Simvastatin Inhibits Cell Proliferation and Migration in Human Anaplastic Thyroid Cancer

Abstract
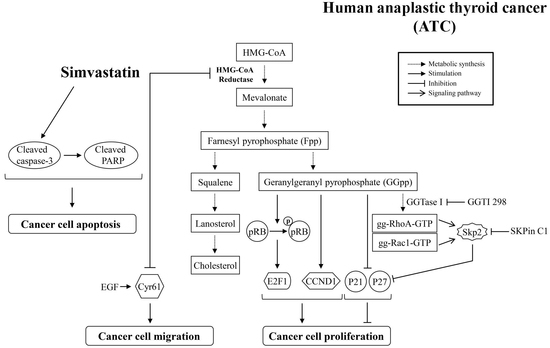
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
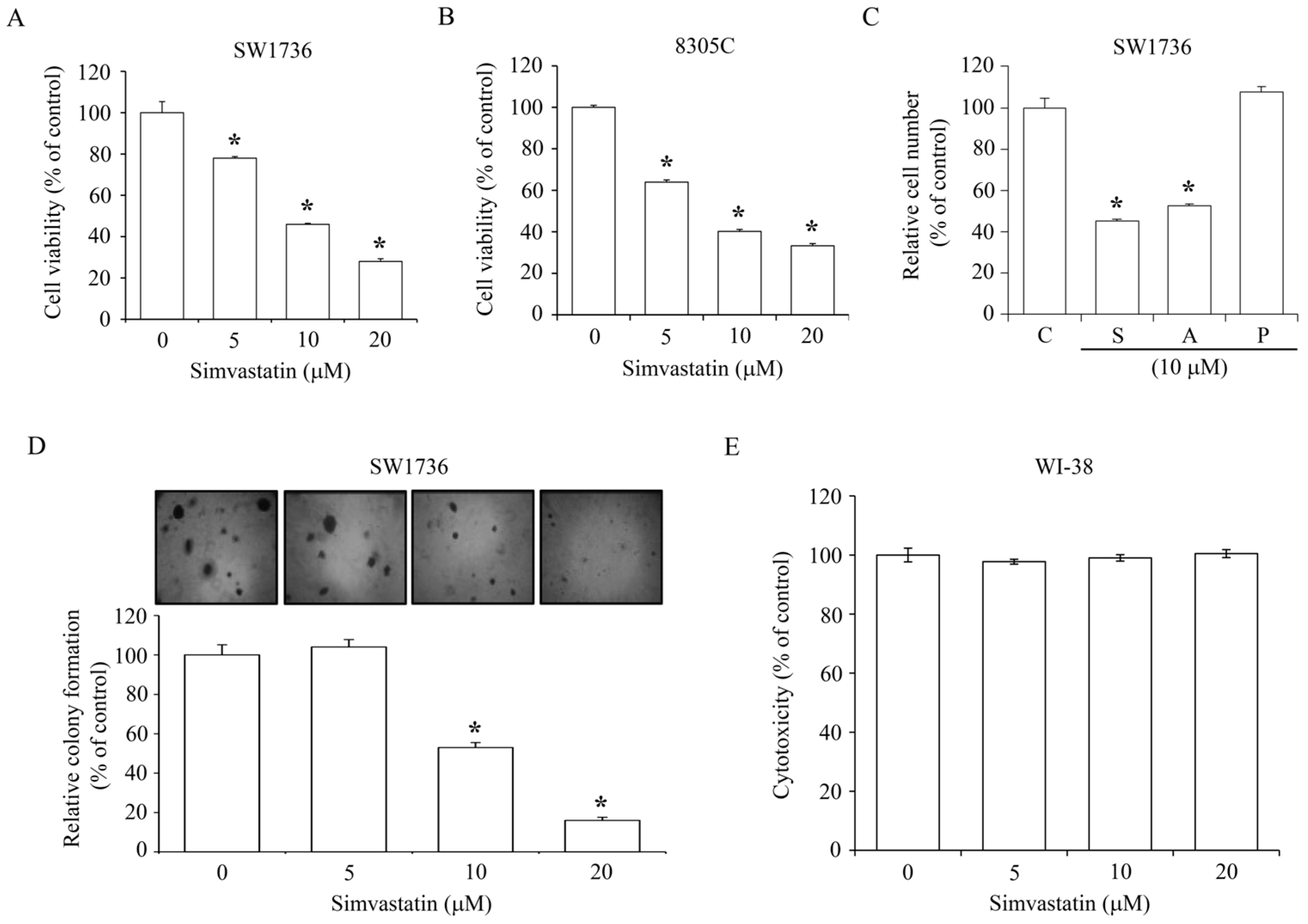
2.1. Inhibition of ATC Cell Growth by Simvastatin
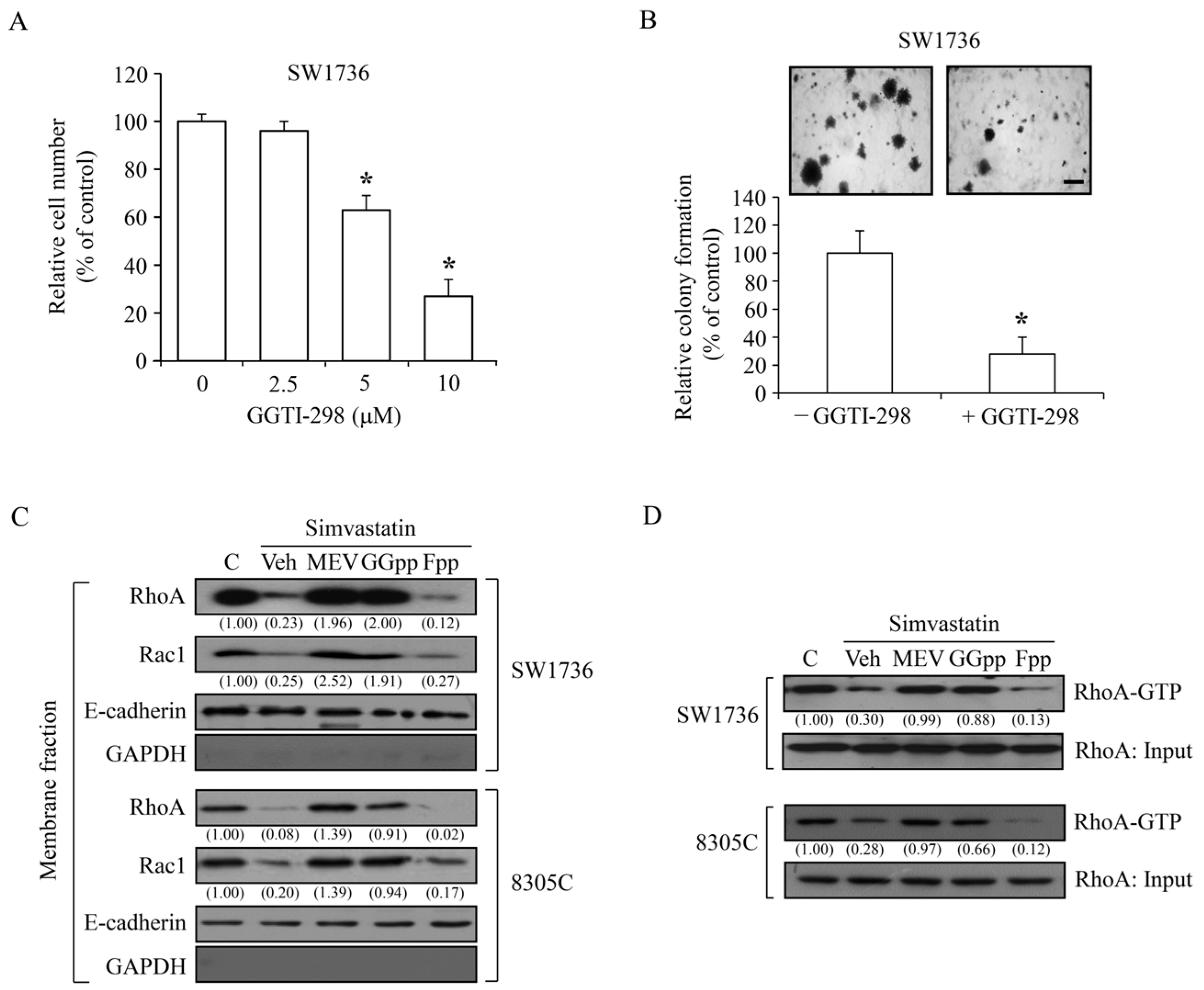
2.2. Effects of MEV and Its Metabolites on the Simvastatin-Induced ATC Cell Proliferation Inhibition
2.3. Simvastatin Reduces the Acitivity of Rho GTPases
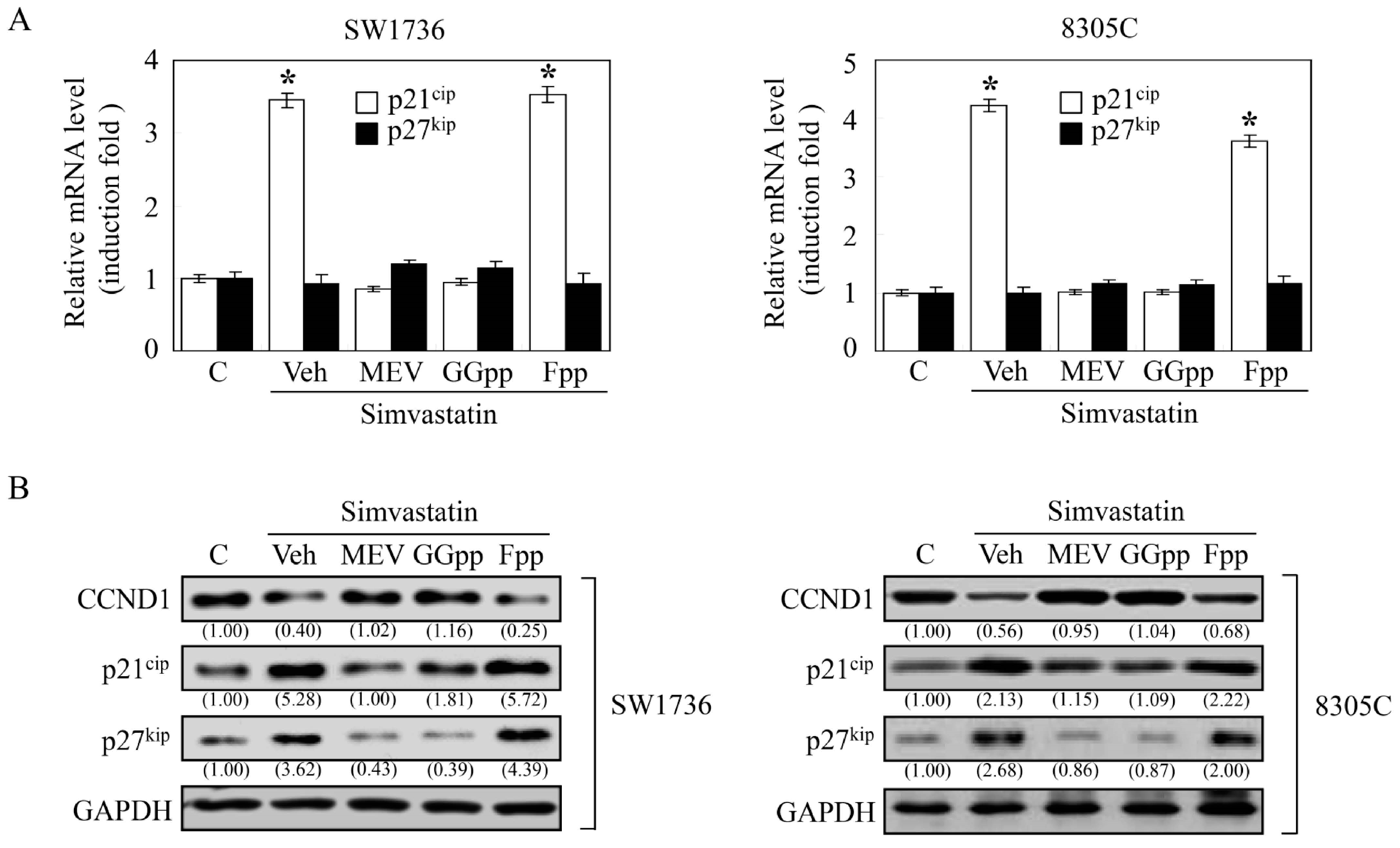
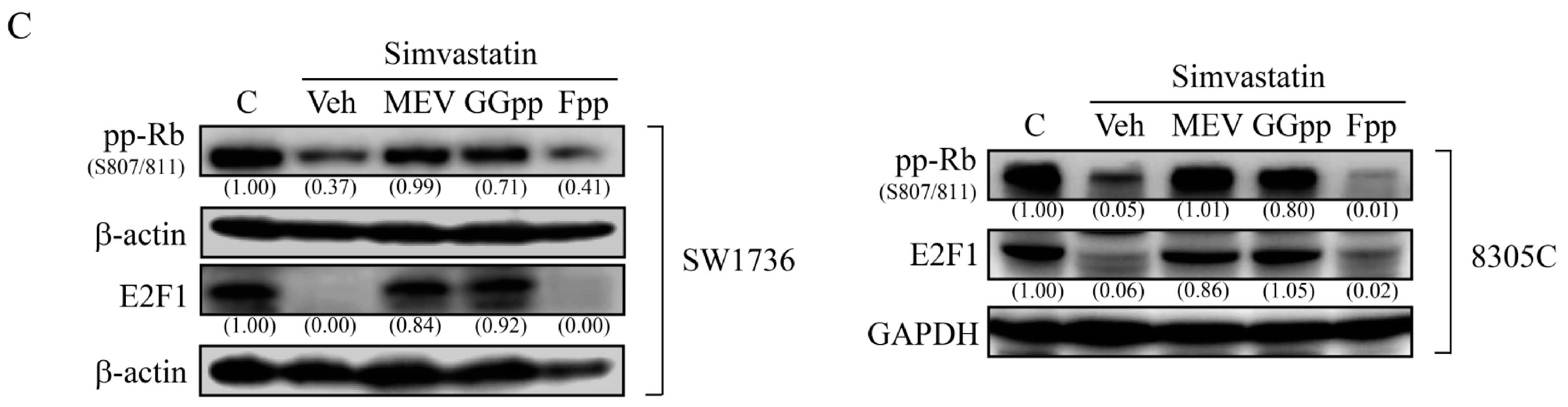
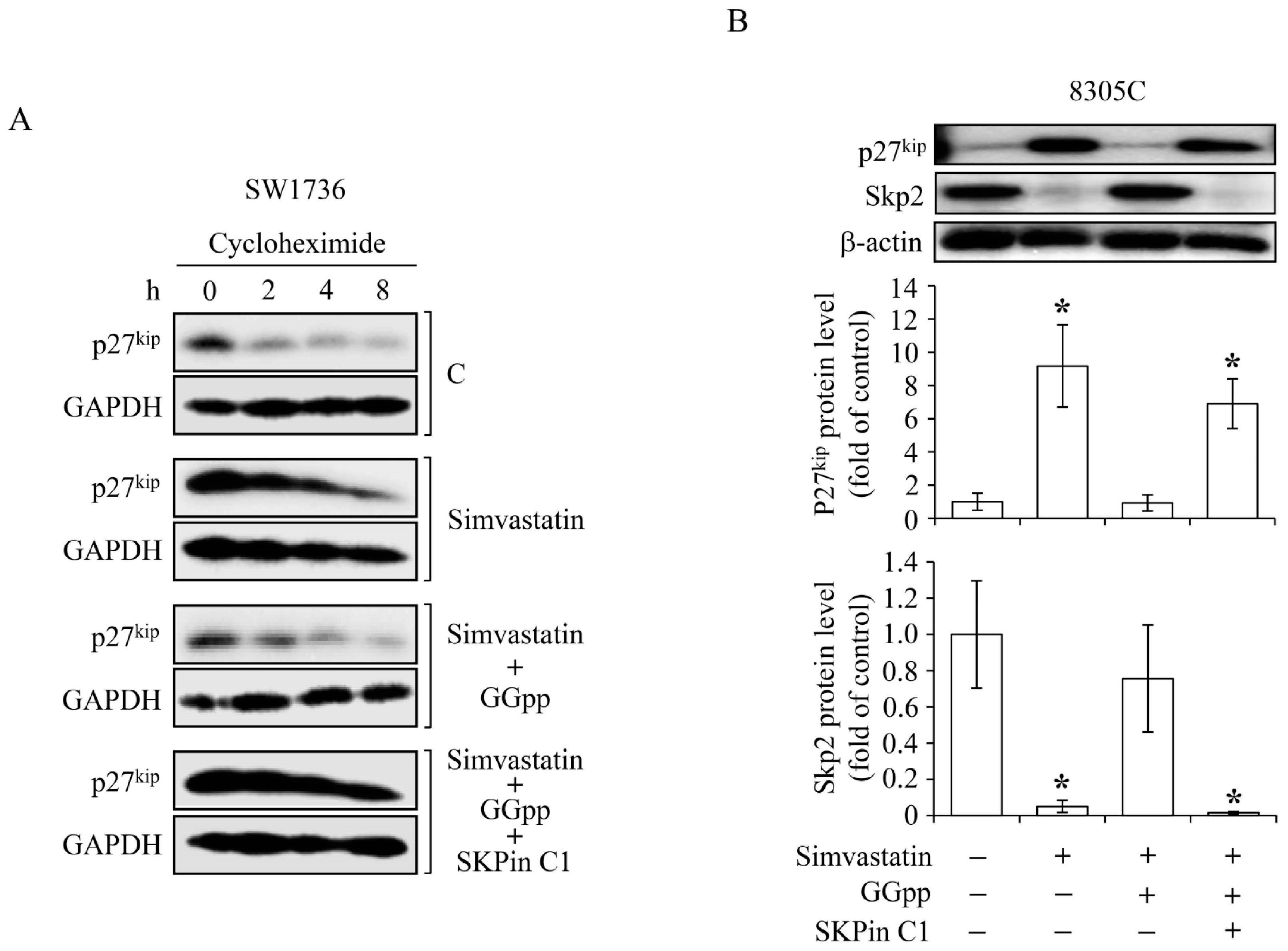
2.4. MEV and GGpp, but Not Fpp, Prevent the Simvastatin-Increased Expression of p21cip and p27kip
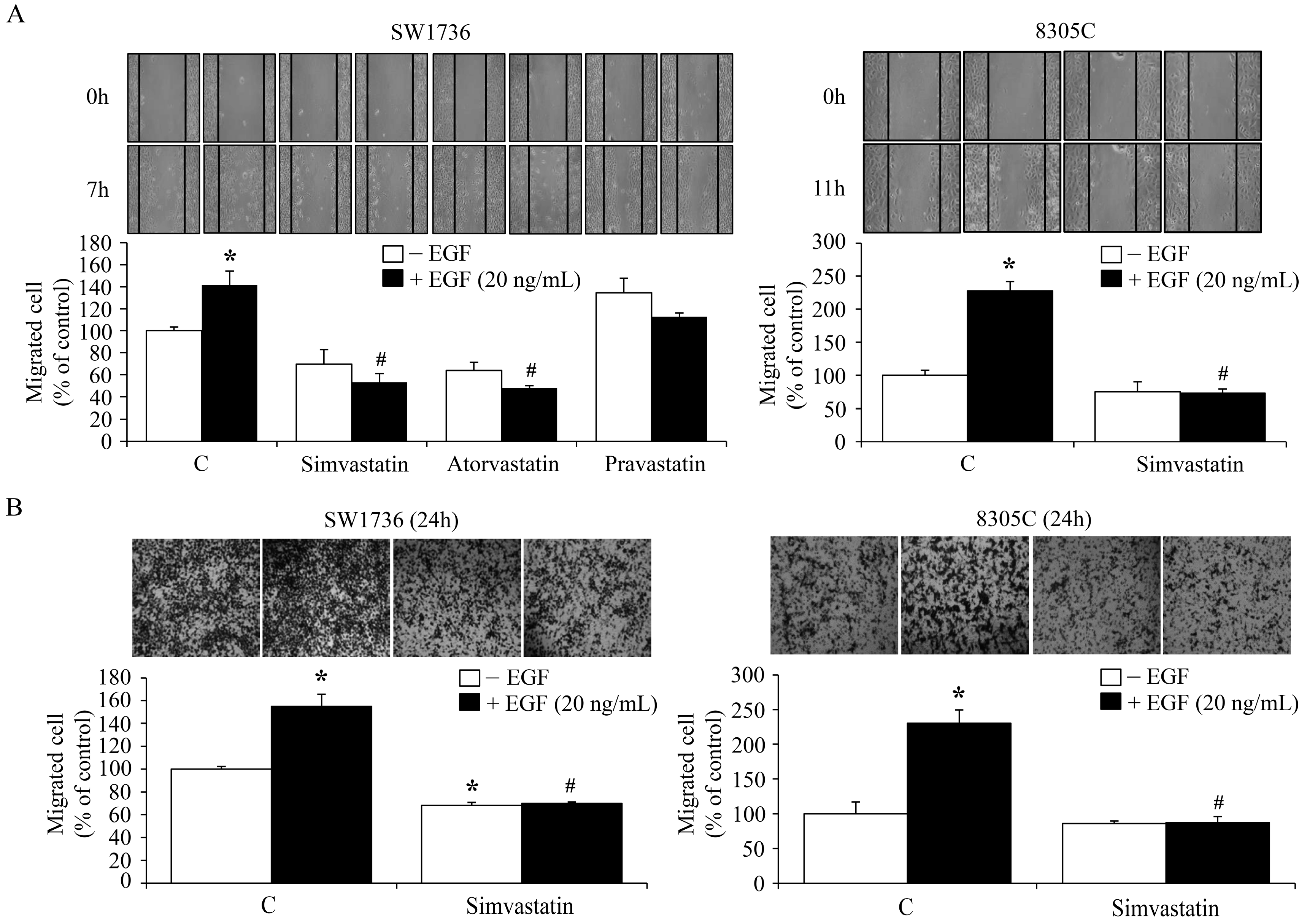
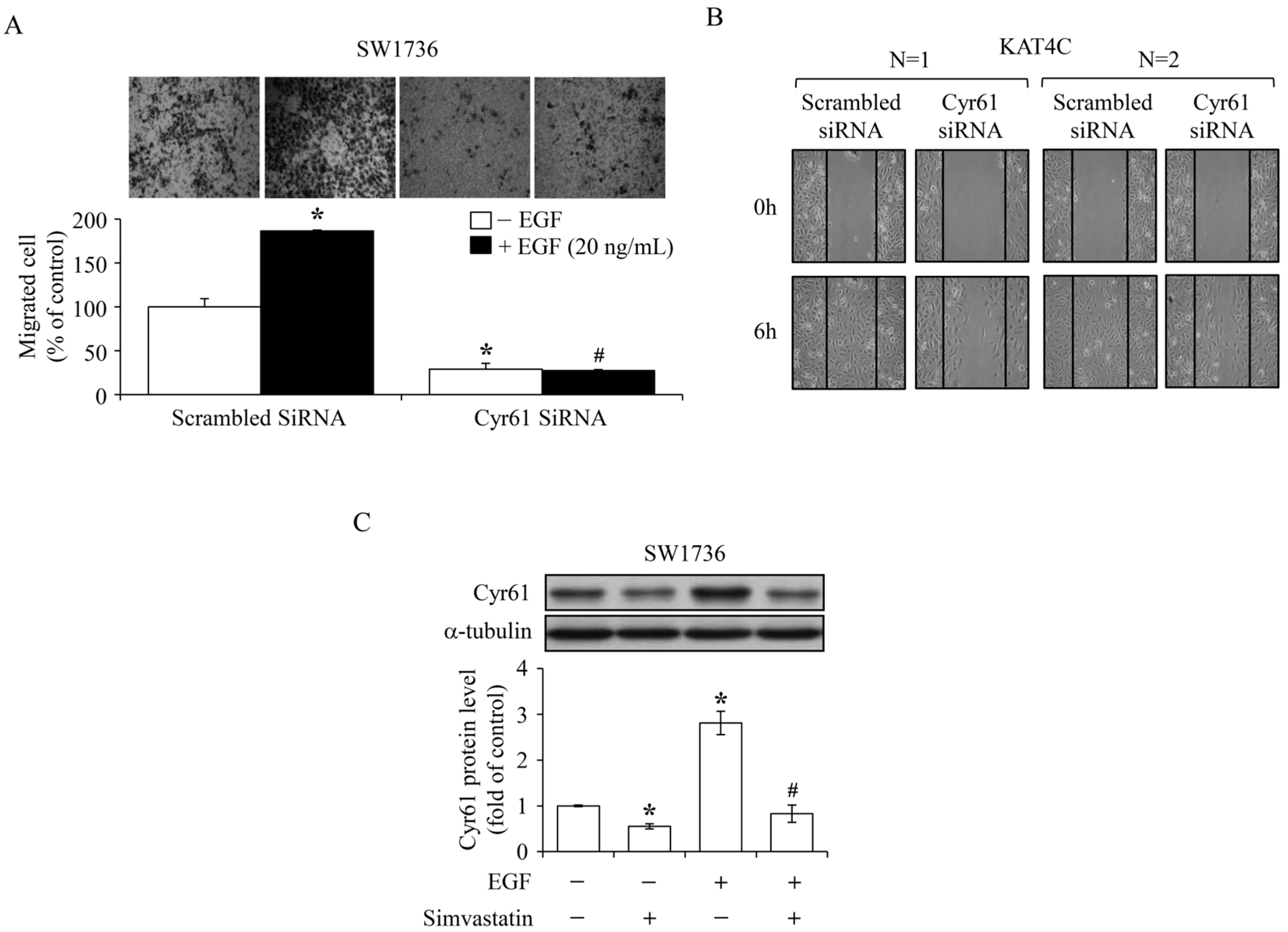
2.5. Inhibition of the EGF-Enhanced ATC Cell Migration by Simvastatin
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents and Antibodies
4.2. Cell Lines and Culture Conditions
4.3. Cell Viability and Proliferation Assay
4.4. Anchorage-Independent Growth Assay
4.5. Cell Cycle Analysis
4.6. Extraction of Membrane-Bound Proteins
4.7. Measurement of RhoA Activity
4.8. Immunoblotting Analysis
4.9. Measurement of mRNA Levels of p21cip and p27kip
4.10. Introduction of Cyr61 Small Interfering RNA (siRNA)
4.11. Scratch Migration Assay
4.12. Transwell Migration Assay
4.13. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviation
| DMSO | Dimethyl sulfoxide |
References
- O’Neill, J.P.; Shaha, A.R. Anaplastic thyroid cancer. Oral Oncol. 2013, 49, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.; Black, M.; Patel, K.; Yoo, J.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. Genomically driven precision medicine to improve outcomes in anaplastic thyroid cancer. J. Oncol. 2014, 2014, 936285. [Google Scholar] [CrossRef] [PubMed]
