Microsomal Prostaglandin E Synthase-1 Facilitates an Intercellular Interaction between CD4+ T Cells through IL-1β Autocrine Function in Experimental Autoimmune Encephalomyelitis


Abstract
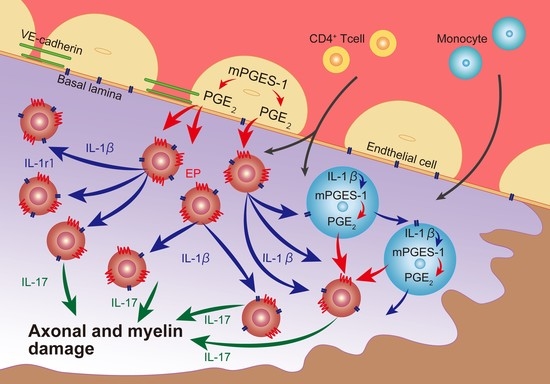
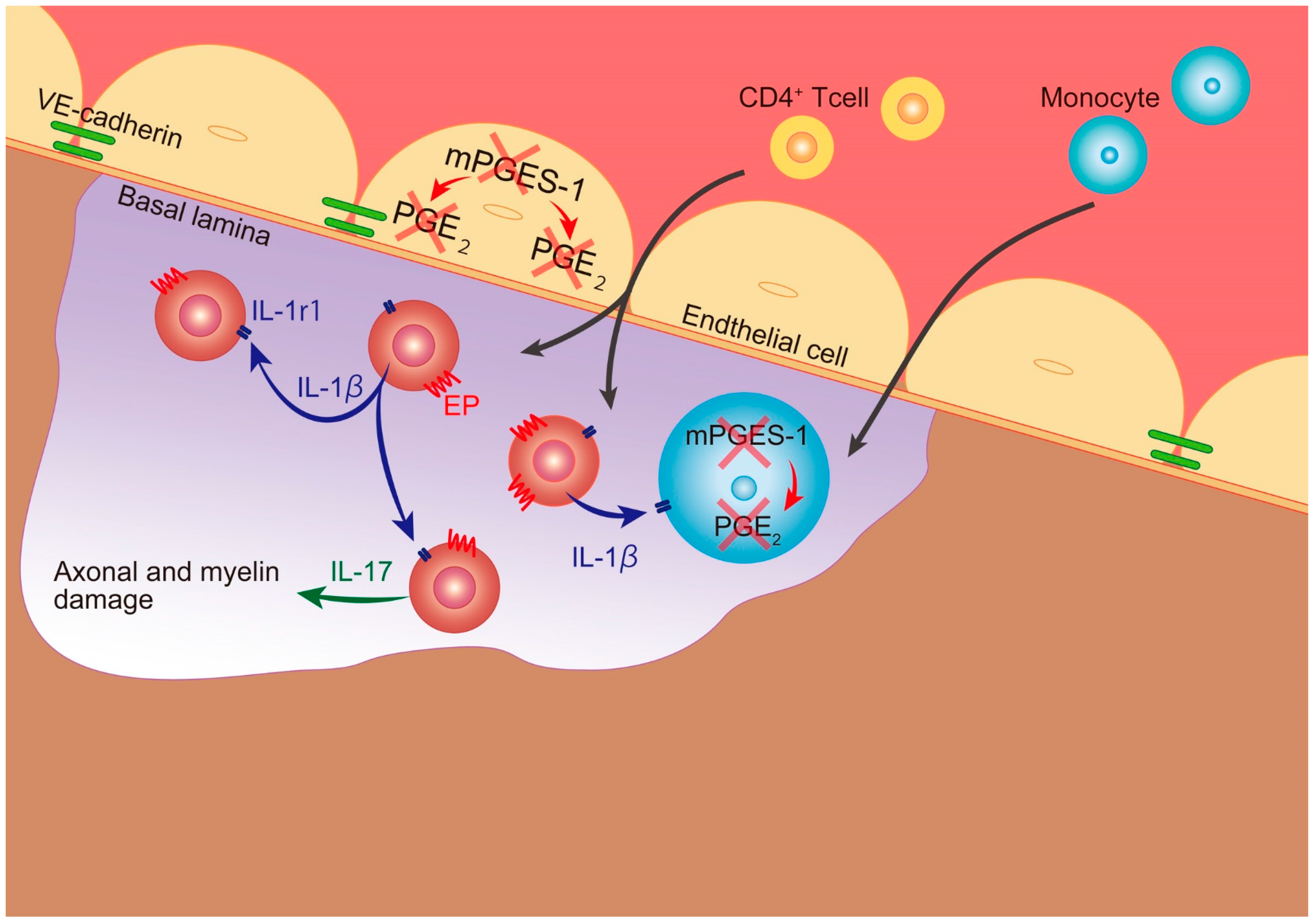
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. mPGES-1 Induced in Endothelial Cells and Macrophages Aggravates EAE Paralysis
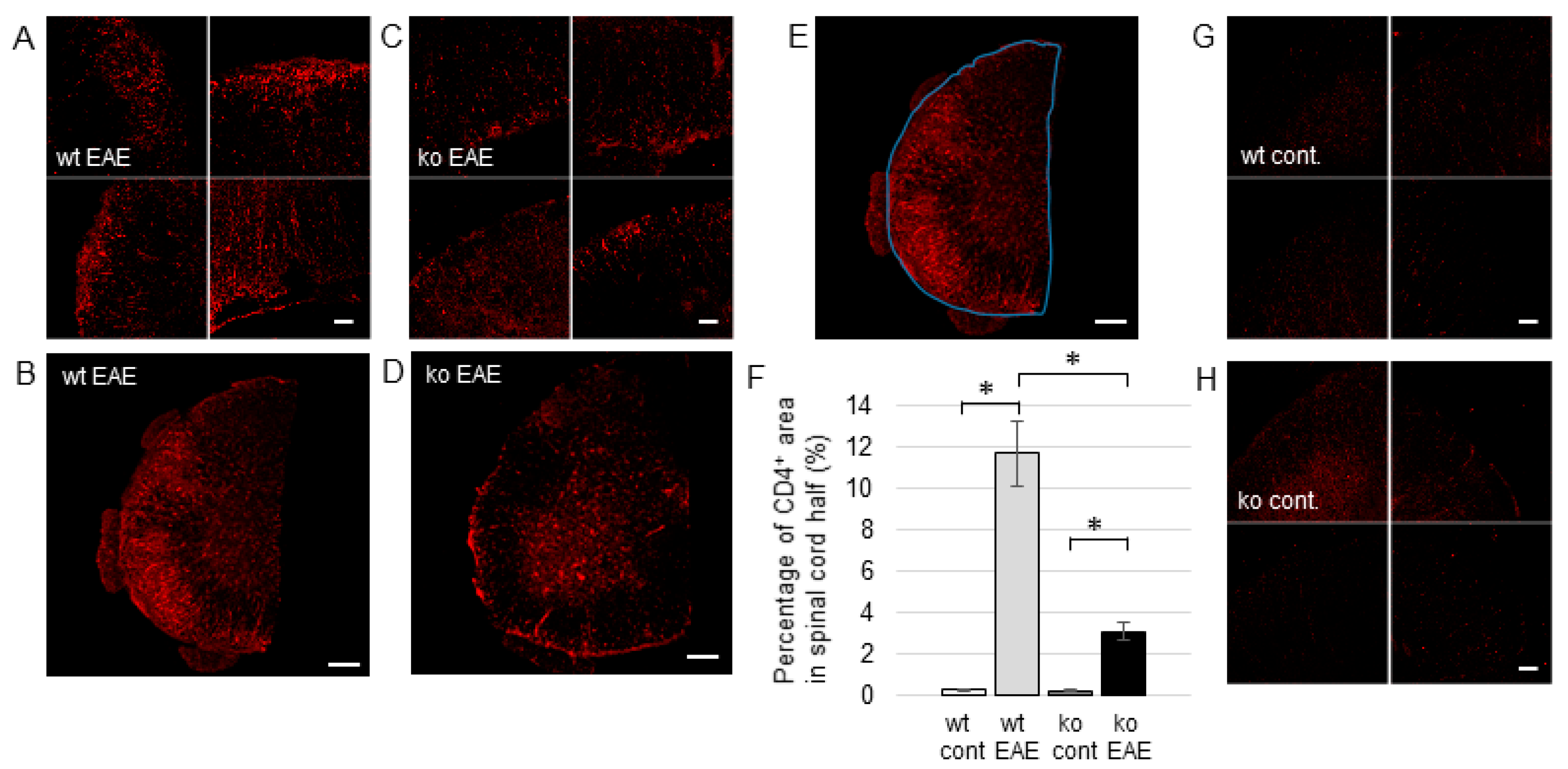
2.2. CD4+ T Cell Invasion in the EAE Spinal Cord is Facilitated by mPGES-1
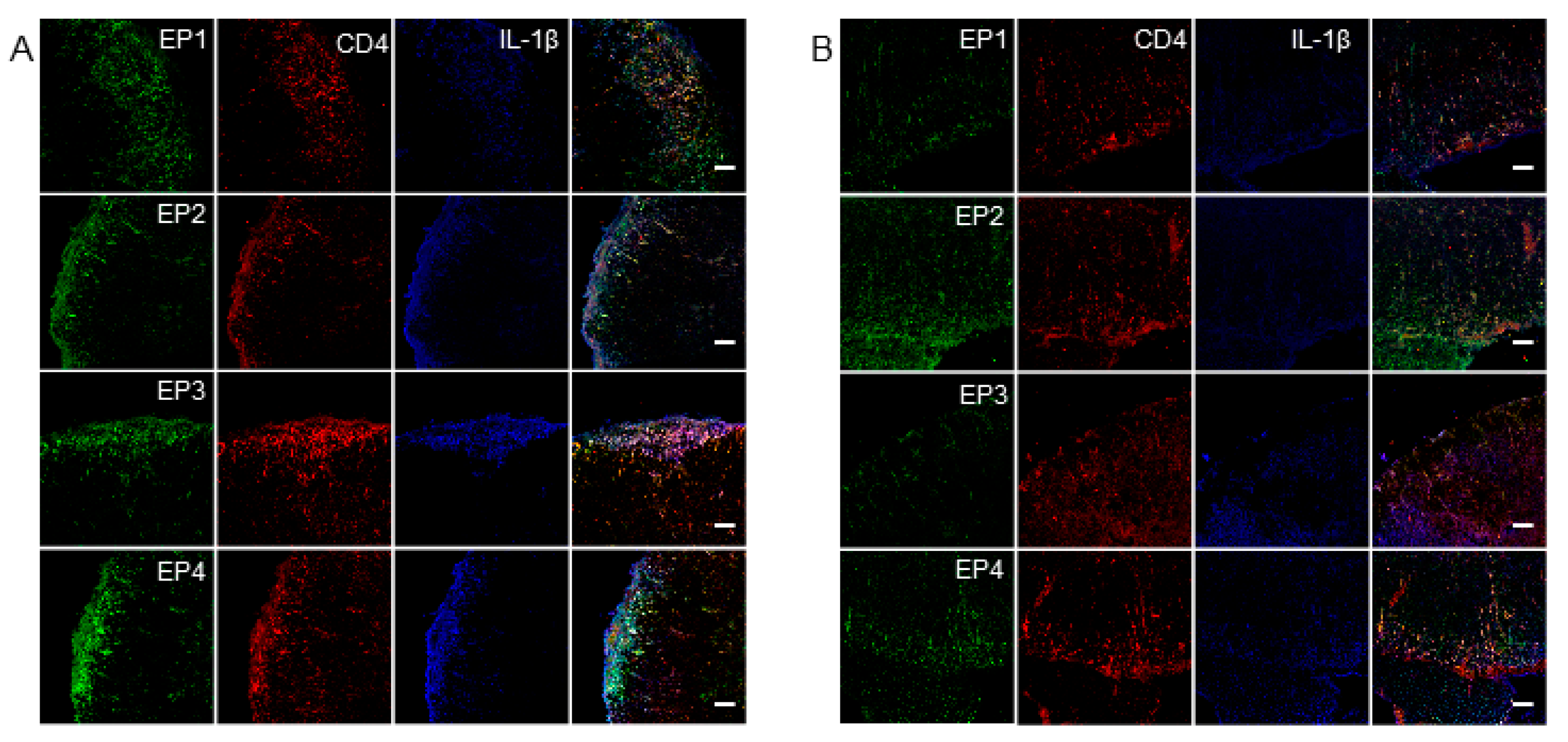
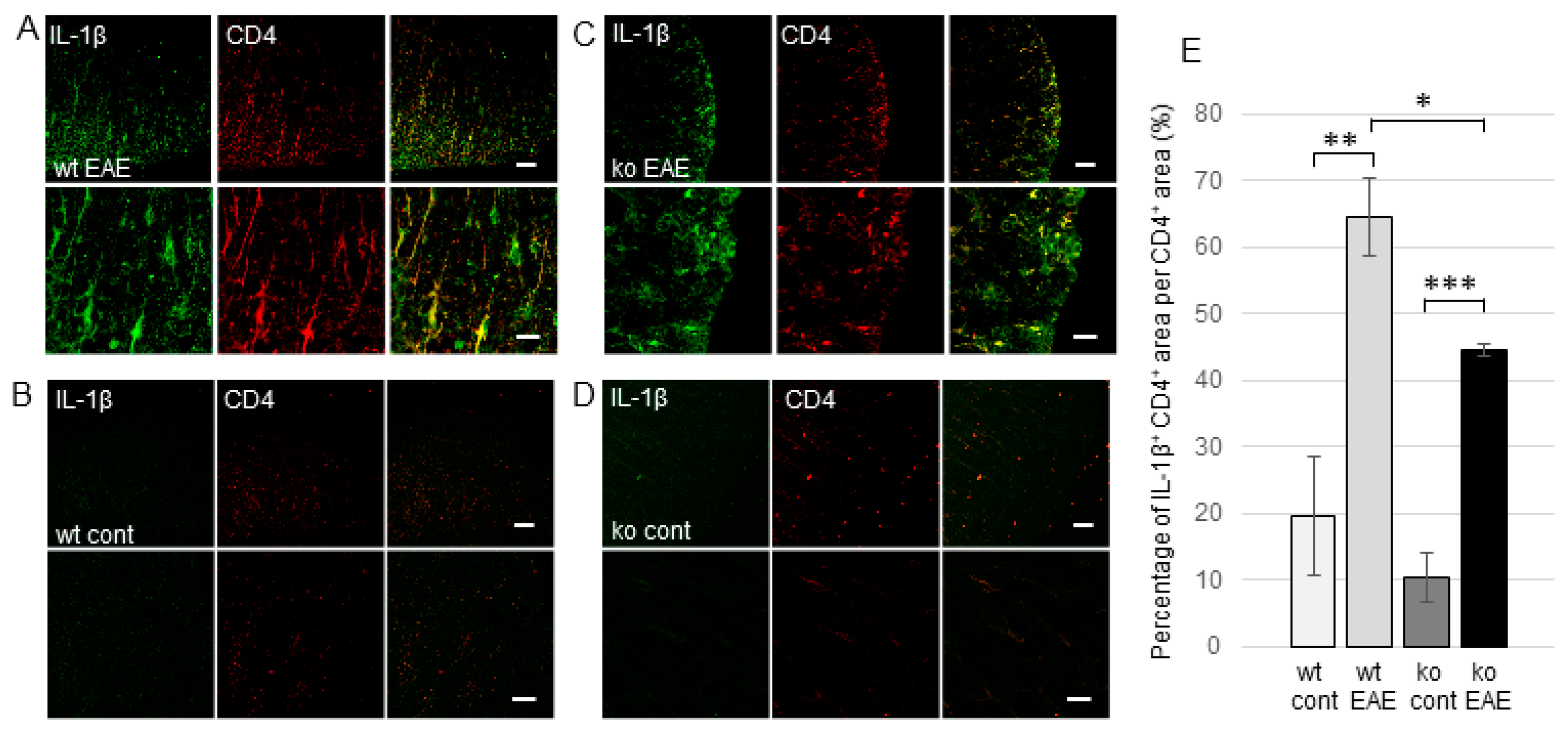
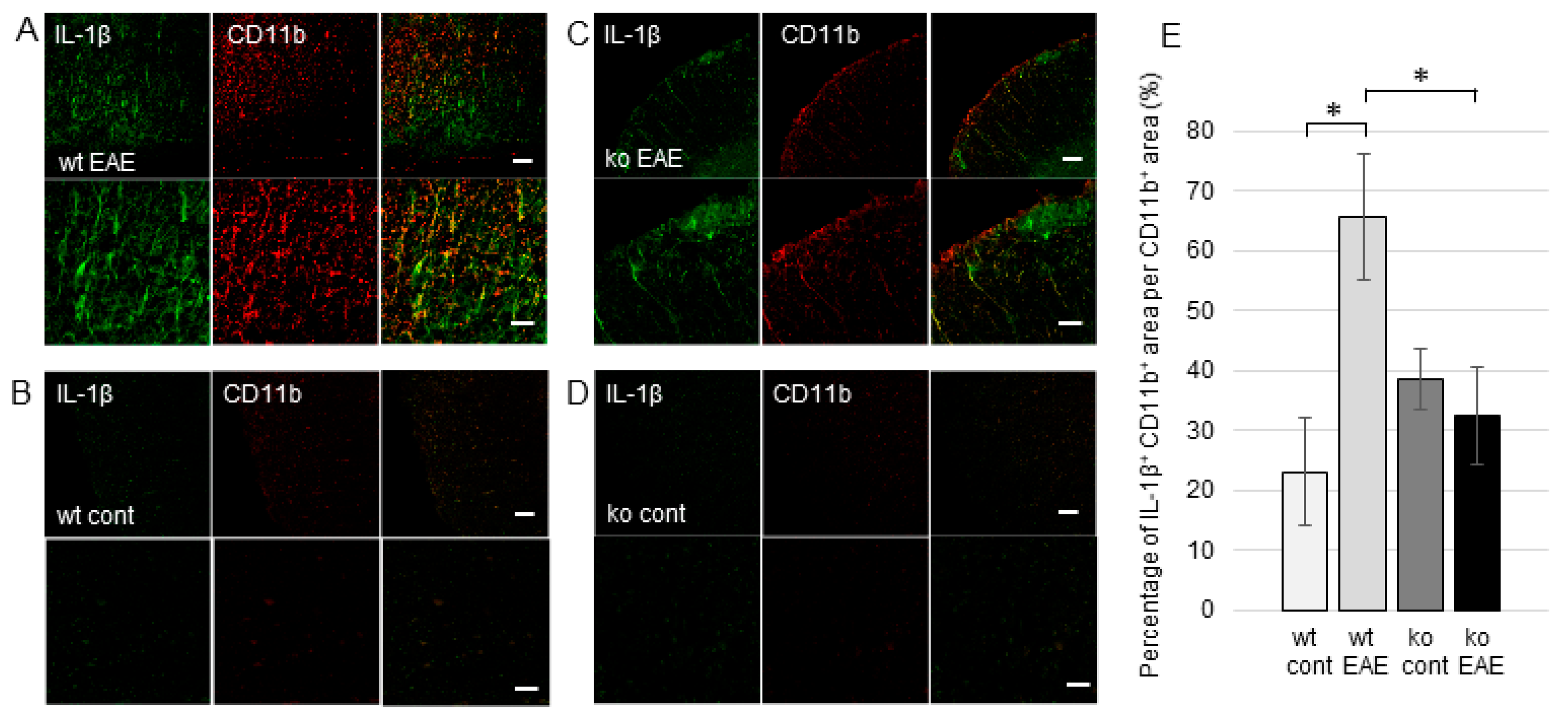
2.3. The Induction of EP Receptors and IL-1β in CD4+ T Cells is Regulated by mPGES-1
2.4. IL-1r1 Expression in CD4+ T Cells is Controlled by mPGES-1
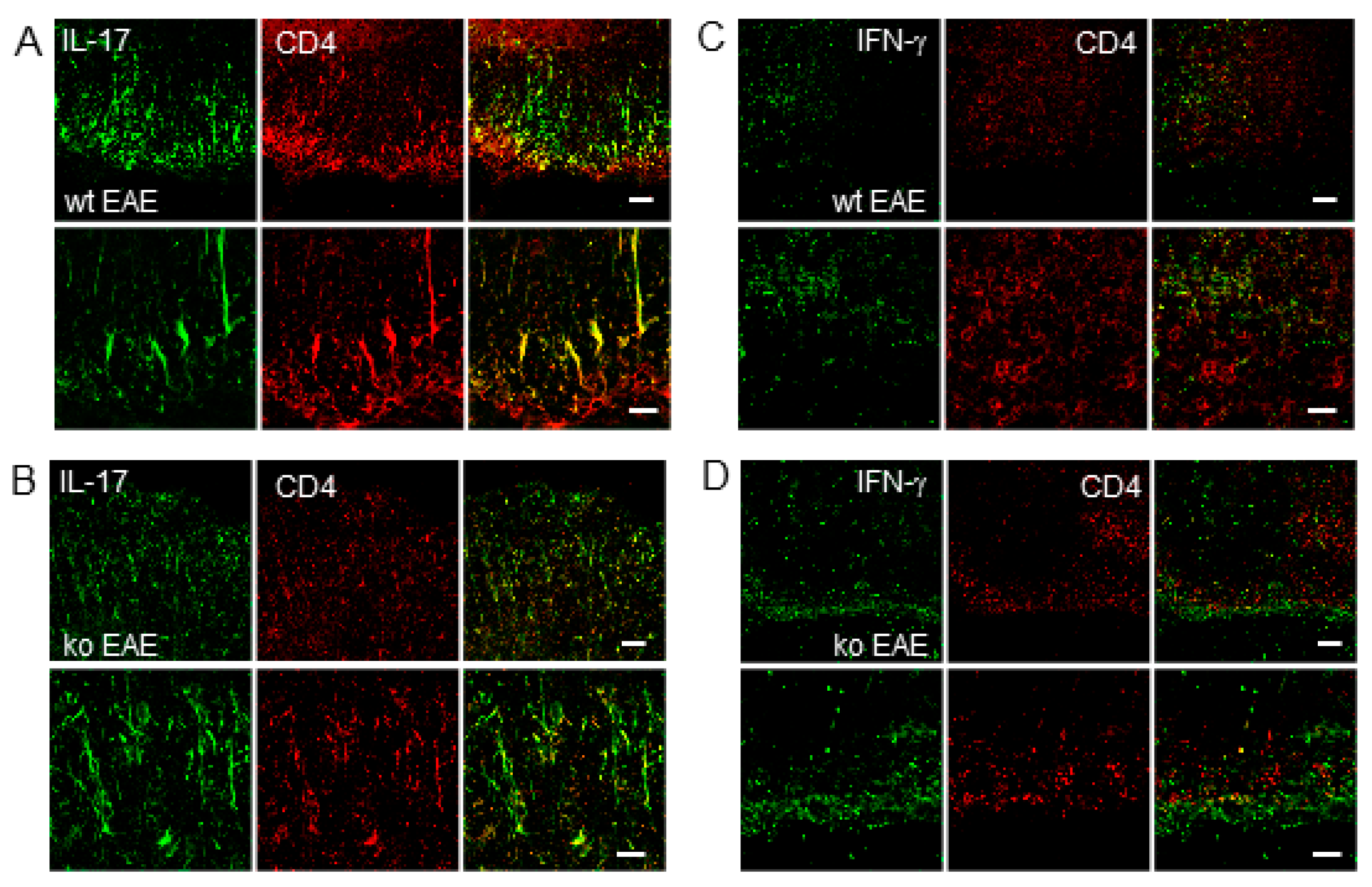
2.5. IL-17 Expression in CD4+ T Cells is Regulated by mPGES-1
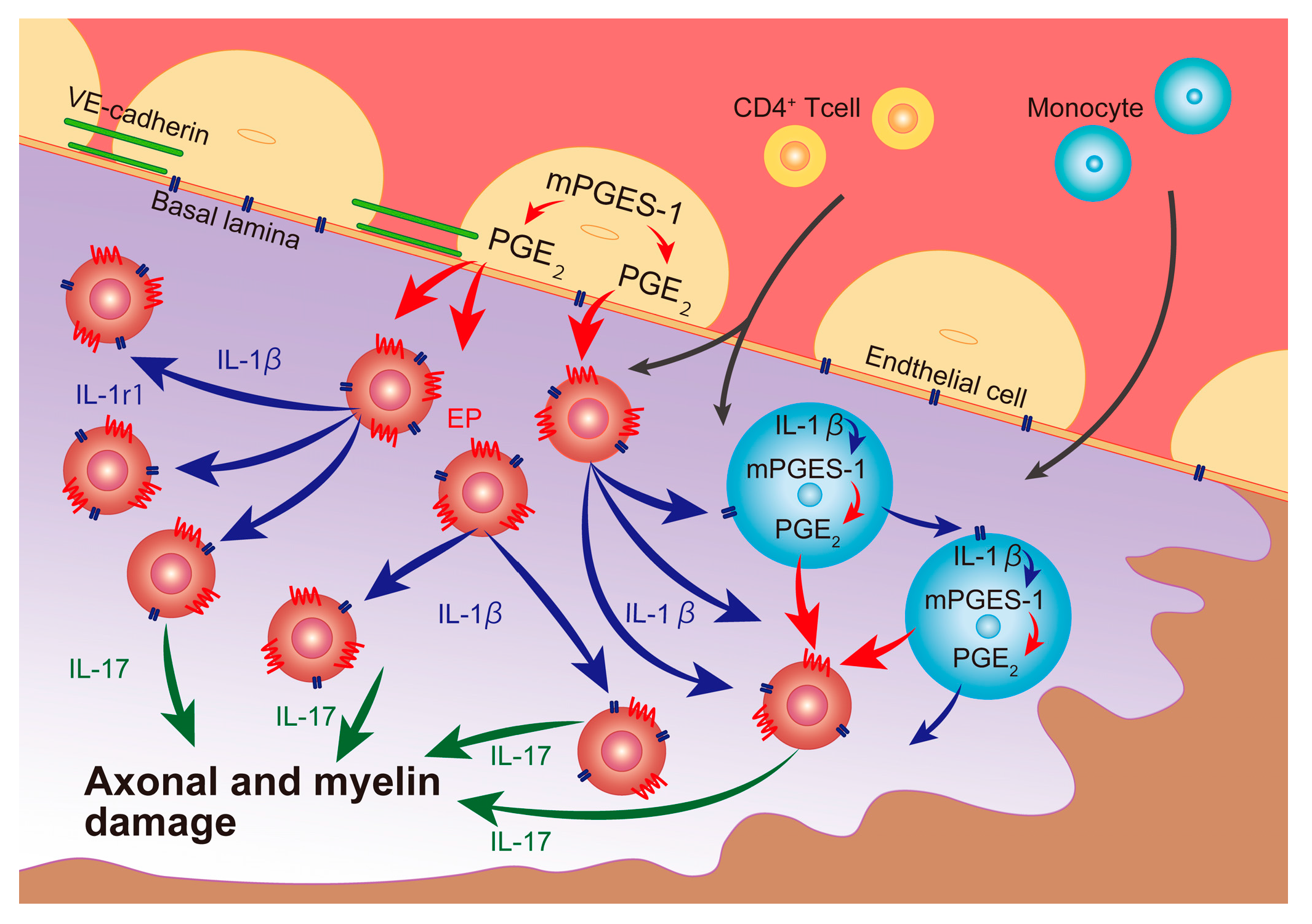
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Induction and Assessment of EAE
4.3. Immunohistochemistry
4.4. Image Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PGE2 | Prostaglandin E2 |
| COX-2 | Cyclooxygenase-2 |
| PGES | PGE2 synthase |
| mPGES-1 | Microsomal prostaglandin synthetase-1 |
| VECs | Vascular endothelial cells |
| KA | Kainic acid |
| MS | Multiple sclerosis |
| EAE | Experimental allergic encephalomyelitis |
| MOG35–55 | Oligodendrocyte glycoprotein35–55 peptide |
| mPGES-1−/− | Microsomal prostaglandin synthetase-1-deficient |
| IL-1β | Interleukin-1β |
| IL-1RN | Interleukin-1 receptor antagonist |
| IL-1r1 | Interleukin-1 receptor 1 |
| IL-17 | Interleukin-17 |
| Th17 | Interleukin-17-producing helper T cell |
| Th1 | Type 1 helper T cell |
| CD4+ T cells | CD4-positive T cells |
| IHC | Immunohistochemistry |
| wt | Wild-type |
| IFN-γ | Interferon-γ |
| cPGES | Cytosolic PGE2 synthase |
| BBB | Blood-brain barrier |
References
- Yamagata, K.; Matsumura, K.; Inoue, W.; Shiraki, T.; Suzuki, K.; Yasuda, S.; Sugiura, H.; Cao, C.; Watanabe, Y.; Kobayashi, S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J. Neurosci. 2001, 21, 2669–2677. [Google Scholar] [PubMed]
