Fibrinogen Gamma Chain Mutations Provoke Fibrinogen and Apolipoprotein B Plasma Deficiency and Liver Storage

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Proband
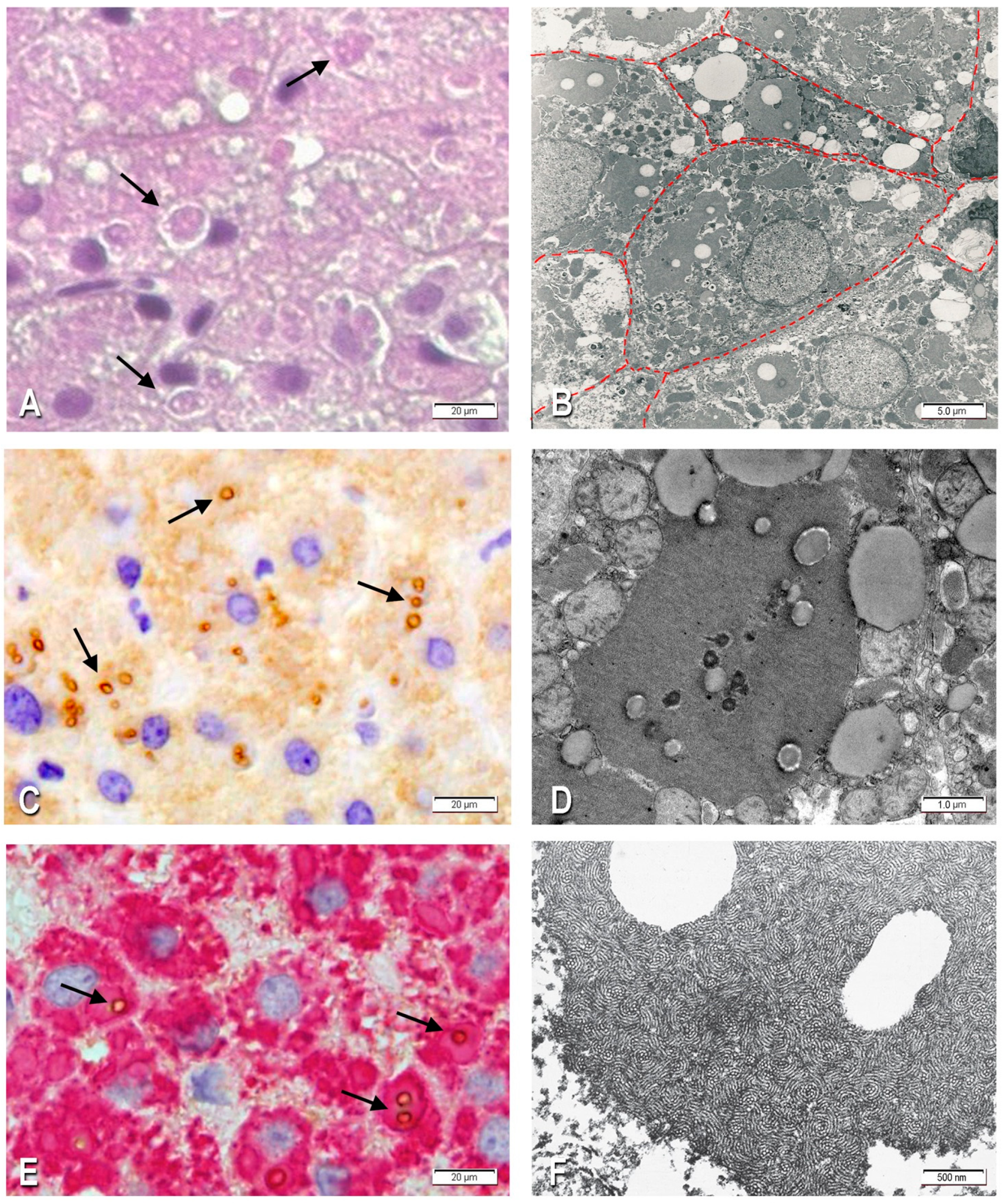
2.1.1. Morphological Studies
2.1.2. Immunohistochemistry
2.1.3. Electron Microscopy
2.1.4. Molecular Genetics Analysis
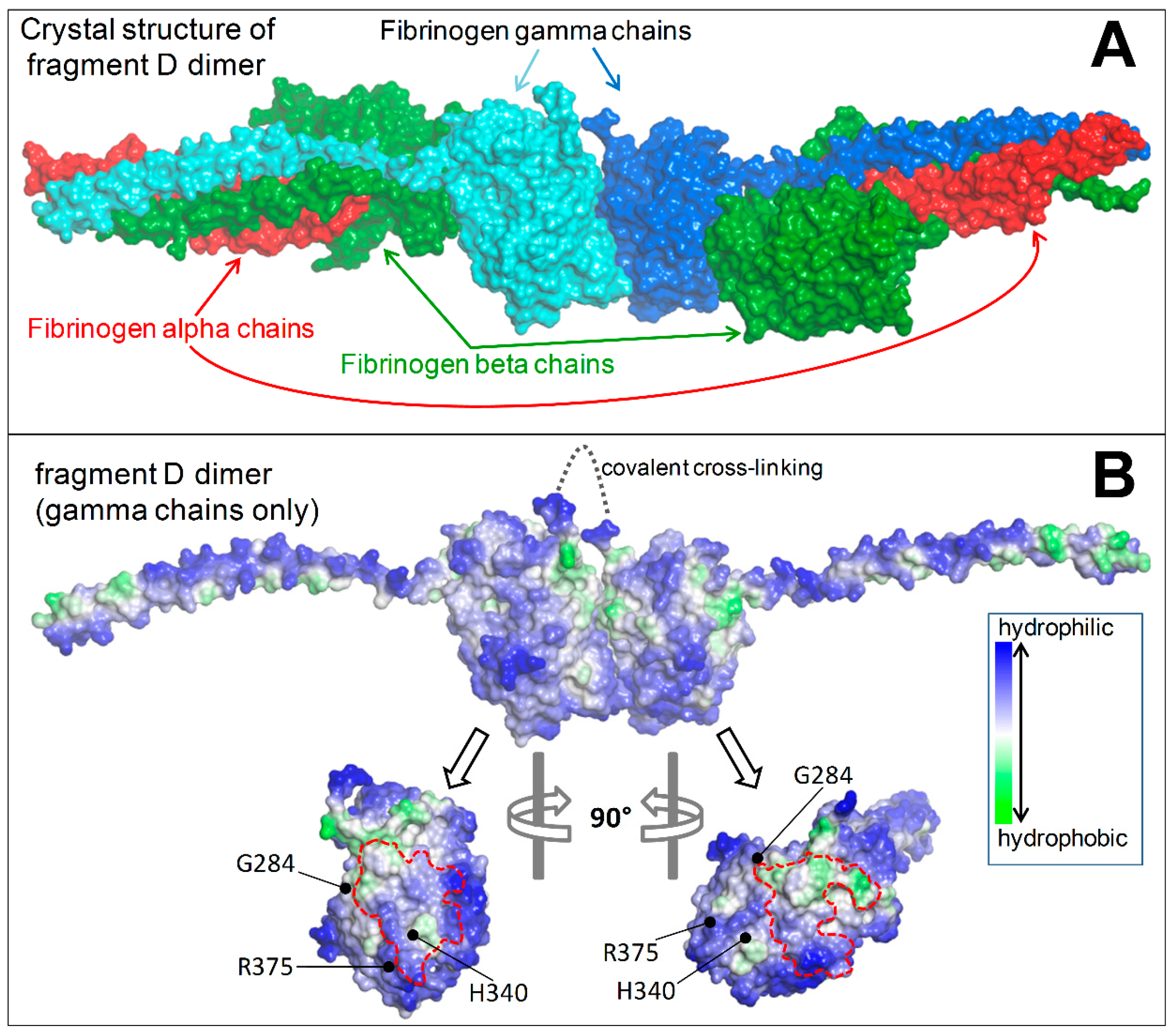
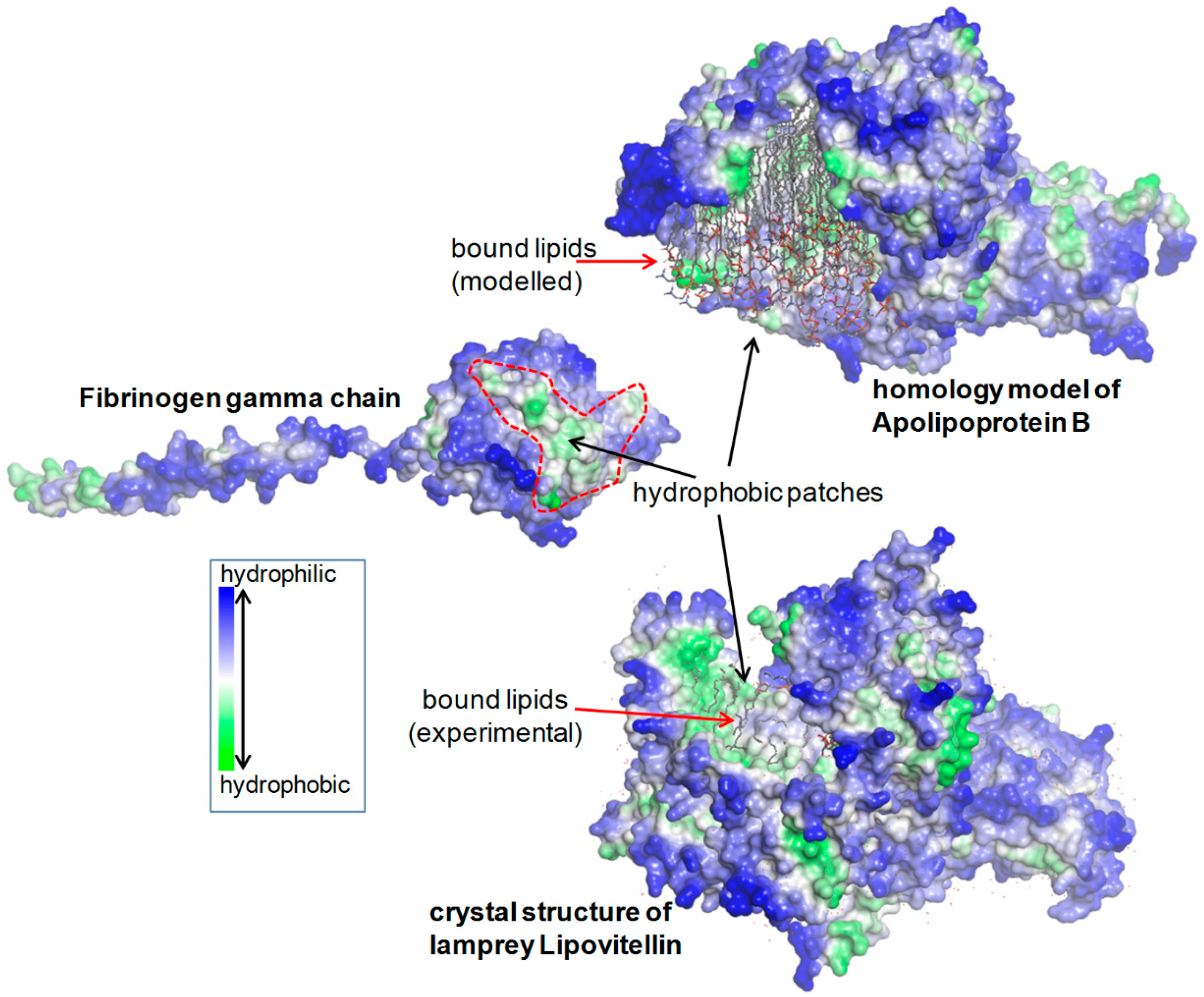
2.1.5. Structural Analysis
2.1.6. Retrospective Analysis
3. Discussion
4. Material and Methods
4.1. Case History (Proband)
4.2. Histology and Immunohistochemistry
4.3. Electron Microscopy
4.4. Molecular Genetics Analysis
4.5. Structural Analysis
4.6. Retrospective Studies
5. Conclusions
Aknowledgments
Author Contributions
Conflict of interest
References
- Callea, F.; De Vos, R.; Pinackat, J. Hereditary Hypofibrinogenemia with Hepatic Storage of Fibrinogen: A New Endoplasmic Reticulum Storage Disease, in Fibrinogen 2; Elsevier: Amsterdam, The Netherlands, 1987; pp. 75–78. [Google Scholar]
- Callea, F.; Brisigotti, M.; Fabbretti, G.; Bonino, F.; Desmet, V.J. Hepatic endoplasmic reticulum storage diseases. Liver 1992, 12, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.O.; Wyatt, J.; Medicina, D.; Callea, F.; George, P.M. Fibrinogen brescia: Hepatic endoplasmic reticulum storage and hypofibrinogenemia because of a gamma284 Gly→Arg mutation. Am. J. Pathol. 2000, 157, 189–196. [Google Scholar] [CrossRef]
- Callea, F.; Tortora, O.; Kojima, T. Hypofibrinogenemia and Fibrinogen Storage Disease, in Fibrinogen 3; Elsevier: Amsterdam, The Netherlands, 1988; pp. 247–250. [Google Scholar]
- Brennan, S.O.; Maghzal, G.; Shneider, B.L.; Gordon, R.; Magid, M.S.; George, P.M. Novel fibrinogen gamma375 Arg→Trp mutation (fibrinogen aguadilla) causes hepatic endoplasmic reticulum storage and hypofibrinogenemia. Hepatology 2002, 36, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Dib, N.; Quelin, F.; Ternisien, C.; Hanss, M.; Michalak, S.; De Mazancourt, P.; Rousselet, M.C.; Cales, P. Fibrinogen angers with a new deletion (gamma GVYYQ 346–350) causes hypofibrinogenemia with hepatic storage. J. Thromb. Haemost. 