- Are, C.; Shaha, A.R. Anaplastic thyroid carcinoma: Biology, pathogenesis, prognostic factors, and treatment approaches. Ann. Surg. Oncol. 2006, 13, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Gandhi, A.; Scott-Coombes, D.; Perros, P. Management of thyroid cancer: United Kingdom national multidisciplinary guidelines. J. Laryngol. Otol. 2016, 130, S150–S160. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.F.; Ducic, Y. Aggressive surgical resection of anaplastic thyroid carcinoma may provide long-term survival in selected patients. Otolaryngol. Head Neck Surg. 2013, 148, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tsang, R.; Asa, S.; Dickson, B.; Arenovich, T.; Brierley, J. Clinical outcome of anaplastic thyroid carcinoma treated with radiotherapy of once- and twice-daily fractionation regimens. Cancer 2006, 107, 1786–1792. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Huddart, R.; Harmer, C. Phase II evaluation of high dose accelerated radiotherapy for anaplastic thyroid carcinoma. Radiother. Oncol. 1999, 50, 33–38. [Google Scholar] [CrossRef]
- Nagaiah, G.; Hossain, A.; Mooney, C.J.; Parmentier, J.; Remick, S.C. Anaplastic thyroid cancer: A review of epidemiology, pathogenesis, and treatment. J. Oncol. 2011, 2011, 542358. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, J.; Sugino, K.; Kitagawa, W.; Nagahama, M.; Kameyama, K.; Shimizu, K.; Ito, K. Prognostic factors and treatment outcomes of 100 cases of anaplastic thyroid carcinoma. Thyroid 2011, 21, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.; Dana, T.; Blazina, I.; Daeges, M.; Jeanne, T.L. Statins for prevention of cardiovascular disease in adults: Evidence report and systematic review for the US preventive services task force. JAMA 2016, 316, 2008–2024. [Google Scholar] [CrossRef] [PubMed]
