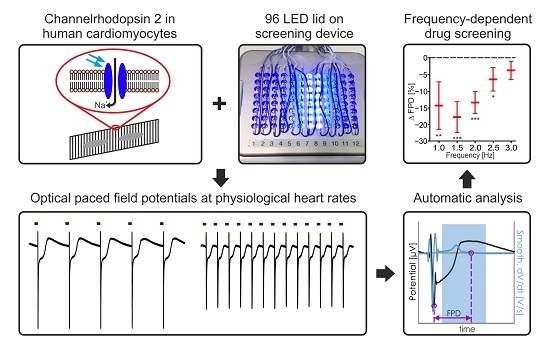
Frequency-Dependent Multi-Well Cardiotoxicity Screening Enabled by Optogenetic Stimulation

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
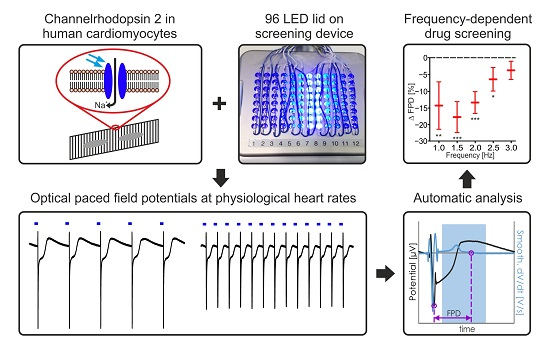
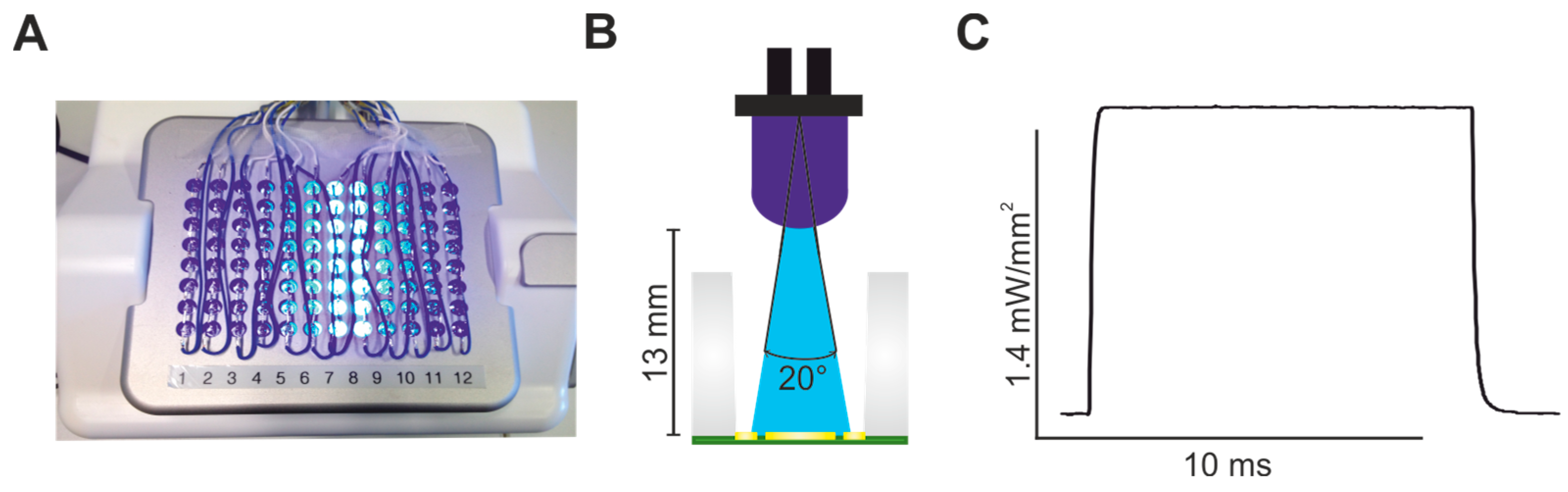
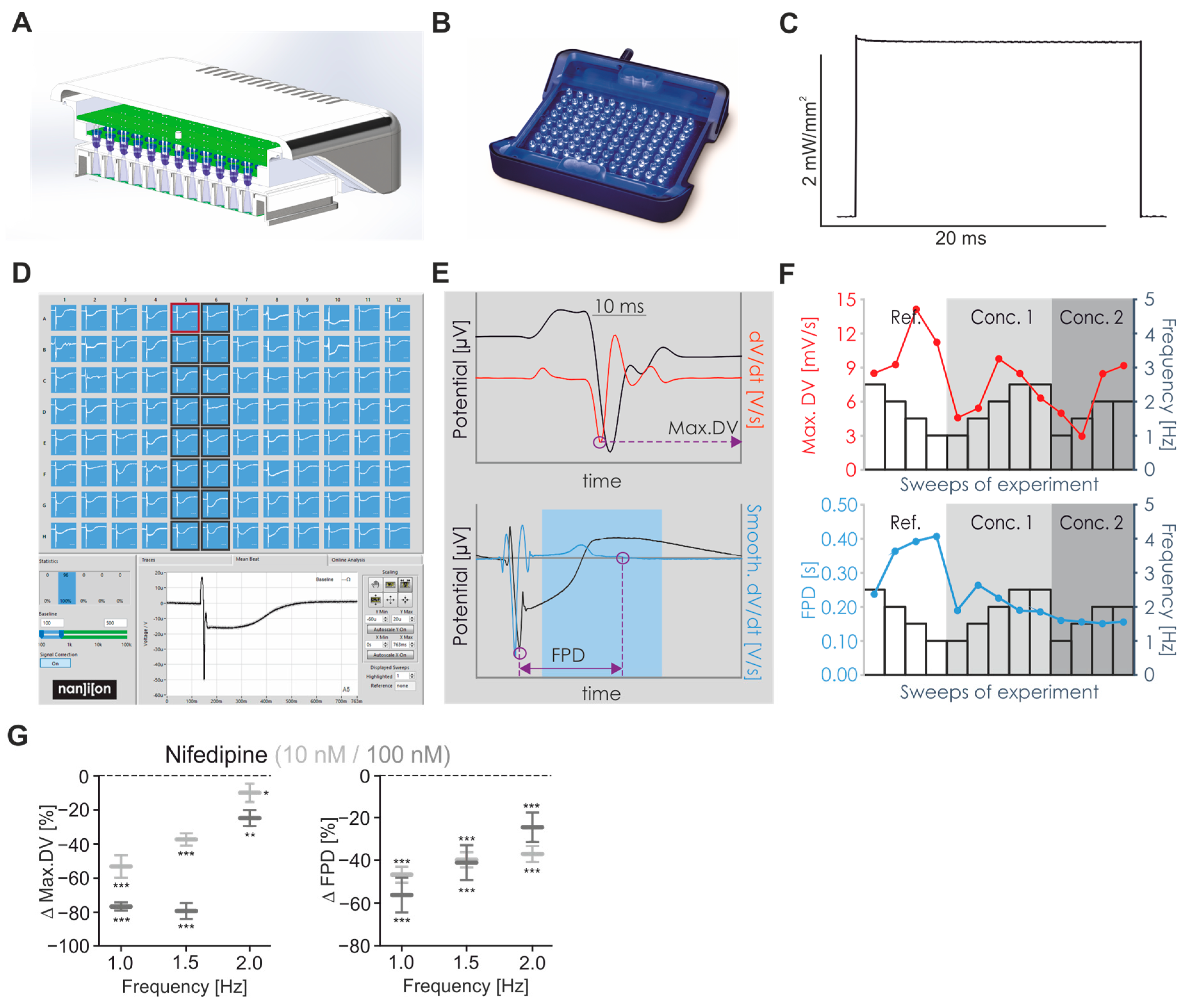
2.1. Establishing LED-Based Optogenetic Pacing on 96-Wells Plates
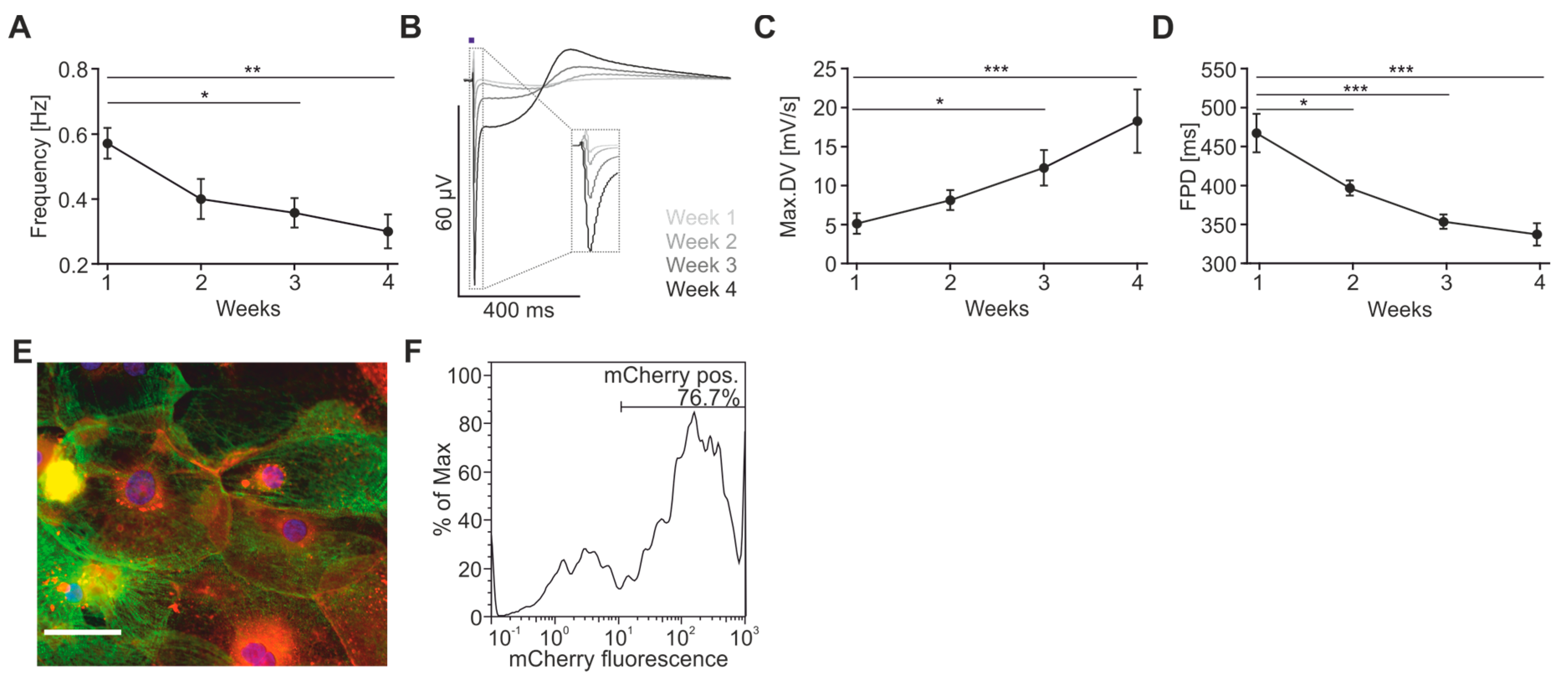
2.2. Maturation and Optical Pacing of Cardiomyocytes
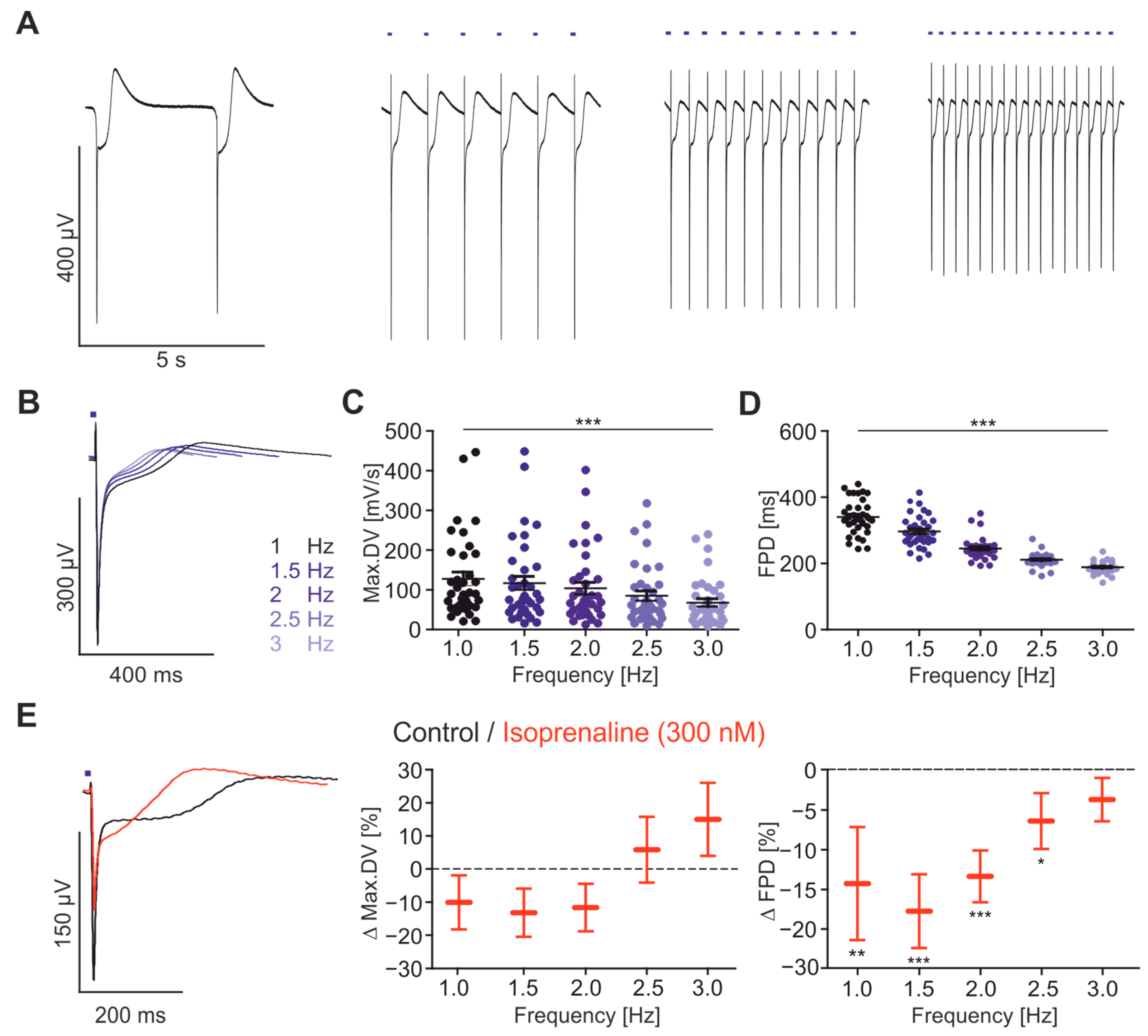
2.3. Frequency Dependence of FP Parameters
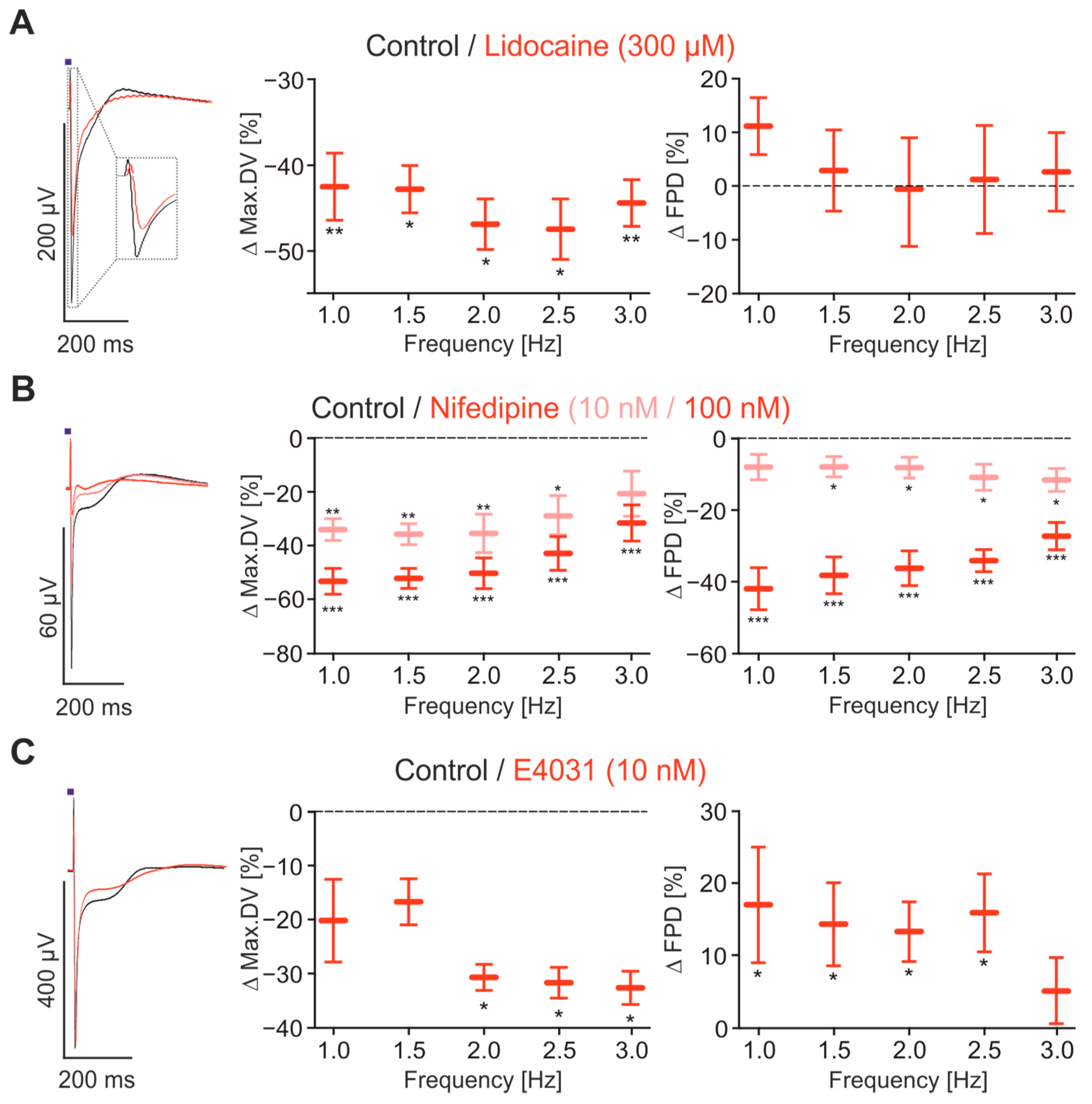
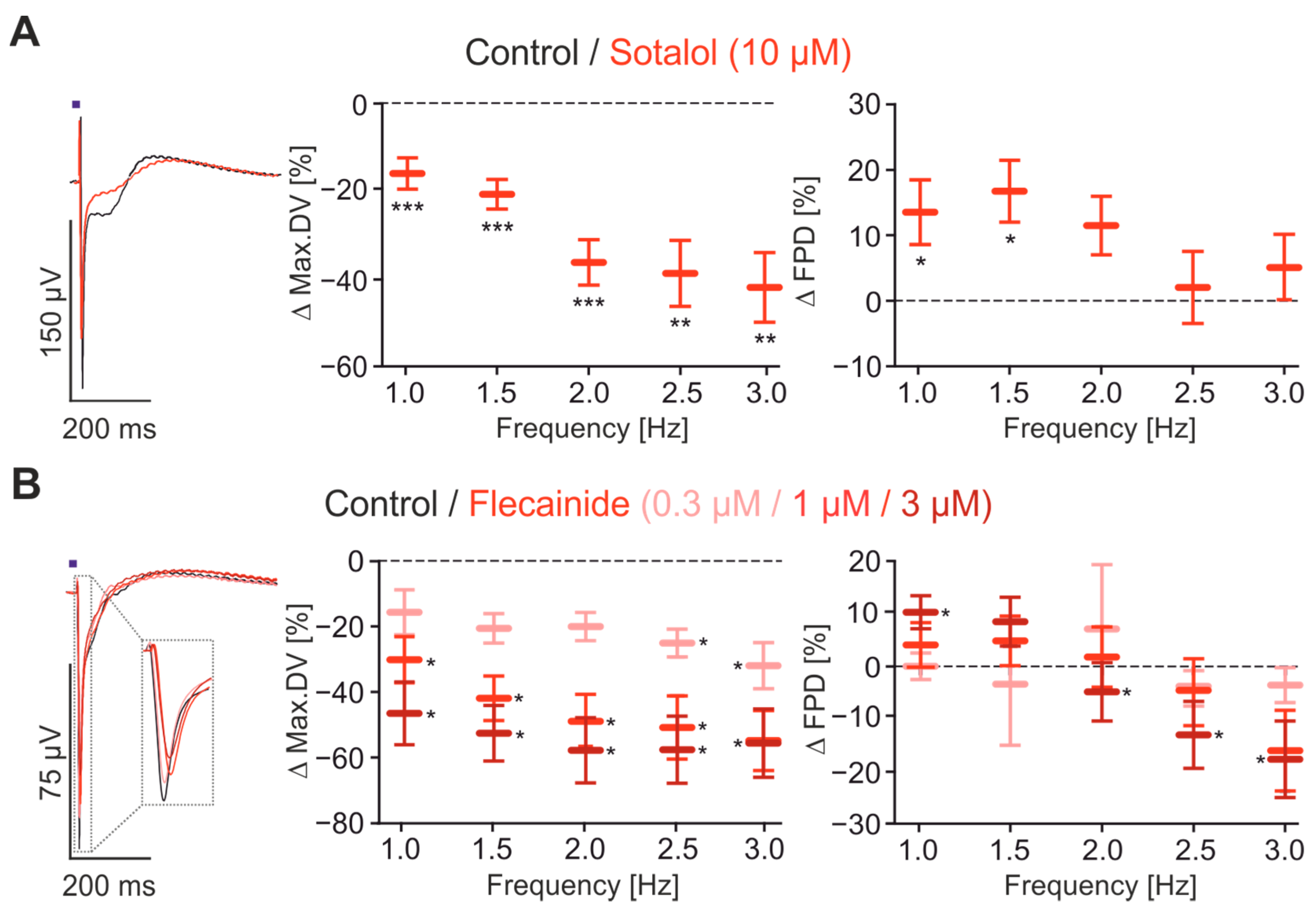
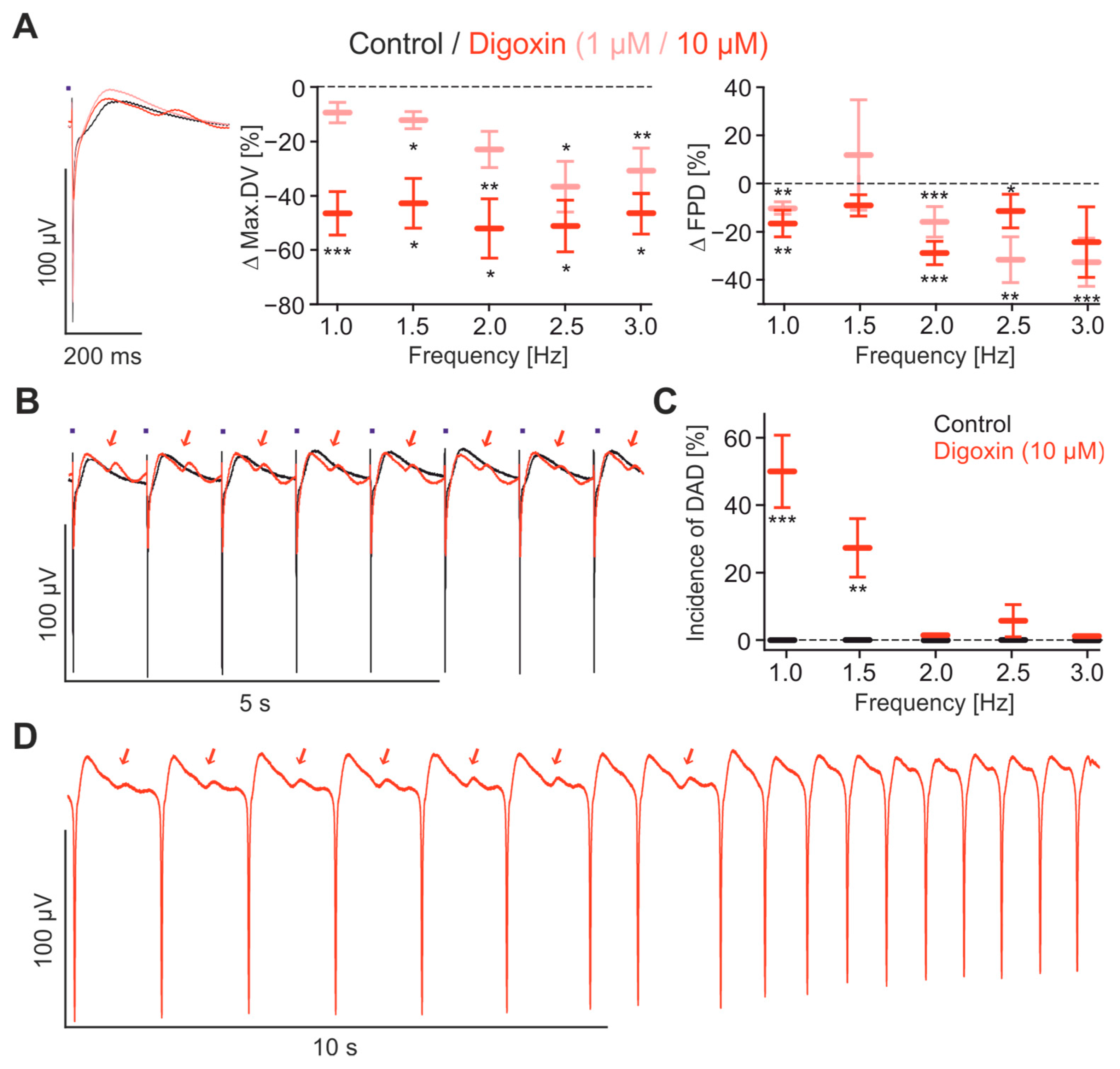
2.4. Pharmacological Characterisation of FP Signals
2.5. Evaluation of Drug Screening with the CardioExcyte96 System
2.6. Xpress.4UTM LightPace for Virus Free ChR2 Expression
2.7. Future Trend and Automatisation with CardioExcyte96 SOL
3. Discussion
4. Materials and Methods
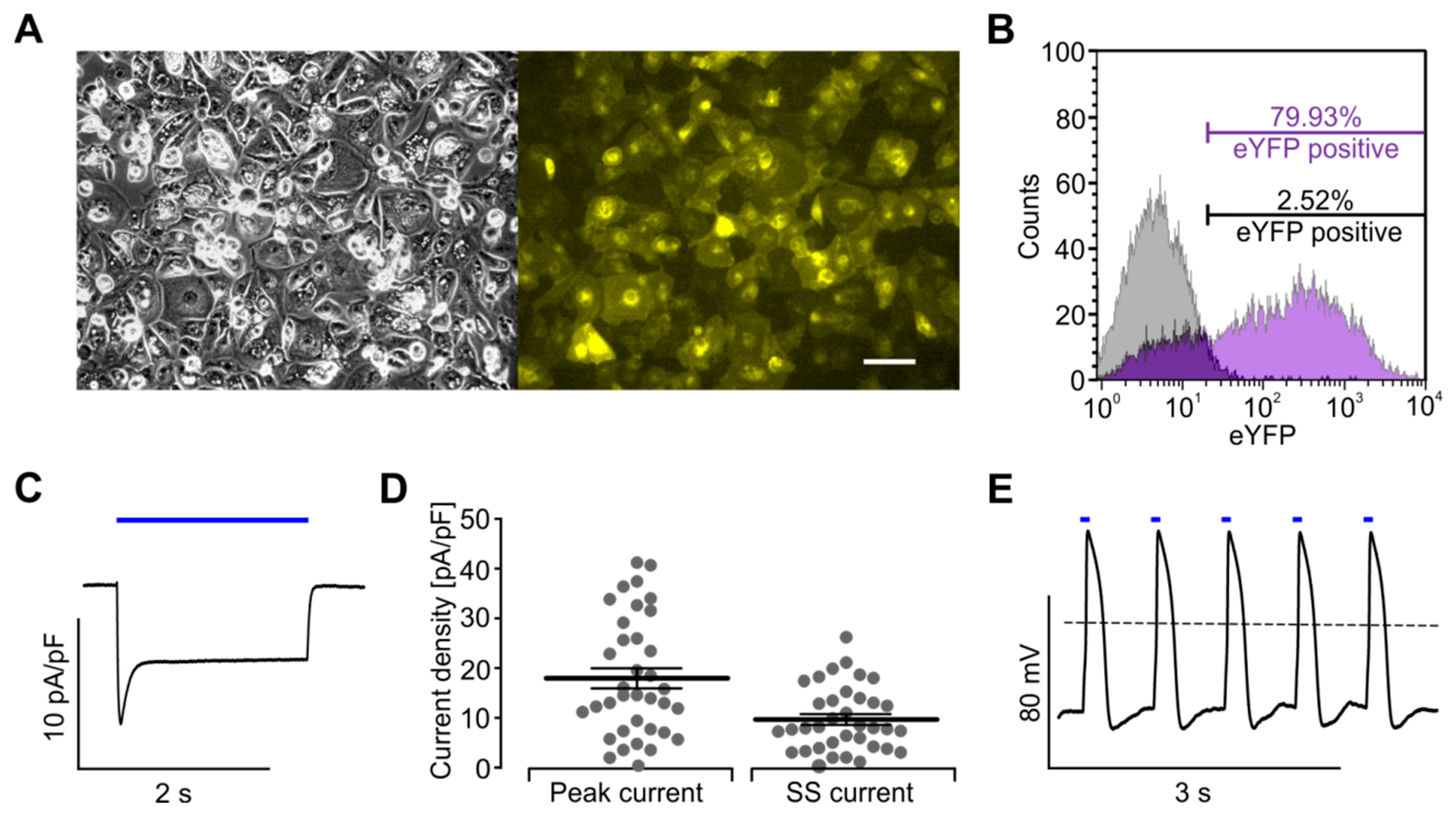
4.1. Cell Culture and AAV-Mediated Transduction
4.2. Determination of ChR2-Expression after AAV-Transduction
4.3. CardioExcyte96 LED Setup and FP Recordings
4.4. Analysis of FP
4.5. Determination of ChR2-Expression in Xpress.4UTM LightPace Transfected Cor.4U® Cells
4.6. Patch Clamp Analysis of Xpress.4UTM LightPace Transfected Cor.4U® Cells
4.7. FP Recordings of Xpress.4UTM LightPace Transfected Cor.4U® Cells with the CardioExcyte96 SOL System
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| AP | action potential |
| APD | action potential duration |
| CAG | chicken β actin |
| ChR2 | Channelrhodopsin-2 |
| CiPA | comprehensive in vitro proarrhythmia assay |
| ECG | electrocardiogram |
| EDTA | Ethylenediaminetetraacetic acid |
| EGTA | Ethylene-bis(oxyethylenenitrilo)tetraacetic acid |
| EYFP | enhanced yellow fluorescent protein |
| FACS | fluorescence-activated cell sorting |
| FCS | fetal calf serum |
| FP | field potential |
| FPD | field potential duration |
| hERG | human Ether-a-go-go Related Gene |
| HEPES | 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-Hydroxyethyl)piperazine-Nesulfonic idhors de acid) |
| hPSC | human induced pluripotent stem cells |
| IMDM | Iscove’s Modified Dulbecco’s Media |
| IKr | rapid potassium current |
| LED | Light-emitted diode |