2007, 5, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.O.; Davis, R.L.; Conard, K.; Savo, A.; Furuya, K.N. Novel fibrinogen mutation gamma314Thr→Pro (fibrinogen AI duPont) associated with hepatic fibrinogen storage disease and hypofibrinogenaemia. Liver Int. 2010, 30, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Robusto, M.; Braidotti, P.; Peyvandi, F.; Nastasio, S.; D’Antiga, L.; Perisic, V.N.; Maggiore, G.; Caccia, S.; Duga, S. Hepatic fibrinogen storage disease: Identification of two novel mutations (p.Asp316Asn, fibrinogen Pisa and p.Gly366Ser, fibrinogen Beograd) impacting on the fibrinogen gamma-module. J. Thromb. Haemost. 2015, 13, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Callea, F.; Giovannoni, I.; Sari, S.; Aksu, A.U.; Esendagly, G.; Dalgic, B.; Boldrini, R.; Akyol, G.; Francalanci, P.; Bellacchio, E. A novel fibrinogen gamma chain mutation (c.1096C>G; p.His340Asp), fibrinogen Ankara, causing hypofibrinogenaemia and hepatic storage. Pathology 2017, 49, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Sari, S.; Yilmaz, G.; Gonul, I.; Dalgic, B.; Akyol, G.; Giovannoni, I.; Francalanci, P.; Callea, F. Fibrinogen storage disease and cirrhosis associated with hypobetalipoproteinemia owing to fibrinogen Aguadilla in a Turkish child. Liver Int. 2015, 35, 2501–2505. [Google Scholar] [CrossRef] [PubMed]
- Sogo, T.; Nagasaka, H.; Komatsu, H.; Inui, A.; Miida, T.; Callea, F.; Francalanci, P.; Hirano, K.; Kitamura, H.; Yorifuji, T.; et al. Fibrinogen storage disease caused by Aguadilla mutation presenting with hypobeta-lipoproteinemia and considerable liver disease. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Callea, F.; Tardanico, R.; Facchetti, E.; Al, E. Pseudo ground-glass hepatocytes immunoreactive for fibrinogen. Occurrence and significance in liver biopsies. Istiocitopatologia 1985, 7, 179–183. [Google Scholar]
