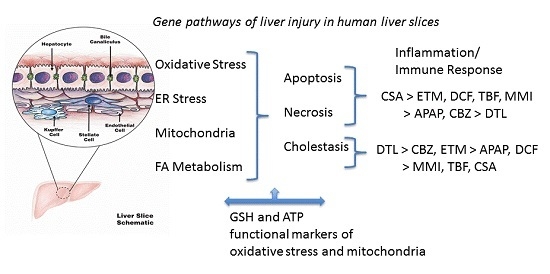
2. Results
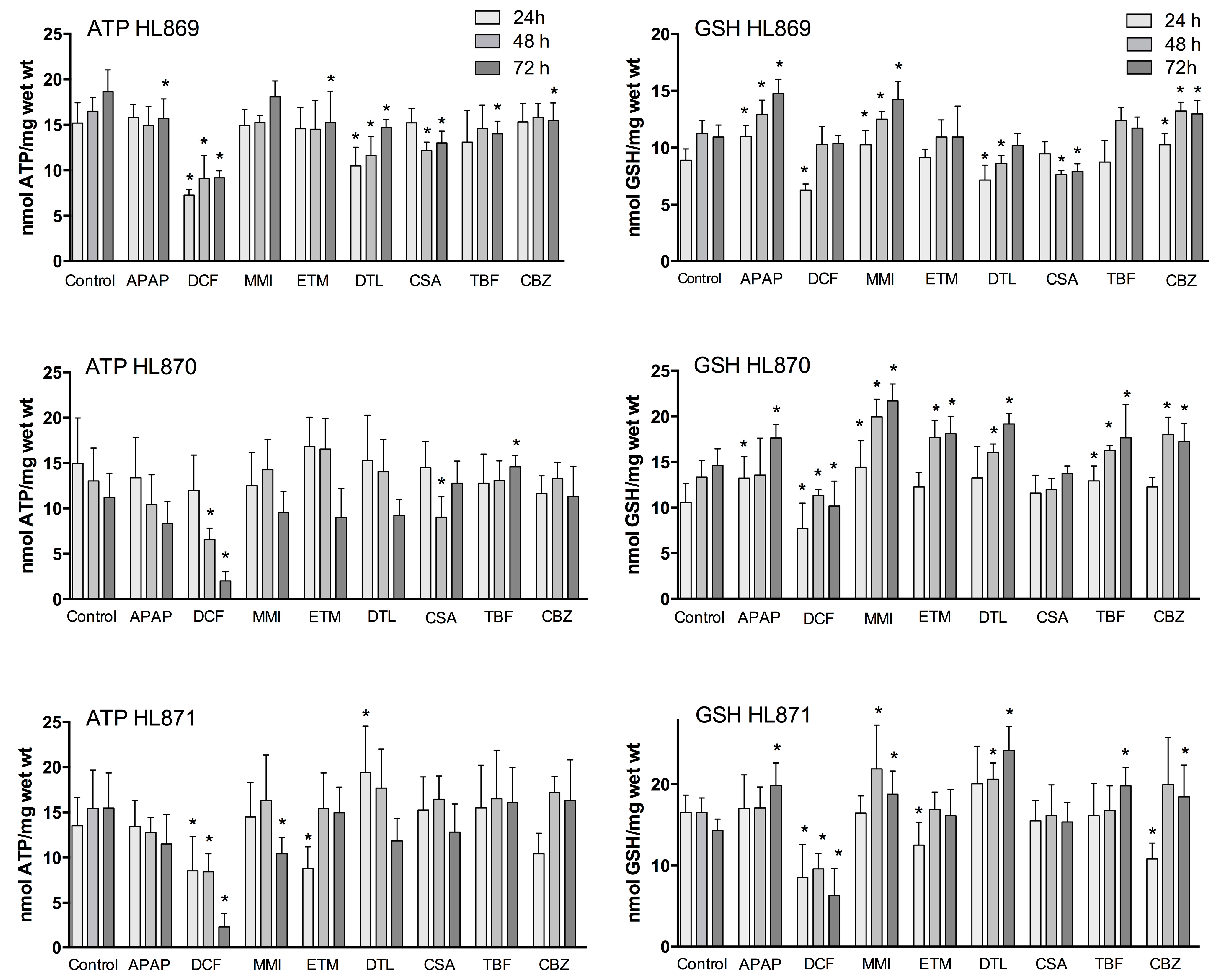
Functional markers of oxidative stress include liver slice ATP and GSH levels. The mitochondria have an essential role in energy metabolism and as a regulator of cell death. GSH is a major liver anti-oxidant and if GSH redox status is compromised reactive metabolites will bind to cell proteins to affect cell function. Utilization of a complete culture medium contributed to the liver slices ability to synthesize ATP and GSH throughout the culture period (
Figure 1). In the control human liver slices, ATP mean values across the three livers were comparable (13–16 nmols/mg wet weight) and remained consistent over the time course, varying 10% for HL869, 14.5% for HL870, and 7% for HL871. GSH mean control values were also comparable across the three livers, ranging from 10–15 nmols/mg wet weight. The variation within each liver across the 72 h incubation period, 12% for HL869, 16% for HL870, and 8% for HL871, paralleled the variation measured for ATP values. The quality of each human liver was initially considered to be very good as determined by high liver slice K