- Takemiya, T.; Matsumura, K.; Sugiura, H.; Maehara, M.; Yasuda, S.; Uematsu, S.; Akira, S.; Yamagata, K. Endothelial microsomal prostaglandin E synthase-1 exacerbates neuronal loss induced by kainate. J. Neurosci. Res. 2010, 88, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.W.; Hill, K.E.; Watt, H.E.; Carlson, N.G. Inflammatory cell expression of cyclooxygenase-2 in the multiple sclerosis lesion. J. Neuroimmunol. 2004, 149, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.H.; Schluesener, H.J. Cyclooxygenases-1 and -2 are differentially localized to microglia and endothelium in rat EAE and glioma. J. Neuroimmunol. 1999, 95, 202–208. [Google Scholar] [CrossRef]
- Mendel, I.; Kerlero de Rosbo, N.; Ben-Nun, A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: Fine specificity and T cell receptor Vβ expression of encephalitogenic T cells. Eur. J. Immunol. 1995, 25, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Kuerten, S.; Kostova-Bales, D.A.; Frenzel, L.P.; Tigno, J.T.; Tary-Lehmann, M.; Angelov, D.N.; Lehmann, P.V. MP4- and MOG:35–55-induced EAE in C57BL/6 mice differentially targets brain, spinal cord and cerebellum. J. Neuroimmunol. 2007, 189, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Goverman, J.M. Active induction of experimental allergic encephalomyelitis. Nat. Protoc. 2006, 1, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Goverman, J.M. Passive induction of experimental allergic encephalomyelitis. Nat. Protoc. 2006, 1, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.; Gordon, D.; Turk, J.L. Prostaglandin and thromboxane levels in central nervous system tissues from rats during the induction and development of experimental allergic encephalomyelitis (EAE). Immunopharmacology 1984, 7, 101–107. [Google Scholar] [CrossRef]