- Richardson, P.E.; Manchekar, M.; Dashti, N.; Jones, M.K.; Beigneux, A.; Young, S.G.; Harvey, S.C.; Segrest, J.P. Assembly of lipoprotein particles containing apolipoprotein-B: Structural model for the nascent lipoprotein particle. Biophys. J. 2005, 88, 2789–2800. [Google Scholar] [CrossRef] [PubMed]
- Francalanci, P.; Santorelli, F.M.; Talini, I.; Boldrini, R.; Devito, R.; Camassei, F.D.; Maggiore, G.; Callea, F. Severe liver disease in early childhood due to fibrinogen storage and de novo gamma375Arg→Trp gene mutation. J. Pediatr. 2006, 148, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Neerman-Arbez, M.; Rougemont, A.L.; Male, P.J.; Spahr, L. Fibrinogen gamma375 arg→trp mutation (fibrinogen aguadilla) causes hereditary hypofibrinogenemia, hepatic endoplasmic reticulum storage disease and cirrhosis. Am. J. Surg. Pathol. 2006, 30, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Puls, F.; Goldschmidt, I.; Bantel, H.; Agne, C.; Brocker, V.; Dammrich, M.; Lehmann, U.; Berrang, J.; Pfister, E.D.; Kreipe, H.H.; et al. Autophagy-enhancing drug carbamazepine diminishes hepatocellular death in fibrinogen storage disease. J. Hepatol. 2013, 59, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, A.; Altalhi, A.; El Hag, I.; AlHussaini, H.; Francalanci, P.; Giovannoni, I.; Callea, F. Hepatic fibrinogen storage disease due to the fibrinogen gamma375 Arg→Trp mutation “fibrinogen Aguadilla” is present in Arabs. Saudi J. Gastroenterol. 2014, 20, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Sokollik, C.; Lukowski, S.W.; Lurz, E.; Rieubland, C.; de Moerloose, P.; Neerman-Arbez, M. Hypofibrinogenemia and liver disease: A new case of Aguadilla fibrinogen and review of the literature. Haemophilia 2015, 21, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.H.; Knisely, A.S.; Wang, N.L.; Gong, J.Y.; Wang, J.S. Fibrinogen storage disease in a Chinese boy with de novo fibrinogen Aguadilla mutation: Incomplete response to carbamazepine and ursodeoxycholic acid. BMC Gastroenterol. 2016, 16, 92. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.P.; Havel, R.J. Disorders of the Biogenesis and Secretion of Lipoproteins Containing the B Apolipoproteins; McGraw-Hill Professional: London, UK, 2000. [Google Scholar]