+ levels at 1 and 4 h and was verified by additional measurements at 24, 48, and 72 h. Liver slice K
+ levels were sustained for 72 h, varying 6.5% for HL869, 4.3% for HL870, and 6.4% for HL870 (
Table 1). The ATP and GSH levels of the individual human liver slice experiments revealed inter-individual differences to drug response and some similar patterns (
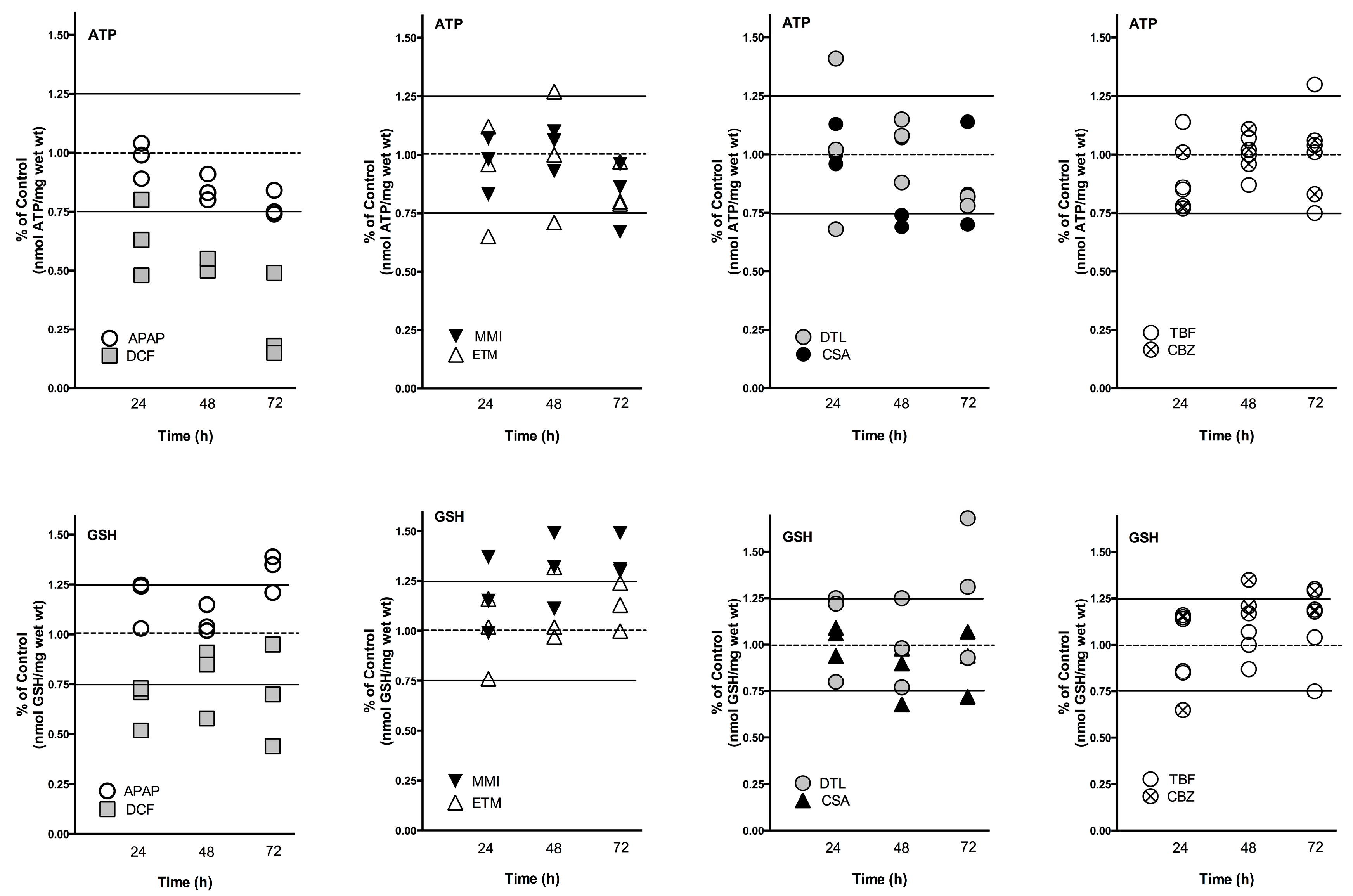
Figure 1). For example, after three doses (72 h) some drugs decreased ATP levels in each liver (APAP, DCF, and DTL), while some drugs reduced ATP levels in two livers (MMI, ETM, CSA) or in one liver (TBF, CBZ). GSH levels after three doses (72 h) were increased in each liver by APAP, MMI, TBF and CBZ, while some drugs increased levels in two livers, ETM and DTL. GSH levels were decreased in two livers by DCF and CSA. To provide an overview, mean ATP and GSH values based on percent of change from time-matched control values are compared across the three livers in
Table 2.