- Marusic, S.; Leach, M.W.; Pelker, J.W.; Azoitei, M.L.; Uozumi, N.; Cui, J.; Shen, M.W.; DeClercq, C.M.; Miyashiro, J.S.; Carito, B.A.; et al. Cytosolic phospholipase A2 α-deficient mice are resistant to experimental autoimmune encephalomyelitis. J. Exp. Med. 2005, 202, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Kihara, Y.; Matsushita, T.; Kita, Y.; Uematsu, S.; Akira, S.; Kira, J.; Ishii, S.; Shimizu, T. Targeted lipidomics reveals mPGES-1-PGE2 as a therapeutic target for multiple sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 21807–21812. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, C.; Matsumoto, Y.; Kohyama, K.; Uematsu, S.; Akira, S.; Yamagata, K.; Takemiya, T. Microsomal prostaglandin E synthase-1 aggravates inflammation and demyelination in a mouse model of multiple sclerosis. Neurochem. Int. 2013, 62, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Miyake, S.; Mizuno, M.; Oka, N.; Kusunoki, S.; Yamamura, T. Selective COX-2 inhibitor celecoxib prevents experimental autoimmune encephalomyelitis through COX-2-independent pathway. Brain 2006, 129, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Muthian, G.; Raikwar, H.P.; Johnson, C.; Rajasingh, J.; Kalgutkar, A.; Marnett, L.J.; Bright, J.J. COX-2 inhibitors modulate IL-12 signaling through JAK-STAT pathway leading to Th1 response in experimental allergic encephalomyelitis. J. Clin. Immunol. 2006, 26, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Shu, Y.Y.; Zhu, Y.N.; Fu, Y.F.; Tang, W.; Zhong, X.G.; Wang, H.; Yang, Y.F.; Ren, J.; Wang, M.W.; et al. COX-2 inhibitors ameliorate experimental autoimmune encephalomyelitis through modulating IFN-γ and IL-10 production by inhibiting T-bet expression. J. Neuroimmunol. 2007, 186, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Near, S.L. Protective effect of the interleukin-1 receptor antagonist (IL-1ra) on experimental allergic encephalomyelitis in rats. J. Neuroimmunol. 1995, 61, 241–245. [Google Scholar] [CrossRef]