- Burnett, J.R.; Zhong, S.; Jiang, Z.G.; Hooper, A.J.; Fisher, E.A.; McLeod, R.S.; Zhao, Y.; Barrett, P.H.; Hegele, R.A.; van Bockxmeer, F.M.; et al. Missense mutations in APOB within the betaalpha1 domain of human APOB-100 result in impaired secretion of ApoB and ApoB-containing lipoproteins in familial hypobetalipoproteinemia. J. Biol. Chem. 2007, 282, 24270–24283. [Google Scholar] [CrossRef] [PubMed]
- Spraggon, G.; Everse, S.J.; Doolittle, R.F. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature 1997, 389, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.R.; Banaszak, L.J. Lipid-protein interactions in lipovitellin. Biochemistry 2002, 41, 9398–9409. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Zhou, H.; Figeys, D.; Wang, Y.; Sundaram, M. Microsome-associated lumenal lipid droplets in the regulation of lipoprotein secretion. Curr. Opin. Lipidol. 2013, 24, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Medicina, D.; Fabbretti, G.; Brennan, S.O.; George, P.M.; Kudryk, B.; Callea, F. Genetic and immunological characterization of fibrinogen inclusion bodies in patients with hepatic fibrinogen storage and liver disease. Ann. N. Y. Acad. Sci. 2001, 936, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.M.; Xia, H. Fibrinogen biosynthesis. Assembly, intracellular degradation, and association with lipid synthesis and secretion. Ann. N. Y. Acad. Sci. 2001, 936, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Kruse, K.B.; Dear, A.; Kaltenbrun, E.R.; Crum, B.E.; George, P.M.; Brennan, S.O.; McCracken, A.A. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: An explanation for liver disease. Am. J. Pathol. 2006, 168, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Shelness, G.S.; Ingram, M.F.; Huang, X.F.; DeLozier, J.A. Apolipoprotein B in the rough endoplasmic reticulum: Translation, translocation and the initiation of lipoprotein assembly. J. Nutr. 1999, 129 (Suppl. 2S), 456S–462S. [Google Scholar]