2.1. Drugs Affecting ATP
A time dependent decrease of ATP levels was evident with 1 mM APAP and 1 mM DCF compared to time-matched control slices (
Figure 2). In the presence of 1 mM APAP, mean liver slice ATP levels decreased significantly at 48 h by about 20% in two livers, and at 72 h by 16%–25% in the three livers. The dose of DCF (1 mM) decreased liver ATP levels dramatically in the three livers, up to 50% at 24 and 48 h and 50%–85% by 72 h.
Several compounds caused fluctuations in ATP levels compared to time-matched controls. MMI (500 μM) increased ATP levels by 10% above control levels at 24 and 48 h in two of three livers, then decreased ATP levels significantly by 14%–33% at 72 h. ETM (100 μM) caused fluctuations in ATP production in all three livers. At 24 h ATP levels were significantly decreased by 35% in one liver, which then readjusted to control values for the remainder of the culture period. In two livers at 48 h, one exhibited increased ATP production (27%) while another liver exhibited a 30% decrease. At 72 h, ATP levels in two of three livers were decreased by 20%.
DTL (10 μM) caused the greatest fluctuation in ATP levels at 24 h, increasing by 41% or decreasing by 32%. At 48 h the liver stabilized to within 12%–15% of control values. At 72 h all three human livers exhibited decreased ATP production, with a significant mean decrease of 19%. CSA (10 μM) stimulated ATP production in one liver at 24 h (13%), then caused decreases in ATP levels in two of three livers at 48 h (26%–31%) and 72 h (17%–30%) and an increase in one liver at 72 h (14%).
TBF (100 μM) initially increased or decreased liver ATP levels by 15% at 24 h, which stabilized to control levels at 48 h, then decreased significantly in one liver by 25% and increased significantly in another liver by 30% at 72 h. CBZ (100 μM) initially decreased ATP levels up to 23% in two livers at 24 h, which then stabilized around control values at 48 h, and one liver exhibited a significant decrease of ATP levels by 17% at 72 h.
2.2. Drugs Affecting GSH
APAP (1 mM) exposure to human liver slices increased GSH levels significantly in two livers at 24 h (up to 25%) and in the three livers significantly at 72 h (21%–39%) compared to time matched controls (
Figure 2). DCF (1 mM) significantly reduced human liver slice GSH levels at 24 h in all three livers, 50%–73%. Recovery of GSH levels was apparent in one liver, with 91% of control values at 48 h, and 95% at 72 h. Another liver exhibited partial recovery at 48 h, 85% of control values, then GSH levels declined significantly to 70% at 72 h. A third liver had significantly reduced GSH levels (about 50%) at all time points.
MMI (500 μM) exposure increased GSH levels significantly at 24 h (15%–37%) in two of three livers. GSH levels remained elevated and were significantly increased in the three livers at 48 h (11%–49%) and at 72 h (30%–49%). ETM (100 μM) increased GSH levels significantly in one liver at 48 h (32%) and 72 h (24%). Another liver decreased ATP levels significantly initially (24%) at 24 h, then exhibited elevated levels at 72 h (13%).
DTL (10 μM) increased GSH levels in two of three livers, up to 25% at 48 h and up to 68% at 72 h. However, in one liver DTL decreased GSH levels significantly 20%–23% at 24 and 48 h, which rebounded to control levels by 72 h. CSA (10 μM) significantly decreased GSH levels in one of three livers, about 30% at 48 and 72 h; while the other livers had values that were equal to control slices.