- Schiffenbauer, J.; Streit, W.J.; Butfiloski, E.; LaBow, M.; Edwards, C., III; Moldawer, L.L. The induction of EAE is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clin. Immunol. 2000, 95, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Napolitani, G.; Acosta-Rodriguez, E.V.; Lanzavecchia, A.; Sallusto, F. Prostaglandin E2 enhances Th17 responses via modulation of IL-17 and IFN-γ production by memory CD4+ T cells. Eur. J. Immunol. 2009, 39, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Bak-Jensen, K.S.; Li, Y.; Blumenschein, W.M.; McGeachy, M.J.; McClanahan, T.K.; McKenzie, B.S.; Kastelein, R.A.; Cua, D.J.; de Waal Malefyt, R. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J. Exp. Med. 2009, 206, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Adamik, J.; Henkel, M.; Ray, A.; Auron, P.E.; Duerr, R.; Barrie, A. The IL17A and IL17F loci have divergent histone modifications and are differentially regulated by prostaglandin E2 in Th17 cells. Cytokine 2013, 64, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy, P.; Donaldson, J.O.; Koff, T.; Longo, M.; Perry, W. Prostaglandin release in multiple sclerosis: Correlation with disease activity. Neurology 1986, 36, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Aberg, J.A.; Demers, L.M.; Romano, P.J.; Tenser, R.B. Prostaglandin production in chronic progressive multiple sclerosis. J. Clin. Lab. Anal. 1990, 4, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, F.; Serafini, B.; Scognamiglio, P.; Di Virgilio, A.; Finocchi, L.; Aloisi, F.; Barnaba, V. Short-lived immunization site inflammation in self-limited active experimental allergic encephalomyelitis. Int. Immunol. 2000, 12, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, P.J.; Thoren, S.; Morgenstern, R.; Samuelsson, B. Identification of human prostaglandin E synthase: A microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA 1999, 96, 7220–7225. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, N.; Ohmiya, Y.; Ohkubo, H.; Hashimoto, K.; Kangawa, K.; Kojima, M.; Ito, S.; Watanabe, K. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem. Biophys. Res. Commun. 2002, 291, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Tanioka, T.; Nakatani, Y.; Semmyo, N.; Murakami, M.; Kudo, I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J. Biol. Chem. 2000, 275, 32775–32782. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Naraba, H.; Tanioka, T.; Semmyo, N.; Nakatani, Y.; Kojima, F.; Ikeda, T.; Fueki, M.; Ueno, A.; Oh, S.; et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 2000, 275, 32783–32792. [Google Scholar] [CrossRef] [PubMed]