- Miziorko, H.M. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 2011, 505, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.L.; Casey, P.J. Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996, 65, 241–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Casey, P.J. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Hooff, G.P.; Wood, W.G.; Muller, W.E.; Eckert, G.P. Isoprenoids, small GTPases and Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.G.; Eckert, G.P.; Igbavboa, U.; Muller, W.E. Statins and neuroprotection: A prescription to move the field forward. Ann. N. Y. Acad. Sci. 2010, 1199, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Goalstone, M.L.; Wall, K.; Leitner, J.W.; Kurowski, T.; Ruderman, N.; Pan, S.J.; Ivy, J.L.; Moller, D.E.; Draznin, B. Increased amounts of farnesylated p21Ras in tissues of hyperinsulinaemic animals. Diabetologia 1999, 42, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Draznin, B.; Miles, P.; Kruszynska, Y.; Olefsky, J.; Friedman, J.; Golovchenko, I.; Stjernholm, R.; Wall, K.; Reitman, M.; Accili, D.; et al. Effects of insulin on prenylation as a mechanism of potentially detrimental influence of hyperinsulinemia. Endocrinology 2000, 141, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Ullah, N.; Mansha, M.; Casey, P.J. Protein geranylgeranyltransferase type 1 as a target in cancer. Curr. Cancer Drug Targets 2016, 16, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-resolution crispr screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Wakelee, H.A.; Aragaki, A.K.; Tang, J.Y.; Kurian, A.W.; Manson, J.E.; Stefanick, M.L. Protective effects of statins in cancer: Should they be prescribed for high-risk patients? Curr. Atheroscler. Rep. 2016, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Freedman, V.H.; Shin, S.I. Cellular tumorigenicity in nude mice: Correlation with cell growth in semi-solid medium. Cell 1974, 3, 355–359. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and cdk inhibitors in human cancer. Adv. Cancer Res. 1996, 68, 67–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lloyd, R.V.; Hutzler, M.J.; Safran, M.S.; Patwardhan, N.A.; Khan, A. The role of cell cycle regulatory protein, cyclin d1, in the progression of thyroid cancer. Mod. Pathol. 