| max.DV | maximum negative downstroke velocity |
| MEA | microelectrode array |
| PBS | Phosphate-buffered saline |
| PFA | paraformaldehyde |
References
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.; Roden, D.M.; Darbar, D. Drug-induced long QT syndrome. Pharmacol. Rev. 2010, 62, 760–781. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Siegl, P.; Corsini, A.; Herrmann, J.; Lerman, A.; Benghozi, R. Drug attrition during pre-clinical and clinical development: Understanding and managing drug-induced cardiotoxicity. Pharmacol. Ther. 2013, 138, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.J.; Roden, D.M. Drug-induced long QT and torsade de pointes: Recent advances. Curr. Opin. Cardiol. 2007, 22, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, E.G.; Liang, P.; Lan, F.; Sanchez-Freire, V.; Simmons, C.; Gong, T.; Sharma, A.; Burridge, P.W.; Patlolla, B.; Lee, A.S.; et al. Screening drug-induced arrhythmia using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays. Circulation 2013, 128, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Leisgen, C.; Gonser, B.; Gunther, E. QT-screen: High-throughput cardiac safety pharmacology by extracellular electrophysiology on primary cardiac myocytes. Assay Drug Dev. Technol. 2004, 2, 507–514. [Google Scholar] [CrossRef] [PubMed]
- ICH. S7b, the Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (Qt Prolongation) by Human Pharmaceuticals. 2005. Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf (accessed on 10 August 2017).
- Meyer, T.; Sartipy, P.; Blind, F.; Leisgen, C.; Guenther, E. New cell models and assays in cardiac safety profiling. Expert Opin. Drug Metab. Toxicol. 2007, 3, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Sager, P.T.; Gintant, G.; Turner, J.R.; Pettit, S.; Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 2014, 167, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Konstantopoulou, A.; Tsikrikas, S.; Asvestas, D.; Korantzopoulos, P.; Letsas, K.P. Mechanisms of drug-induced proarrhythmia in clinical practice. World J. Cardiol. 2013, 5, 175–185. [Google Scholar] [CrossRef] [PubMed]