- Kunori, S.; Matsumura, S.; Okuda-Ashitaka, E.; Katano, T.; Audoly, L.P.; Urade, Y.; Ito, S. A novel role of prostaglandin E2 in neuropathic pain: Blockade of microglial migration in the spinal cord. Glia 2011, 59, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Kleine, T.O.; Zwerenz, P.; Graser, C.; Zofel, P. Approach to discriminate subgroups in multiple sclerosis with cerebrospinal fluid (CSF) basic inflammation indices and TNF-α, IL-1β, IL-6, IL-8. Brain Res. Bull. 2003, 61, 327–346. [Google Scholar] [CrossRef]
- Levesque, S.A.; Pare, A.; Mailhot, B.; Bellver-Landete, V.; Kebir, H.; Lecuyer, M.A.; Alvarez, J.I.; Prat, A.; de Rivero Vaccari, J.P.; Keane, R.W.; et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1β-driven neuroinflammation during autoimmune encephalomyelitis in mice. J. Exp. Med. 2016, 213, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Matsumura, K.; Yamagata, K.; Watanabe, Y. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1β: A possible site of prostaglandin synthesis responsible for fever. Brain Res. 1996, 733, 263–272. [Google Scholar] [CrossRef]
- Serou, M.J.; DeCoster, M.A.; Bazan, N.G. Interleukin-1β activates expression of cyclooxygenase-2 and inducible nitric oxide synthase in primary hippocampal neuronal culture: Platelet-activating factor as a preferential mediator of cyclooxygenase-2 expression. J. Neurosci. Res. 1999, 58, 593–598. [Google Scholar] [CrossRef]
- Samad, T.A.; Moore, K.A.; Sapirstein, A.; Billet, S.; Allchorne, A.; Poole, S.; Bonventre, J.V.; Woolf, C.J. Interleukin-1β-mediated induction of COX-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 2001, 410, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Favrais, G.; Schwendimann, L.; Gressens, P.; Lelievre, V. Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1β on excitotoxic brain lesions in newborn mice. Neurobiol. Dis. 2007, 25, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Casos, K.; Siguero, L.; Fernandez-Figueras, M.T.; Leon, X.; Sarda, M.P.; Vila, L.; Camacho, M. Tumor cells induce COX-2 and mPGES-1 expression in microvascular endothelial cells mainly by means of IL-1 receptor activation. Microvasc. Res. 2011, 81, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Anacker, C.; Suarez-Mier, G.B.; Wang, Q.; Andreasson, K. Function of prostaglandin E2 EP receptors in the acute outcome of rodent hypoxic ischemic encephalopathy. Neurosci. Lett. 2011, 504, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Sugata, Y.; Fujiwara, T.; Matsumoto, R.; Nishibori, M.; Shimizu, K.; Maeda, M.; Kimura, Y.; Kariya, S.; Hattori, H.; et al. E prostanoid 2 (EP2)/EP4-mediated suppression of antigen-specific human T-cell responses by prostaglandin E2. Immunology 2006, 118, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Maslanka, T.; Spodniewska, A.; Barski, D.; Jasiecka, A.; Zuska-Prot, M.; Ziolkowski, H.; Markiewicz, W.; Jaroszewski, J.J. Prostaglandin E2 down-regulates the expression of CD25 on bovine T cells, and this effect is mediated through the EP4 receptor. Vet. Immunol. Immunopathol. 2014, 160, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Sreeramkumar, V.; Hons, M.; Punzon, C.; Stein, J.V.; Sancho, D.; Fresno, M.; Cuesta, N. Efficient T-cell priming and activation requires signaling through prostaglandin E2 (EP) receptors. Immunol. Cell Biol. 2016, 94, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Genc, A.; Zhang, X.; Zhang, J.; Bazan, N.G.; Chen, C. Heterogeneous expression and regulation of hippocampal prostaglandin E2 receptors. J. Neurosci. Res. 2005, 81, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.N.; Wang, C.; Zhang, C.J.; Kang, Z.; Gulen, M.F.; Zepp, J.A.; Zhao, J.; Bian, G.; Do, J.S.; Min, B.; et al. T cell-intrinsic ASC critically promotes Th17-mediated experimental autoimmune encephalomyelitis. Nat. Immunol. 