenesis. Matrix Biol. 2014, 35, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mittal, A.; Makonchuk, D.Y.; Bhatnagar, S.; Kumar, A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 2584–2598. [Google Scholar] [CrossRef] [PubMed]
- Macri, L.; Silverstein, D.; Clark, R.A. Growth factor binding to the pericellular matrix and its importance in tissue engineering. Adv. Drug Deliv. Rev. 2007, 59, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Kinsella, M.G.; Evanko, S.P.; Potter-Perigo, S.; Merrilees, M.J. Versican and the regulation of cell phenotype in disease. Biochim. Biophys. Acta 2014, 1840, 2441–2451. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Mikami, T.; Uyama, T.; Mizuguchi, S.; Nomura, K.; Kitagawa, H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 2003, 13, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.E.; Sun, Y.Y.; Vranka, J.A.; Hayashi, L.; Acott, T.S. Inhibition of hyaluronan synthesis reduces versican and fibronectin levels in trabecular meshwork cells. PLoS ONE 2012, 7, e48523. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Morgan, J.L.; Buchberg, A.M.; Siracusa, L.D.; Iozzo, R.V. Expression pattern and mapping of the murine versican gene (Cspg2) to chromosome 13. Genomics 1995, 29, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Calve, S.; Isaac, J.; Gumucio, J.P.; Mendias, C.L. Hyaluronic acid, HAS1, and HAS2 are significantly upregulated during muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2012, 303, C577–C588. [Google Scholar] [CrossRef] [PubMed]
- Velleman, S.G.; Sporer, K.R.; Ernst, C.W.; Reed, K.M.; Strasburg, G.M. Versican, matrix gla protein, and death-associated protein expression affect muscle satellite cell proliferation and differentiation. Poult. Sci. 2012, 91, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Carrino, D.A.; Sorrell, J.M.; Caplan, A.I. Dynamic expression of proteoglycans during chicken skeletal muscle development and maturation. Poult. Sci. 1999, 78, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Sandy, J.D.; Westling, J.; Kenagy, R.D.; Iruela-Arispe, M.L.; Verscharen, C.; Rodriguez-Mazaneque, J.C.; Zimmermann, D.R.; Lemire, J.M.; Fischer, J.W.; Wight, T.N.; et al. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J. Biol. Chem. 2001, 276, 13372–13378. [Google Scholar] [CrossRef] [PubMed]
- Dancevic, C.M.; Fraser, F.W.; Smith, A.D.; Stupka, N.; Ward, A.C.; McCulloch, D.R. Biosynthesis and expression of a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats-15: A novel versican-cleaving proteoglycanase. J. Biol. Chem. 2013, 288, 37267–37276. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Maurice, S.B.; Chahal, B.; Schaeffer, D.F.; Winwood, P.J. Versican: A novel modulator of hepatic fibrosis. Lab. Investig. 2016, 96, 361–374. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.R.; Nelson, C.M.; Dixon, L.J.; Silver, D.L.; Wylie, J.D.; Lindner, V.; Sasaki, T.; Cooley, M.A.; Argraves, W.S.; Apte, S.S. ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev. Cell 2009, 17, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Hope, C.; Foulcer, S.; Jagodinsky, J.; Chen, S.X.; Jensen, J.L.; Patel, S.; Leith, C.; Maroulakou, I.; Callander, N.; Miyamoto, S.; et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 2016, 128, 680. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Mendell, J.R. Emerging drugs for duchenne muscular dystrophy. Expert Opin. Emerg. Drugs 2012, 17, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Guerron, A.D.; Gordish-Dressman, H.; Spurney, C.F.; Iantorno, M.; Hoffman, E.P.; Nagaraju, K. Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse. PLoS ONE 2012, 7, e34204. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Peterle, E. Old and new therapeutic developments in steroid treatment in duchenne muscular dystrophy. Acta Myol. 2012, 31, 9–15. [Google Scholar] [PubMed]
- Anderson, J.E.; McIntosh, L.M.; Poettcker, R. Deflazacort but not prednisone improves both muscle repair and fiber growth in diaphragm and limb muscle in vivo in the mdx dystrophic mouse. Muscle Nerve 1996, 19, 1576–1585. [Google Scholar] [CrossRef]