2000, 13, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kobayashi, T.; Takeda, T.; Komoike, Y.; Wakasugi, E.; Tamaki, Y.; Tsujimoto, M.; Matsuura, N.; Monden, M. Expression of p21 (waf1/cip1) protein in clinical thyroid tissues. Br. J. Cancer 1996, 74, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.J.; Crist, H.S.; Durvesh, S.; Bruggeman, R.D.; Goldenberg, D. A comparative study of cell cycle mediator protein expression patterns in anaplastic and papillary thyroid carcinoma. Cancer Biol. Ther. 2012, 13, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Erickson, L.A.; Jin, L.; Wollan, P.C.; Thompson, G.B.; van Heerden, J.; Lloyd, R.V. Expression of p27kip1 and ki-67 in benign and malignant thyroid tumors. Mod. Pathol. 1998, 11, 169–174. [Google Scholar] [PubMed]
- Slingerland, J.; Pagano, M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 2000, 183, 10–17. [Google Scholar] [CrossRef]
- Kossatz, U.; Dietrich, N.; Zender, L.; Buer, J.; Manns, M.P.; Malek, N.P. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004, 18, 2602–2607. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Miyake, S.; Ishida, N.; Hatakeyama, S.; Kitagawa, M.; Iemura, S.; Natsume, T.; Nakayama, K.I. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 2004, 6, 661–672. [Google Scholar] [CrossRef]
- Chin, L.H.; Hsu, S.P.; Zhong, W.B.; Liang, Y.C. Involvement of cysteine-rich protein 61 in the epidermal growth factor-induced migration of human anaplastic thyroid cancer cells. Mol. Carcinog. 2016, 55, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Željko, R. Statins in the primary prevention of cardiovascular disease. Nat. Rev. Cardiol. 2013, 10, 453–464. [Google Scholar] [CrossRef]
- Kanugula, A.K.; Dhople, V.M.; Volker, U.; Ummanni, R.; Kotamraju, S. Fluvastatin mediated breast cancer cell death: A proteomic approach to identify differentially regulated proteins in mda-mb-231 cells. PLoS ONE 2014, 9, e108890. [Google Scholar] [CrossRef] [PubMed]