2016, 17, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Vinet, J.; Brouwer, N.; Brendecke, S.; Biagini, G.; Biber, K.; Boddeke, H.W.; Eggen, B.J. In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia 2014, 62, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Amiable, N.; de Rivero Vaccari, J.P.; Chae, J.J.; Keane, R.W.; Lacroix, S.; Vallieres, L. The inflammasome pyrin contributes to pertussis toxin-induced IL-1β synthesis, neutrophil intravascular crawling and autoimmune encephalomyelitis. PLoS Pathog. 2014, 10, e1004150. [Google Scholar] [CrossRef] [PubMed]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Pare, A.; Mailhot, B.; Levesque, S.A.; Lacroix, S. Involvement of the IL-1 system in experimental autoimmune encephalomyelitis and multiple sclerosis: Breaking the vicious cycle between IL-1β and GM-CSF. Brain Behav. Immun. 2017, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef] [PubMed]
- Blamire, A.M.; Anthony, D.C.; Rajagopalan, B.; Sibson, N.R.; Perry, V.H.; Styles, P. Interleukin-1β-induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: A magnetic resonance study. J. Neurosci. 2000, 20, 8153–8159. [Google Scholar] [PubMed]
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1α regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J. Immunol. 2006, 177, 5574–5584. [Google Scholar] [CrossRef] [PubMed]
- McCandless, E.E.; Budde, M.; Lees, J.R.; Dorsey, D.; Lyng, E.; Klein, R.S. IL-1r signaling within the central nervous system regulates CXCL12 expression at the blood-brain barrier and disease severity during experimental autoimmune encephalomyelitis. J. Immunol. 2009, 183, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Powell, N.; Zhang, H.; Belevych, N.; Ching, S.; Chen, Q.; Sheridan, J.; Whitacre, C.; Quan, N. Endothelial IL-1r1 is a critical mediator of EAE pathogenesis. Brain Behav. Immun. 2011, 25, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krady, J.K.; O’Malley, M.; Styren, S.D.; DeKosky, S.T.; Levison, S.W. The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J. Neurosci. 2002, 22, 6071–6082. [Google Scholar] [PubMed]