- Guiraud, S.; Davies, K.E. Pharmacological advances for treatment in duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Belanto, J.J.; Diaz-Perez, S.V.; Magyar, C.E.; Maxwell, M.M.; Yilmaz, Y.; Topp, K.; Boso, G.; Jamieson, C.H.; Cacalano, N.A.; Jamieson, C.A. Dexamethasone induces dysferlin in myoblasts and enhances their myogenic differentiation. Neuromuscul. Disord. 2010, 20, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Passaquin, A.C.; Metzinger, L.; Leger, J.J.; Warter, J.M.; Poindron, P. Prednisolone enhances myogenesis and dystrophin-related protein in skeletal muscle cell cultures from mdx mouse. J. Neurosci. Res. 1993, 35, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Hunt, L.C.; Gorman, C.; Kintakas, C.; McCulloch, D.R.; Mackie, E.J.; White, J.D. Hyaluronan synthesis and myogenesis: A requirement for hyaluronan synthesis during myogenic differentiation independent of pericellular matrix formation. J. Biol. Chem. 2013, 288, 13006–13021. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Sasamura, H.; Shimizu-Hirota, R.; Mifune, M.; Nakaya, H.; Kobayashi, E.; Hayashi, M.; Saruta, T. Glucocorticoid regulation of proteoglycan synthesis in mesangial cells. Kidney Int. 2002, 62, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Todorova, L.; Gurcan, E.; Miller-Larsson, A.; Westergren-Thorsson, G. Lung fibroblast proteoglycan production induced by serum is inhibited by budesonide and formoterol. Am. J. Respir. Cell Mol. Biol. 2006, 34, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Averbeck, M.; Diedenhofen, N.; Willenberg, A.; Anderegg, U.; Sleeman, J.P.; Simon, J.C. Dermal hyaluronan is rapidly reduced by topical treatment with glucocorticoids. J. Investig. Dermatol. 2010, 130, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Watson, C.E.; Liu, C.; Williams, K.J.; Werth, V.P. Glucocorticoids induce a near-total suppression of hyaluronan synthase mRNA in dermal fibroblasts and in osteoblasts: A molecular mechanism contributing to organ atrophy. Biochem. J. 2000, 349, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Radley, H.G.; Lynch, G.S.; Nagaraju, K.; De Luca, A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of duchenne muscular dystrophy. Neurobiol. Dis. 2008, 31, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.S.; Hinkle, R.T.; Chamberlain, J.S.; Brooks, S.V.; Faulkner, J.A. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J. Physiol. 2001, 535, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Stupka, N.; Michell, B.J.; Kemp, B.E.; Lynch, G.S. Differential calcineurin signalling activity and regeneration efficacy in diaphragm and limb muscles of dystrophic mdx mice. Neuromuscul. Disord. 2006, 16, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Cheng, G.; Lu, H.; Aronica, M.; Ransohoff, R.M.; Zhou, L. Impaired respiratory function in mdx and mdx/utrn+/− mice. Muscle Nerve 2011, 43, 263–267. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.S.; Steeds, C.M.; Wiseman, R.W.; Pavlath, G.K. Phosphocreatine as an energy source for actin cytoskeletal rearrangements during myoblast fusion. J. Physiol. 2008, 586, 2841–2853. [Google Scholar] [CrossRef] [PubMed]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Horsley, V.; Pavlath, G.K. Forming a multinucleated cell: Molecules that regulate myoblast fusion. Cells Tissues Organs 2004, 176, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Pavlath, G.K. Spatial and functional restriction of regulatory molecules during mammalian myoblast fusion. Exp. Cell Res. 2010, 316, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Horsley, V.; Jansen, K.M.; Mills, S.T.; Pavlath, G.K. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell 2003, 113, 483–494. [Google Scholar] [CrossRef]