- Fisher, E.; Lake, E.; McLeod, R.S. Apolipoprotein B100 quality control and the regulation of hepatic very low density lipoprotein secretion. J. Biomed. Res. 2014, 28, 178–193. [Google Scholar] [PubMed]
- Maggiore, G.; Nastasio, S.; Sciveres, M. Long-term outcome of liver disease-related fibrinogen aguadilla storage disease in a child. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 699. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Case | Age | Sex | Ethn | Mutation | Fibrinogen (Normal Value-n.v. 200–400) | APO-B | TChol | LDLChol | Triglyc | Lipid Inclusion | Liver Path |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 Proband | 2 y | F | Turkey | Aguadilla | 74 | <24.5 | 69.3 | 14 | 32.6 | +++ | Portal and septal fibrosis |
| 2 | 5 y | F | Turkey | Aguadilla | 48 | <22.1 | 69 | 37 | 28 | ++ | Mild |
| 3 | 4 y | M | Italy | Aguadilla | 43 | NA | NA | NA | NA | ++ | Septal fibrosis |
| 4 | 2 y | M | Japan | Aguadilla | 37.6 | 22 | 76 | 32 | 45 | +++ | Early cirrhosis |
| 5 | 3 y | F | Saudi A. | Aguadilla | 89 | NA | NA | NA | NA | +++ | Mild |
| 6 | 5.5 y | F | Turkey | Ankara | 49 | 75 | 169 | 99 | 100 | ++ | Mild |
| 7 | 49 y | F | Italy | Brescia | 20 | NA | NA | NA | NA | +++ | Cirrhosis |
| Reference | Mutant Fibrinogen | Age (Year) | Gender | Clinical Presentation | Liver Disease |
|---|---|---|---|---|---|
| [3] | |||||
| Index | Brescia | 64 | M | Elevated ALT/AST | Severe |
| 6 adult family members | 5 F & 1 M | Mild | |||
| [5] | |||||
| Index | Aguadilla | 3 | F | Elevated ALT/AST | Mild |
| Sibling | 7 | F | Elevated ALT/AST | Mild | |
| Father | NA | M | Mild | ||
| [14] | |||||
| Index | Aguadilla | 6 | M | Elevated ALT/AST | Severe |
| [15] | |||||
| Index | Aguadilla | 61 | M | Elevated ALT/AST | Severe |
| Son | 28 | M | Normal ALT/AST | Mild | |
| Son | 22 | M | Normal ALT/AST | ||
| [6] | |||||
| Index | Anger | 35 | F | Elevated ALT/AST | Severe |