- De Ponti, F.; Poluzzi, E.; Cavalli, A.; Recanatini, M.; Montanaro, N. Safety of non-antiarrhythmic drugs that prolong the QT interval or induce torsade de pointes: An overview. Drug Saf. 2002, 25, 263–286. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Liang, P.; Wu, J.C. Induced pluripotent stem cells as a disease modeling and drug screening platform. J. Cardiovasc. Pharmacol. 2012, 60, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Mordwinkin, N.M.; Burridge, P.W.; Wu, J.C. A review of human pluripotent stem cell-derived cardiomyocytes for high-throughput drug discovery, cardiotoxicity screening, and publication standards. J. Cardiovasc. Transl. Res. 2013, 6, 22–30. [Google Scholar] [CrossRef]
- Belles, B.; Malecot, C.O.; Hescheler, J.; Trautwein, W. “Run-down” of the Ca current during long whole-cell recordings in guinea pig heart cells: Role of phosphorylation and intracellular calcium. Pflugers Arch. 1988, 411, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Boven, K.H.; Gunther, E.; Fejtl, M. Micro-electrode arrays in cardiac safety pharmacology: A novel tool to study QT interval prolongation. Drug Saf. 2004, 27, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Halbach, M.; Egert, U.; Hescheler, J.; Banach, K. Estimation of action potential changes from field potential recordings in multicellular mouse cardiac myocyte cultures. Cell. Physiol. Biochem. 2003, 13, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Lapp, H.; Bruegmann, T.; Malan, D.; Friedrichs, S.; Kilgus, C.; Heidsieck, A.; Sasse, P. Frequency-dependent drug screening using optogenetic stimulation of human iPSC-derived cardiomyocytes. Sci. Rep. 2017, 7, 9629. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Vargas, H.M. Proarrhythmia Risk Assessment in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Using the Maestro MEA Platform. Toxicol. Sci. 2015, 147, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Kuusela, J.; Kujala, V.J.; Kiviaho, A.; Ojala, M.; Swan, H.; Kontula, K.; Aalto-Setala, K. Effects of cardioactive drugs on human induced pluripotent stem cell derived long QT syndrome cardiomyocytes. Springerplus 2016, 5, 234. [Google Scholar] [CrossRef] [PubMed]