- Pavlath, G.K.; Horsley, V. Cell fusion in skeletal muscle: Central role of NFATC2 in regulating muscle cell size. Cell Cycle 2003, 2, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Mayer, G.; Zaid, A.; Rousselet, E.; Nassoury, N.; Poirier, S.; Essalmani, R.; Prat, A. The activation and physiological functions of the proprotein convertases. Int. J. Biochem. Cell Biol. 2008, 40, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Mylona, E.; Jones, K.A.; Mills, S.T.; Pavlath, G.K. CD44 regulates myoblast migration and differentiation. J. Cell. Physiol. 2006, 209, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Goetsch, K.P.; Myburgh, K.H.; Niesler, C.U. In vitro myoblast motility models: Investigating migration dynamics for the study of skeletal muscle repair. J. Muscle Res. Cell Motil. 2013, 34, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, S.; Cheng, T. TGF-β1 promotes osteosarcoma cell migration and invasion through the miR-143-versican pathway. Cell. Physiol. Biochem. 2014, 34, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Yang, P. MicroRNA-203 inhibits malignant melanoma cell migration by targeting versican. Exp. Ther. Med. 2014, 8, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.J.; Ybot-Gonzalez, P.; Copp, A.J. Over-expression of the chondroitin sulphate proteoglycan versican is associated with defective neural crest migration in the pax3 mutant mouse (splotch). Mech. Dev. 1997, 69, 39–51. [Google Scholar] [CrossRef]
- Wight, T.N. Versican: A versatile extracellular matrix proteoglycan in cell biology. Curr. Opin. Cell Biol. 2002, 14. [Google Scholar] [CrossRef]
- Carthy, J.M.; Abraham, T.; Meredith, A.J.; Boroomand, S.; McManus, B.M. Versican localizes to the nucleus in proliferating mesenchymal cells. Cardiovasc. Pathol. 2015, 24, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Munoz-Canoves, P. Understanding the process of fibrosis in duchenne muscular dystrophy. Biomed. Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Haslett, J.N.; Sanoudou, D.; Kho, A.T.; Bennett, R.R.; Greenberg, S.A.; Kohane, I.S.; Beggs, A.H.; Kunkel, L.M. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 15000–15005. [Google Scholar] [CrossRef] [PubMed]
- Dours-Zimmermann, M.T.; Zimmermann, D.R. A novel glycosaminoglycan attachment domain identified in two alternative splice variants of human versican. J. Biol. Chem. 1994, 269, 32992–32998. [Google Scholar] [PubMed]
- Pallafacchina, G.; Francois, S.; Regnault, B.; Czarny, B.; Dive, V.; Cumano, A.; Montarras, D.; Buckingham, M. An adult tissue-specific stem cell in its niche: A gene profiling analysis of in vivo quiescent and activated muscle satellite cells. Stem Cell Res. 2010, 4, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Kang, I.; Merrilees, M.J. Versican and the control of inflammation. Matrix Biol. 2014, 35, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, D.R.; Ruoslahti, E. Multiple domains of the large fibroblast proteoglycan, versican. EMBO J. 1989, 8, 2975–2981. [Google Scholar] [PubMed]
- Carrino, D.A.; Oron, U.; Pechak, D.G.; Caplan, A.I. Reinitiation of chondroitin sulphate proteoglycan synthesis in regenerating skeletal muscle. Development 1988, 103, 641–656. [Google Scholar] [PubMed]
- Stupka, N.; Schertzer, J.D.; Bassel-Duby, R.; Olson, E.N.; Lynch, G.S. Stimulation of calcineurin Aα activity attenuates muscle pathophysiology in mdx dystrophic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R983–R992. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Koyama, S.; Yabuta, Y.; Kitagawa, H. Chondroitin sulfate is a crucial determinant for skeletal muscle development/regeneration and improvement of muscular dystrophies. J. Biol. Chem. 2012, 287, 38531–38542. [Google Scholar] [CrossRef] [PubMed]