| 3 adult family members | 2 F & 1 M | Mild | |||
| [11] | |||||
| Index | Aguadilla | 2 | M | Elevated ALT/AST and hypo-β-lipoproteinemia | Moderate–Severe |
| Father | Normal ALT/AST | ||||
| [7] | |||||
| Index | Al duPont | 4 | M | Elevated ALT/AST and mild coagulopathy | Mild |
| [16] | |||||
| Index | Aguadilla | 6 | F | Elevated ALT/AST | Mild |
| [16] | |||||
| Index | Brescia | 5 | M | Elevated ALT/AST | Mild |
| [17] | |||||
| Index | Aguadilla | 3 | F | Elevated ALT/AST | Mild |
| Father | 42 | ||||
| [10] | |||||
| Index | Aguadilla | 7 | F | Elevated ALT/AST and hypo-β-lipoproteinemia | Mild |
| Mother | 33 | F | Normal ALT/AST | ||
| Brother | 11 | M | Normal ALT/AST | ||
| Sister I | 9 | F | Normal ALT/AST | ||
| Sister II | 5 | F | Elevated ALT/AST | ||
| [18] | |||||
| Index | Aguadilla | 4.6 | F | Fibrinogen = 70 mg/dL Elevated ALT/AST | NA |
| Mother | Elevated ALT/AST | NA | |||
| Grandmother | Elevated ALT/AST | NA | |||
| [8] | |||||
| Index | Aguadilla | 3 | F | Fibrinogen (immunological 117 (n.v. 160–400) | Normal Liver |
| Mother | |||||
| Maternal grandfather | |||||
| [8] | |||||
| Index | Aguadilla | 3 | M | Fibrinogen 66 mg/dL Elevated ALT/AST | Normal Liver |
| Mother | |||||
| Brother | |||||
| [9] | |||||
| Index | Ankara | 5.5 | F | Elevated ALT/AST Hypo-APOB-lipoproteinemia | Mild |
| Father | |||||
| [19] | |||||
| Index | Aguadilla (de novo) | 2 | M | Fibrinogen 29mg/dL | Portal fibrosis and mild hepatitis |
| Present Case | |||||
| Index | Aguadilla | 3 | F | Elevated ALT/AST hypo-APOB-lipoprotereinemia | Mild |
| Mother | 24 | F | Normal ALT/AST | NA |
| Proband | Sibling | Mother | Father | |
|---|---|---|---|---|
| Age | 2 years | 4 years | 24 years | 36 years |
| Gender | F | M | F | M |
| Hepatomegaly | - | - | - | - |
| Splenomegaly | - | - | - | - |
| AST, U/L (n.v. 20–60) | 77 | 25 | 15 | 30 |
| ALT, U/L (n.v. 5–45) | 151 | 13 | 13 | 47 |
| GGT, U/L (n.v. 5–32) | 67 | 10 | 10 | 35 |
| ALP, U/L (n.v. 145–420) | 316 | 216 | 83 | 67 |