- Kitaguchi, T.; Moriyama, Y.; Taniguchi, T.; Ojima, A.; Ando, H.; Uda, T.; Otabe, K.; Oguchi, M.; Shimizu, S.; Saito, H.; et al. CSAHi study: Evaluation of multi-electrode array in combination with human iPS cell-derived cardiomyocytes to predict drug-induced QT prolongation and arrhythmia--Effects of 7 reference compounds at 10 facilities. J. Pharmacol. Toxicol. Methods 2016, 78, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Nomura, F.; Hamada, T.; Abe, Y.; Takamori, H.; Sakakura, T.; Takasuna, K.; Sanbuissho, A.; Hyllner, J.; Sartipy, P.; et al. On-chip in vitro cell-network pre-clinical cardiac toxicity using spatiotemporal human cardiomyocyte measurement on a chip. Sci. Rep. 2014, 4, 4670. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Aylott, M.; Cui, Y.; Louttit, J.B.; McMahon, N.C.; Sridhar, A. Comparison of electrophysiological data from human-induced pluripotent stem cell-derived cardiomyocytes to functional preclinical safety assays. Toxicol. Sci. 2013, 134, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, K.H.; Lewis, G.F.; Gay, E.A.; Sellgren, K.L.; Grego, S. High-throughput cardiac safety evaluation and multi-parameter arrhythmia profiling of cardiomyocytes using microelectrode arrays. Toxicol. Appl. Pharmacol. 2015, 288, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.; Thomas, N. High-throughput multi-parameter profiling of electrophysiological drug effects in human embryonic stem cell derived cardiomyocytes using multi-electrode arrays. Toxicol. Sci. 2014, 140, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.; Millar, V.; Williams, A.S.; Kalinka, S. Bridging Functional and Structural Cardiotoxicity Assays Using Human Embryonic Stem Cell-Derived Cardiomyocytes for a More Comprehensive Risk Assessment. Toxicol. Sci. 2015, 148, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Braam, S.R.; Tertoolen, L.; van de Stolpe, A.; Meyer, T.; Passier, R.; Mummery, C.L. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010, 4, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Klimas, A.; Ambrosi, C.M.; Yu, J.; Williams, J.C.; Bien, H.; Entcheva, E. OptoDyCE as an automated system for high-throughput all-optical dynamic cardiac electrophysiology. Nat. Commun. 2016, 7, 11542. [Google Scholar] [CrossRef] [PubMed]
- Hortigon-Vinagre, M.P.; Zamora, V.; Burton, F.L.; Green, J.; Gintant, G.A.; Smith, G.L. The Use of Ratiometric Fluorescence Measurements of the Voltage Sensitive Dye Di-4-ANEPPS to Examine Action Potential Characteristics and Drug Effects on Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Sci. 2016, 154, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, G.T.; Chaudhary, K.W.; Atwater, N.; Nguyen, C.; Brown, B.S.; McNeish, J.D.; Cohen, A.E.; Kralj, J.M. Cardiotoxicity screening with simultaneous optogenetic pacing, voltage imaging and calcium imaging. J. Pharmacol. Toxicol. Methods 2016, 81, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Doerr, L.; Thomas, U.; Guinot, D.R.; Bot, C.T.; Stoelzle-Feix, S.; Beckler, M.; George, M.; Fertig, N. New Easy-to-Use Hybrid System for Extracellular Potential and Impedance Recordings. J. Lab. Autom. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, D.A.; Potter, S.M. Real-time multi-channel stimulus artifact suppression by local curve fitting. J. Neurosci. Methods 2002, 120, 113–120. [Google Scholar] [CrossRef]
- Merrill, D.R.; Bikson, M.; Jefferys, J.G. Electrical stimulation of excitable tissue: Design of efficacious and safe protocols. J. Neurosci. Methods 2005, 141, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Hondeghem, L.M.; Snyders, D.J. Class III antiarrhythmic agents have a lot of potential but a long way to go. Reduced effectiveness and dangers of reverse use dependence. Circulation 1990, 81, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Priori, S.G. Genetics of sudden death: Focus on inherited channelopathies. Eur. Heart J. 2011, 32, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Masse, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Lieu, D.K.; Fu, J.D.; Chiamvimonvat, N.; Tung, K.C.; McNerney, G.P.; Huser, T.; Keller, G.; Kong, C.W.; Li, R.A. Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Arrhythm. Electrophysiol. 2013, 6, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Herron, T.J.; Rocha, A.M.; Campbell, K.F.; Ponce-Balbuena, D.; Willis, B.C.; Guerrero-Serna, G.; Liu, Q.; Klos, M.; Musa, H.; Zarzoso, M.; et al. Extracellular Matrix-Mediated Maturation of Human Pluripotent Stem Cell-Derived Cardiac Monolayer Structure and Electrophysiological Function. Circ. Arrhythm. Electrophysiol. 2016, 9, e003638. [Google Scholar] [CrossRef] [PubMed]