TBF (100 μM) increased GSH levels, reaching significance, 20% 24–72 h in one liver, and 39% at 72 h in a second liver. CBZ (100 μM) overall increased GSH levels significantly, up to 16% at 24 h and 20%–30% at 48 and 72 h in two livers. There was an initial decrease of GSH levels at 24 h (35%) in one liver, which then increased to 30% above control values at 72 h.
2.3. Fluctuations in Drug Response
Fluctuations in ATP and GSH levels occurred across the time-course to the drugs in each liver, which is indicative of a system adjusting to drug exposure. Additionally, inter-individual variation in response to drug exposure was apparent across the livers (
Figure 2,
Table 2). For example, ETM and DTL exposure caused an initial decrease in ATP levels at 24 h which was followed by an increase at 48 h, which then decreased by 72 h. A considerable spread in response across the three livers was also measured at 24 h with DTL (0.68–1.41 percent of control), and at 48 h with ETM exposure (0.7–1.27 percent of control). The doses of APAP, DCF, MMI, CSA, TBF, and CBZ caused less fluctuation in response across time-points; however inter-individual variation was apparent.
Human liver slice GSH levels were increased substantially by APAP (up to 39%), MMI (up to 49%), and DTL (up to 68%), and substantially decreased by DCF (50%) exposure. Fluctuations in GSH levels were evident across the time course. An initial decrease of GSH levels (24 h) was followed by an increase at 48 h compared to time-matched controls for DCF, and in one liver for ETM and CBZ. In contrast, APAP induced an initial increase at 24 h in GSH levels followed by a decrease at 48 h, and then an increase at 72 h. MMI caused a general increase in GSH levels across the time course, while CBZ increased GSH levels at 48 and 72 h. Inter-individual response was most evident with DCF and DTL exposure, particularly at 72 h. Drug exposure exhibiting a tighter response included CBZ, CSA, ETM and APAP.
2.4. Gene Expression
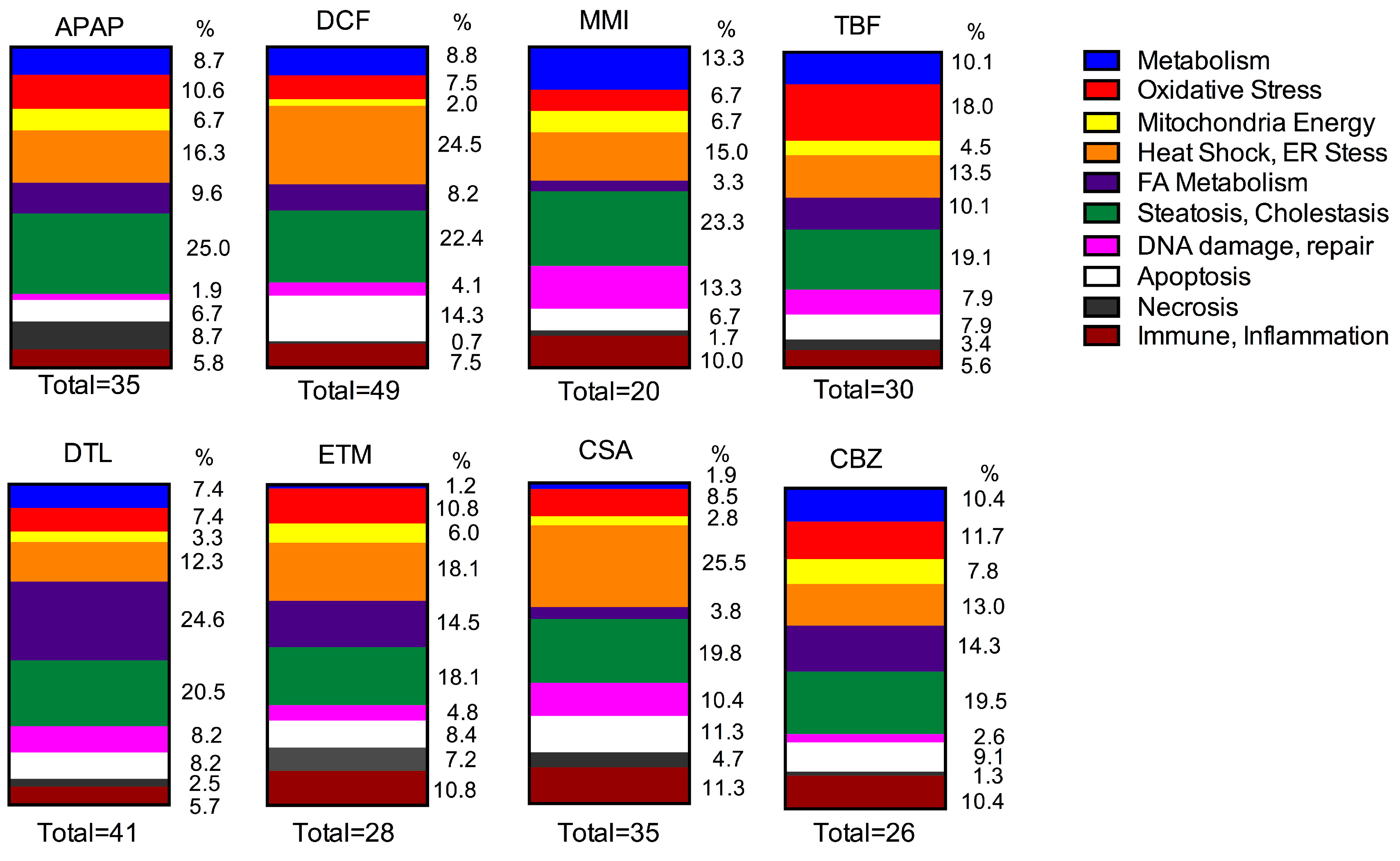
Genes linked to organ injury were interrogated following two doses of each drug and compared to time-matched control liver slice RNA with the molecular toxicology pathway finder human PCR array (PANZ-3401, 370 genes). Exposure of the eight drugs to three independent human livers reveals differences across the drugs in effects as well as differences in human response across the livers. Changes in gene expression reflect a perturbation of organelles and tissue stresses. A comparison of the gene categories altered by each drug is shown in