- Schointuch, M.N.; Gilliam, T.P.; Stine, J.E.; Han, X.; Zhou, C.; Gehrig, P.A.; Kim, K.; Bae-Jump, V.L. Simvastatin, an Hmg-Coa reductase inhibitor, exhibits anti-metastatic and anti-tumorigenic effects in endometrial cancer. Gynecol. Oncol. 2014, 134, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Warita, K.; Warita, T.; Beckwitt, C.H.; Schurdak, M.E.; Vazquez, A.; Wells, A.; Oltvai, Z.N. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci. Rep. 2014, 4, 7593. [Google Scholar] [CrossRef] [PubMed]
- Quesney-Huneeus, V.; Wiley, M.H.; Siperstein, M.D. Essential role for mevalonate synthesis in DNA replication. Proc. Natl. Acad. Sci. USA 1979, 76, 5056–5060. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Copaja, M.; Venegas, D.; Aranguiz, P.; Canales, J.; Vivar, R.; Avalos, Y.; Garcia, L.; Chiong, M.; Olmedo, I.; Catalan, M.; et al. Simvastatin disrupts cytoskeleton and decreases cardiac fibroblast adhesion, migration and viability. Toxicology 2012, 294, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Meredith, J.E., Jr.; Fazeli, B.; Schwartz, M.A. The extracellular matrix as a cell survival factor. Mol. Biol. Cell 1993, 4, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Juliano, R. Mitogenic signal transduction by integrin- and growth factor receptor-mediated pathways. Mol. Cells 2004, 17, 188–202. [Google Scholar] [PubMed]
- Frisch, S.M.; Ruoslahti, E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997, 9, 701–706. [Google Scholar] [CrossRef]
- Gassmann, P.; Haier, J. The tumor cell-host organ interface in the early onset of metastatic organ colonisation. Clin. Exp. Metastasis 2008, 25, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.P.; Philips, M.R. Thematic review series: Lipid posttranslational modifications. Caax modification and membrane targeting of Ras. J. Lipid Res. 2006, 47, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.B.; Hsu, S.P.; Ho, P.Y.; Liang, Y.C.; Chang, T.C.; Lee, W.S. Lovastatin inhibits proliferation of anaplastic thyroid cancer cells through up-regulation of p27 by interfering with the Rho/ROCK-mediated pathway. Biochem. Pharmacol. 