- Foulcer, S.J.; Nelson, C.M.; Quintero, M.V.; Kuberan, B.; Larkin, J.; Dours-Zimmermann, M.T.; Zimmermann, D.R.; Apte, S.S. Determinants of versican-V1 proteoglycan processing by the metalloproteinase ADAMTS5. J. Biol. Chem. 2014, 289, 27859–27873. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Damsker, J.M.; Yu, Q.; Dillingham, B.C.; Huynh, T.; Van der Meulen, J.H.; Sali, A.; Miller, B.K.; Phadke, A.; Scheffer, L.; et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med. 2013, 5, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Reeves, E.; Damsker, J.; Nagaraju, K.; McCall, J.M.; Connor, E.M.; Bushby, K. Novel approaches to corticosteroid treatment in duchenne muscular dystrophy. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sivasankar, M.; Kraus, D.H.; Sandulache, V.C.; Amin, M.; Branski, R.C. Glucocorticoids regulate extracellular matrix metabolism in human vocal fold fibroblasts. Laryngoscope 2011, 121, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Hartel, J.V.; Granchelli, J.A.; Hudecki, M.S.; Pollina, C.M.; Gosselin, L.E. Impact of prednisone on TGF-β1 and collagen in diaphragm muscle from mdx mice. Muscle Nerve 2001, 24, 428–432. [Google Scholar] [CrossRef]
- Ma, Z.; Zhong, Z.; Zheng, Z.; Shi, X.M.; Zhang, W. Inhibition of glycogen synthase kinase-3β attenuates glucocorticoid-induced suppression of myogenic differentiation in vitro. PLoS ONE 2014, 9, e105528. [Google Scholar] [CrossRef] [PubMed]
- Bolkenius, U.; Hahn, D.; Gressner, A.M.; Breitkopf, K.; Dooley, S.; Wickert, L. Glucocorticoids decrease the bioavailability of TGF-β which leads to a reduced Tgf-β signaling in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2004, 325, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.Q.; Kohyama, T.; Skold, C.M.; Zhu, Y.K.; Liu, X.; Romberger, D.J.; Stoner, J.; Rennard, S.I. Glucocorticoids modulate TGF-β production by human fetal lung fibroblasts. Inflammation 2003, 27, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Schoepe, S.; Schacke, H.; May, E.; Asadullah, K. Glucocorticoid therapy-induced skin atrophy. Exp. Dermatol. 2006, 15, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Snow, H.E.; Riccio, L.M.; Mjaatvedt, C.H.; Hoffman, S.; Capehart, A.A. Versican expression during skeletal/joint morphogenesis and patterning of muscle and nerve in the embryonic mouse limb. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2005, 282, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Hirose, M.; Hirose, J.; Nagakubo, D.; Plaas, A.H.; Miyasaka, M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J. Biol. Chem. 2000, 275, 35448–35456. [Google Scholar] [CrossRef] [PubMed]
- Aruffo, A.; Stamenkovic, I.; Melnick, M.; Underhill, C.B.; Seed, B. CD44 is the principal cell surface receptor for hyaluronate. Cell 1990, 61, 1303–1313. [Google Scholar] [CrossRef]
- Nikitovic, D.; Zafiropoulos, A.; Katonis, P.; Tsatsakis, A.; Theocharis, A.D.; Karamanos, N.K.; Tzanakakis, G.N. Transforming growth factor-β as a key molecule triggering the expression of versican isoforms V0 and V1, hyaluronan synthase-2 and synthesis of hyaluronan in malignant osteosarcoma cells. IUBMB Life 2006, 58, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N. Arterial remodeling in vascular disease: A key role for hyaluronan and versican. Front. Biosci. 2008, 13, 4933–4937. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.A.; Chandrasekharan, S.; Jokonya, N.; Fowles, A.; Hamdy, F.C.; Buttle, D.J.; Eaton, C.L. The expression and regulation of ADAMTS-1, -4, -5, -9, and -15, and TIMP-3 by TGFβ1 in prostate cells: Relevance to the accumulation of versican. Prostate 2005, 63, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M.; Meredith, A.J.; Boroomand, S.; Abraham, T.; Luo, Z.; Knight, D.; McManus, B.M. Versican V1 overexpression induces a myofibroblast-like phenotype in cultured fibroblasts. PLoS ONE 2015, 10, e0133056. [Google Scholar] [CrossRef] [PubMed]
- Choocheep, K.; Hatano, S.; Takagi, H.; Watanabe, H.; Kimata, K.; Kongtawelert, P.; Watanabe, H. Versican facilitates chondrocyte differentiation and regulates joint morphogenesis. J. Biol. Chem. 2010, 285, 21114–21125. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Frevert, C.W.; Debley, J.S.; Reeves, S.R.; Parks, W.C.; Ziegler, S.F. Interplay of extracellular matrix and leukocytes in lung inflammation. Cell. Immunol. 2017, 312, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Harten, I.A.; Chang, M.Y.; Braun, K.R.; Sheih, A.; Nivison, M.P.; Johnson, P.Y.; Workman, G.; Kaber, G.; Evanko, S.P.; et al. Versican deficiency significantly reduces lung inflammatory response induced by polyinosine-polycytidylic acid stimulation. J. Biol. Chem. 2017, 292, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, T.R. Lectin binding and desmin staining during bupivicaine-induced necrosis and regeneration in rat skeletal muscle. J. Pathol. 