- Nagel, G.; Szellas, T.; Huhn, W.; Kateriya, S.; Adeishvili, N.; Berthold, P.; Ollig, D.; Hegemann, P.; Bamberg, E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 2003, 100, 13940–13945. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Scharnhorst, K.S.; Stieg, A.Z.; Gimzewski, J.K.; Minami, I.; Nakatsuji, N.; Nakano, H.; Nakano, A. Two dimensional electrophysiological characterization of human pluripotent stem cell-derived cardiomyocyte system. Sci. Rep. 2017, 7, 43210. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, T.; Rudy, Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1023–H1030. [Google Scholar] [CrossRef] [PubMed]
- Seed, W.A.; Noble, M.I.; Oldershaw, P.; Wanless, R.B.; Drake-Holland, A.J.; Redwood, D.; Pugh, S.; Mills, C. Relation of human cardiac action potential duration to the interval between beats: Implications for the validity of rate corrected QT interval (QTc). Heart J. 1987, 57, 32–37. [Google Scholar] [CrossRef]
- Yokoo, N.; Baba, S.; Kaichi, S.; Niwa, A.; Mima, T.; Doi, H.; Yamanaka, S.; Nakahata, T.; Heike, T. The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 2009, 387, 482–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.; Marshall, R.J.; Winslow, E. Effects of selective channel blocking agents on contractions and action potentials in K+-depolarized guinea-pig atria. Br. J. Pharmacol. 1985, 86, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Hayashi, S.; Ojima, A.; Taniguchi, T.; Miyamoto, N.; Nakamori, C.; Nagasawa, C.; Kitamura, T.; Osada, T.; Honda, Y.; et al. Improvement of acquisition and analysis methods in multi-electrode array experiments with iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 2015, 75, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Niwa, R.; Mitcheson, J.S.; Sanguinetti, M.C. Molecular determinants of HERG channel block. Mol. Pharmacol. 2006, 69, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, T.; Aoshima, J. A compilation of analytical data from inhibition studies on DNA polymerases and some of its implications. J. Biochem. 1985, 97, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.R. Cellular electrophysiology of digitalis toxicity. J. Am. Coll. Cardiol. 1985, 5, 22A–34A. [Google Scholar] [CrossRef]
- Saner, H.E.; Lange, H.W.; Pierach, C.A.; Aeppli, D.M. Relation between serum digoxin concentration and the electrocardiogram. Clin. Cardiol. 1988, 11, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Ahnve, S. QT interval prolongation in acute myocardial infarction. Eur. Heart J. 1985, 6, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Qian, J.Y.; Abrams, R.; Tang, H.M.; Weiser, T.; Sanders, M.J.; Kolaja, K.L. The electrophysiological effects of cardiac glycosides in human iPSC-derived cardiomyocytes and in guinea pig isolated hearts. Cell. Physiol. Biochem. 