2011, 82, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, C.; Giordano, A. Rb and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Guerra, A.; Di Crescenzo, V.; Garzi, A.; Cinelli, M.; Carlomagno, C.; Tonacchera, M.; Zeppa, P.; Vitale, M. Genetic mutations in the treatment of anaplastic thyroid cancer: A systematic review. BMC Surg. 2013, 13, S44. [Google Scholar] [CrossRef] [PubMed]
- Latteyer, S.; Tiedje, V.; Konig, K.; Ting, S.; Heukamp, L.C.; Meder, L.; Schmid, K.W.; Fuhrer, D.; Moeller, L.C. Targeted next-generation sequencing for TP53, RAS, BRAF, ALK and NF1 mutations in anaplastic thyroid cancer. Endocrine 2016, 54, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Abukhdeir, A.M.; Park, B.H. P21 and p27: Roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Tam, S.W.; Theodoras, A.M.; Beer-Romero, P.; del Sal, G.; Chau, V.; Yew, P.R.; Draetta, G.F.; Rolfe, M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995, 269, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.; Pagano, M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin. Cancer Biol. 2003, 13, 41–47. [Google Scholar] [CrossRef]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. Skp2 is required for ubiquitin-mediated degradation of the cdk inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, A.; Huang, S.; Moore, K.; Oh, P.; Ingber, D.E. Role of RhoA, mDia, and ROCK in cell shape-dependent control of the Skp2-p27kip1 pathway and the G1/S transition. J. Biol. Chem. 2004, 279, 26323–26330. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.; Wu, Y.J.; Sala-Newby, G.B.; Newby, A.C. Rho GTPase, Rac1, regulates Skp2 levels, vascular smooth muscle cell proliferation, and intima formation in vitro and in vivo. Cardiovasc. Res. 2008, 80, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, G.; Bloom, J.; Sitry-Shevah, D.; Nakayama, K.; Pagano, M.; Hershko, A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem. 2003, 278, 25752–25757. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. P21 governs p53’s repressive side. Cell Cycle 2016, 15, 2852–2853. [Google Scholar] [CrossRef] [PubMed]
- Ros, P.; Rossi, D.L.; Acebron, A.; Santisteban, P. Thyroid-specific gene expression in the multi-step process of thyroid carcinogenesis. Biochimie 1999, 81, 389–396. [Google Scholar] [CrossRef]
- Haugen, B.R. Redifferentiation therapy in advanced thyroid cancer. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2004, 4, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Zhong, W.B.; Chang, T.C.; Lai, S.M.; Tsai, Y.F. Lovastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, induces apoptosis and differentiation in human anaplastic thyroid carcinoma cells. J. Clin. Endocrinol. Metab. 2003, 88, 3021–3026. [Google Scholar] [CrossRef] [PubMed]
- Zeybek, N.D.; Gulcelik, N.E.; Kaymaz, F.F.; Sarisozen, C.; Vural, I.; Bodur, E.; Canpinar, H.; Usman, A.; Asan, E. Rosuvastatin induces apoptosis in cultured human papillary thyroid cancer cells. J. Endocrinol. 2011, 210, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.A.; Puig-Domingo, M.; Lomena, F.; Estorch, M.; Camacho Marti, V.; Bittini, A.L.; Marazuela, M.; Santamaria, J.; Castro, J.; Martinez de Icaya, P.; et al. Effectiveness of retinoic acid treatment for redifferentiation of thyroid cancer in relation to recovery of radioiodine uptake. J. Endocrinol. Investig. 2009, 32, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.B.; Wang, C.Y.; Chang, T.C.; Lee, W.S. Lovastatin induces apoptosis of anaplastic thyroid cancer cells via inhibition of protein geranylgeranylation and de novo protein synthesis. Endocrinology 2003, 144, 3852–3859. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.B.; Liang, Y.C.; Wang, C.Y.; Chang, T.C.; Lee, W.S. Lovastatin suppresses invasiveness of anaplastic thyroid cancer cells by inhibiting RHO geranylgeranylation and RHOA/rock signaling. Endocr.-Relat. Cancer 2005, 12, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Mullen, P.J.; Zahno, A.; Lindinger, P.; Maseneni, S.; Felser, A.; Krahenbuhl, S.; Brecht, K. Susceptibility to simvastatin-induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of AKT. Biochim. Biophys. Acta 2011, 1813, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Kunter, I.; Erdal, E.; Nart, D.; Yilmaz, F.; Karademir, S.; Sagol, O.; Atabey, N. Active form of AKT controls cell proliferation and response to apoptosis in hepatocellular carcinoma. Oncol. Rep. 2014, 31, 573–580. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.-C.; Tsai, Y.-C.; Tseng, J.-H.; Liou, J.-J.; Horng, S.; Wen, H.-C.; Fan, Y.-C.; Zhong, W.-B.; Hsu, S.-P. Simvastatin Inhibits Cell Proliferation and Migration in Human Anaplastic Thyroid Cancer. Int. J. Mol. Sci. 2017, 18, 2690. https://doi.org/10.3390/ijms18122690
Chen M-C, Tsai Y-C, Tseng J-H, Liou J-J, Horng S, Wen H-C, Fan Y-C, Zhong W-B, Hsu S-P. Simvastatin Inhibits Cell Proliferation and Migration in Human Anaplastic Thyroid Cancer. International Journal of Molecular Sciences. 2017; 18(12):2690. https://doi.org/10.3390/ijms18122690
Chicago/Turabian StyleChen, Mei-Chieh, Yuan-Chin Tsai, Jen-Ho Tseng, Jr-Jiun Liou, Steve Horng, Heng-Ching Wen, Yu-Ching Fan, Wen-Bin Zhong, and Sung-Po Hsu. 2017. "Simvastatin Inhibits Cell Proliferation and Migration in Human Anaplastic Thyroid Cancer" International Journal of Molecular Sciences 18, no. 12: 2690. https://doi.org/10.3390/ijms18122690
APA StyleChen, M. -C., Tsai, Y. -C., Tseng, J. -H., Liou, J. -J., Horng, S., Wen, H. -C., Fan, Y. -C., Zhong, W. -B., & Hsu, S. -P. (2017). Simvastatin Inhibits Cell Proliferation and Migration in Human Anaplastic Thyroid Cancer. International Journal of Molecular Sciences, 18(12), 2690. https://doi.org/10.3390/ijms18122690