1988, 155, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Garry, G.A.; Li, S.; Bezprozvannaya, S.; Sanchez-Ortiz, E.; Chen, B.; Shelton, J.M.; Jaichander, P.; Bassel-Duby, R.; Olson, E.N. A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat. Cell Biol. 2017, 19, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Emde, B.; Heinen, A.; Godecke, A.; Bottermann, K. Wheat germ agglutinin staining as a suitable method for detection and quantification of fibrosis in cardiac tissue after myocardial infarction. Eur. J. Histochem. 2014, 58, 2448. [Google Scholar] [CrossRef] [PubMed]
- Blau, H.M.; Pavlath, G.K.; Hardeman, E.C.; Chiu, C.P.; Silberstein, L.; Webster, S.G.; Miller, S.C.; Webster, C. Plasticity of the differentiated state. Science 1985, 230, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Bains, W.; Ponte, P.; Blau, H.; Kedes, L. Cardiac actin is the major actin gene product in skeletal muscle cell differentiation in vitro. Mol. Cell. Biol. 1984, 4, 1449–1453. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, X.; Zheng, Y.; Li, F.; Lu, Z.; Chen, C.; Liu, J.; Wang, Y.; Peng, Y.; Shen, Z.; et al. MiR-23a inhibits myogenic differentiation through down regulation of fast myosin heavy chain isoforms. Exp. Cell Res. 2012, 318, 2324–2334. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | Name | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) |
|---|---|---|---|
| NM_009621.5 | Adamts1 | CCTGTGAAGCCAAAGGCATTG | TGCACACAGACAGAGGTAGAGT |
| NM_011782.2 | Adamts5 | GCTACTGCACAGGGAAGAGG | GCCAGGACACCTGCATATTT |
| NM_001329420.1 | Adamts15 | GCTCATCTGCCGAGCCAAT | CAGCCAGCCTTGATGCACTT |
| NM_007710.2 | Ckm 1 | CCGTGTCACCTCTGCTGCTG | TCCTTCATATTGCCTCCCTTCTCC |
| XM_011250822.2 | Pcsk3 2 | CAGCGAGACCTGAATGTGAA | CAGGGTCATAATTGCCTGCT |
| NM_008216.3 | Has2 | GGGACCTGGTGAGACAGAAG | ATGAGGCAGGGTCAAGCATA |
| XM_006511645.3 | Hyal2 | AGCCGCAACTTTGTCAGTTT | GAGTCCTCGGGTGTATGTGG |
| XM_017314318.1 | Myh1 3 | GCTCAAAGCCCTGTGTTACC | CATAGACGGCTTTGGCTAGG |
| NM_001291184.1 | Pcsk6 4 | ATTTCCCCAACCTCGTCTCT | AGCTGAGTCCTTGCCACCTA |
| NM_011577.2 | Tgfb1 | GCCTGAGTGGCTGTCTTTTGA | CACAAGAGCAGTGAGCGCTGAA |
| NM_001081249.1 (V0) | V0/V1 Vcan 5 | ACCAAGGAGAAGTTCGAGCA | CTTCCCAGGTAGCCAAATCA |
| NM_019389.2 (V1) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McRae, N.; Forgan, L.; McNeill, B.; Addinsall, A.; McCulloch, D.; Van der Poel, C.; Stupka, N. Glucocorticoids Improve Myogenic Differentiation In Vitro by Suppressing the Synthesis of Versican, a Transitional Matrix Protein Overexpressed in Dystrophic Skeletal Muscles. Int. J. Mol. Sci. 2017, 18, 2629. https://doi.org/10.3390/ijms18122629
McRae N, Forgan L, McNeill B, Addinsall A, McCulloch D, Van der Poel C, Stupka N. Glucocorticoids Improve Myogenic Differentiation In Vitro by Suppressing the Synthesis of Versican, a Transitional Matrix Protein Overexpressed in Dystrophic Skeletal Muscles. International Journal of Molecular Sciences. 2017; 18(12):2629. https://doi.org/10.3390/ijms18122629
Chicago/Turabian StyleMcRae, Natasha, Leonard Forgan, Bryony McNeill, Alex Addinsall, Daniel McCulloch, Chris Van der Poel, and Nicole Stupka. 2017. "Glucocorticoids Improve Myogenic Differentiation In Vitro by Suppressing the Synthesis of Versican, a Transitional Matrix Protein Overexpressed in Dystrophic Skeletal Muscles" International Journal of Molecular Sciences 18, no. 12: 2629. https://doi.org/10.3390/ijms18122629
APA StyleMcRae, N., Forgan, L., McNeill, B., Addinsall, A., McCulloch, D., Van der Poel, C., & Stupka, N. (2017). Glucocorticoids Improve Myogenic Differentiation In Vitro by Suppressing the Synthesis of Versican, a Transitional Matrix Protein Overexpressed in Dystrophic Skeletal Muscles. International Journal of Molecular Sciences, 18(12), 2629. https://doi.org/10.3390/ijms18122629