2011, 27, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.H.; Wu, J.; Chung, Y.Y.; Liu, Z.; Zhang, R.R.; Chong, K.; Korzh, V.; Ting, S.; Oh, S.; Shim, W.; et al. Erratum: Identification of Na+/K+-ATPase inhibition-independent proarrhythmic ionic mechanisms of cardiac glycosides. Sci. Rep. 2017, 7, 11498. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.T.; Cunningham, P.M.; January, C.T. Digoxin-induced delayed afterdepolarizations: Biphasic effects of digoxin on action potential duration and the Q-T interval in cardiac Purkinje fibers. Methods Find. Exp. Clin. Pharmacol. 1995, 17, 113–120. [Google Scholar] [PubMed]
- Bruegmann, T.; Boyle, P.M.; Vogt, C.C.; Karathanos, T.V.; Arevalo, H.J.; Fleischmann, B.K.; Trayanova, N.A.; Sasse, P. Optogenetic defibrillation terminates ventricular arrhythmia in mouse hearts and human simulations. J. Clin. Investig. 2016, 126, 3894–3904. [Google Scholar] [CrossRef] [PubMed]
- Vogt, C.C.; Bruegmann, T.; Malan, D.; Ottersbach, A.; Roell, W.; Fleischmann, B.K.; Sasse, P. Systemic gene transfer enables optogenetic pacing of mouse hearts. Cardiovasc. Res. 2015, 106, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.J.; Wu, C.Y.C.; Jiang, Y.P.; Ballou, L.M.; Clausen, C.; Cohen, I.S.; Lin, R.Z. Suppression of Phosphoinositide 3-Kinase Signaling and Alteration of Multiple Ion Currents in Drug-Induced Long QT Syndrome. Sci. Transl. Med. 2012, 4, 131ra50. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chun, Y.W.; Stroud, D.M.; Mosley, J.D.; Knollmann, B.C.; Hong, C.; Roden, D.M. Screening for Acute I-Kr Block Is Insufficient to Detect Torsades de Pointes Liability Role of Late Sodium Current. Circulation 2014, 130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Honda, Y.; Tsujimoto, S.; Watanabe, H.; Kunimatsu, T.; Funabashi, H. Availability of human induced pluripotent stem cell-derived cardiomyocytes in assessment of drug potential for QT prolongation. Toxicol. Appl. Pharmacol. 2014, 278, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Moak, J.P. Developmental cellular electrophysiologic effects of d-sotalol on canine cardiac Purkinje fibers. Pediatr. Res. 1991, 29, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, E. Electrophysiologic and voltage clamp analysis of the effects of sotalol on isolated cardiac muscle and Purkinje fibers. J. Pharmacol. Exp. Ther. 1985, 232, 817–825. [Google Scholar] [PubMed]