- Domingues, H.S.; Mues, M.; Lassmann, H.; Wekerle, H.; Krishnamoorthy, G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS ONE 2010, 5, e15531. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, A.; Bauer, J.; Litzenburger, T.; Schubart, A.; Linington, C. T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia 2001, 36, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Hjelmstrom, P.; Juedes, A.E.; Fjell, J.; Ruddle, N.H. B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J. Immunol. 1998, 161, 4480–4483. [Google Scholar] [PubMed]
- Uematsu, S.; Matsumoto, M.; Takeda, K.; Akira, S. Lipopolysaccharide-dependent prostaglandin E2 production is regulated by the glutathione-dependent prostaglandin E2 synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J. Immunol. 2002, 168, 5811–5816. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takemiya, T.; Takeuchi, C.; Kawakami, M. Microsomal Prostaglandin E Synthase-1 Facilitates an Intercellular Interaction between CD4+ T Cells through IL-1β Autocrine Function in Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2017, 18, 2758. https://doi.org/10.3390/ijms18122758
Takemiya T, Takeuchi C, Kawakami M. Microsomal Prostaglandin E Synthase-1 Facilitates an Intercellular Interaction between CD4+ T Cells through IL-1β Autocrine Function in Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2017; 18(12):2758. https://doi.org/10.3390/ijms18122758
Chicago/Turabian StyleTakemiya, Takako, Chisen Takeuchi, and Marumi Kawakami. 2017. "Microsomal Prostaglandin E Synthase-1 Facilitates an Intercellular Interaction between CD4+ T Cells through IL-1β Autocrine Function in Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 18, no. 12: 2758. https://doi.org/10.3390/ijms18122758
APA StyleTakemiya, T., Takeuchi, C., & Kawakami, M. (2017). Microsomal Prostaglandin E Synthase-1 Facilitates an Intercellular Interaction between CD4+ T Cells through IL-1β Autocrine Function in Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences, 18(12), 2758. https://doi.org/10.3390/ijms18122758