| Triglyceride, mg/dL (n.v. 34–112) | 32.6 | 75 | 39.8 | 282.6 |
| Cholesterol, mg/dL (n.v. 112–200) | 69.3 | 176.8 | 167.7 | 245.7 |
| LDL-Cholesterol, mg/dL (n.v. 63–129) | 14 | 103 | 95 | 152 |
| APOB mg/dL (n.v. 55–135) | <24.5 | NA | NA | 128 |
| PT, s. (n.v. 12.1–14.5) | 14.8 | NA | NA | 11.3 |
| INR (n.v. 0.92–1.14) | 1.27 | NA | NA | 0.95 |
| PTT, s. 1 (n.v. 25–34) | 26 | NA | NA | 25 |
| Fibrinogen, mg/dL (n.v. 200–400) | 74 | NA | 140 | 292 |
| Liver biopsy | + | NA | NA | NA |
| Molecular analysis | Aguadilla | - | Aguadilla | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callea, F.; Giovannoni, I.; Sari, S.; Guldal, E.; Dalgic, B.; Akyol, G.; Sogo, T.; Al-Hussaini, A.; Maggiore, G.; Bartuli, A.; et al. Fibrinogen Gamma Chain Mutations Provoke Fibrinogen and Apolipoprotein B Plasma Deficiency and Liver Storage. Int. J. Mol. Sci. 2017, 18, 2717. https://doi.org/10.3390/ijms18122717
Callea F, Giovannoni I, Sari S, Guldal E, Dalgic B, Akyol G, Sogo T, Al-Hussaini A, Maggiore G, Bartuli A, et al. Fibrinogen Gamma Chain Mutations Provoke Fibrinogen and Apolipoprotein B Plasma Deficiency and Liver Storage. International Journal of Molecular Sciences. 2017; 18(12):2717. https://doi.org/10.3390/ijms18122717
Chicago/Turabian StyleCallea, Francesco, Isabella Giovannoni, Sinan Sari, Esendagli Guldal, Buket Dalgic, Gulen Akyol, Tsuyoshi Sogo, Abdulrahman Al-Hussaini, Giuseppe Maggiore, Andrea Bartuli, and et al. 2017. "Fibrinogen Gamma Chain Mutations Provoke Fibrinogen and Apolipoprotein B Plasma Deficiency and Liver Storage" International Journal of Molecular Sciences 18, no. 12: 2717. https://doi.org/10.3390/ijms18122717
APA StyleCallea, F., Giovannoni, I., Sari, S., Guldal, E., Dalgic, B., Akyol, G., Sogo, T., Al-Hussaini, A., Maggiore, G., Bartuli, A., Boldrini, R., Francalanci, P., & Bellacchio, E. (2017). Fibrinogen Gamma Chain Mutations Provoke Fibrinogen and Apolipoprotein B Plasma Deficiency and Liver Storage. International Journal of Molecular Sciences, 18(12), 2717. https://doi.org/10.3390/ijms18122717