- Izumi-Nakaseko, H.; Nakamura, Y.; Wada, T.; Ando, K.; Kanda, Y.; Sekino, Y.; Sugiyama, A. Characterization of human iPS cell-derived cardiomyocyte sheets as a model to detect drug-induced conduction disturbance. J. Toxicol. Sci. 2017, 42, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Melgari, D.; Zhang, Y.; El Harchi, A.; Dempsey, C.E.; Hancox, J.C. Molecular basis of hERG potassium channel blockade by the class Ic antiarrhythmic flecainide. J. Mol. Cell. Cardiol. 2015, 86, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, E.; O’Leary, M.E. State-dependent trapping of flecainide in the cardiac sodium channel. J. Physiol. 2004, 560, 37–49. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehnelt, S.; Malan, D.; Juhasz, K.; Wolters, B.; Doerr, L.; Beckler, M.; Kettenhofen, R.; Bohlen, H.; Bruegmann, T.; Sasse, P. Frequency-Dependent Multi-Well Cardiotoxicity Screening Enabled by Optogenetic Stimulation. Int. J. Mol. Sci. 2017, 18, 2634. https://doi.org/10.3390/ijms18122634
Rehnelt S, Malan D, Juhasz K, Wolters B, Doerr L, Beckler M, Kettenhofen R, Bohlen H, Bruegmann T, Sasse P. Frequency-Dependent Multi-Well Cardiotoxicity Screening Enabled by Optogenetic Stimulation. International Journal of Molecular Sciences. 2017; 18(12):2634. https://doi.org/10.3390/ijms18122634
Chicago/Turabian StyleRehnelt, Susanne, Daniela Malan, Krisztina Juhasz, Benjamin Wolters, Leo Doerr, Matthias Beckler, Ralf Kettenhofen, Heribert Bohlen, Tobias Bruegmann, and Philipp Sasse. 2017. "Frequency-Dependent Multi-Well Cardiotoxicity Screening Enabled by Optogenetic Stimulation" International Journal of Molecular Sciences 18, no. 12: 2634. https://doi.org/10.3390/ijms18122634
APA StyleRehnelt, S., Malan, D., Juhasz, K., Wolters, B., Doerr, L., Beckler, M., Kettenhofen, R., Bohlen, H., Bruegmann, T., & Sasse, P. (2017). Frequency-Dependent Multi-Well Cardiotoxicity Screening Enabled by Optogenetic Stimulation. International Journal of Molecular Sciences, 18(12), 2634. https://doi.org/10.3390/ijms18122634