Figure 3 and
Figure 4. Furthermore, an overview of the number of genes altered as well as the list of the genes significantly altered by each drug is shown in
Table 3 and
Table 4.
2.5. Metabolism
Several drugs altered the expression of the drug metabolism genes including enzymes that play a role in their own metabolism. DCF, TBF, CBZ, APAP, MMI and DTL affected several genes, while ETM and CSA had minimal effects on the metabolism genes. CYP3A4 was up-regulated by TBF (1.7–10.5-fold) and CBZ (1.9–3.0-fold) in two different livers, and in one liver by APAP (8.4-fold), and CSA (4.7-fold). A down-regulation of CYP3A4 occurred with DCF (−11-fold), ETM (−8.7-fold) and DTL (−5.5-fold) exposure. CYP2C19 gene expression, was down-regulated by DCF (−2.1 to −28.6-fold) in all three livers; while up-regulated by CBZ (3-fold) in two livers. DTL altered CYP2C19 expression differently in two livers. MMI up-regulated CYP2C19 and CYP2B6 in the same liver and down-regulated these genes in a second liver. APAP up-regulated CYP2B6 (1.7–2.4-fold) in two livers, and down-regulated CYP2E1 in the one liver. DCF, additionally, down-regulated CYP2B6 (−22-fold) gene expression in one liver, and caused down-regulation of FMO4 (up to −6.4-fold) in the three livers and FMO5 (−7.2-fold) in one liver. DTL down-regulated FMO3 (up to −6.3-fold) in two livers. CSA also caused down-regulation of CYP2C9 in one liver.
The cytochrome P450 reductase (POR) gene, which encodes for the enzyme that transfers electrons from NADPH to the various cytochrome P450s, was up-regulated by DCF (2.5 to 3.5-fold) in all three livers, and by TBF (2.8-fold) in one liver. Several drugs caused an up-regulation of CYP1A1 and CYP1A2 gene expression. TBF up-regulated both genes (2.2 to 4.5-fold) in all three livers; while MMI (2.6 to 7.8-fold) and DTL (7.4 to 9.6-fold) caused up-regulation of these genes in two livers. APAP and CBZ up-regulated CYP1A1 in one liver and down-regulated it in another liver. DCF exposure down-regulated CYP1A2 (−70-fold) in one liver.
2.6. Oxidative Stress
Oxidative stress is often an initial consequence of reactive intermediate formation, and several of the drugs in this study undergo metabolic conversion; hence, many oxidative stress genes were affected. DCF altered 3 of 5 genes (7.5%) in all three livers. The same gene changes in two livers occurred with TBF, 6 of 10 genes (18%), CBZ, 3 of 6 genes (11.7%), DTL, 2 of 6 genes (7.4%), CSA, 2 of 7 genes (8.5%), ETM, 1 of 8 genes (10.8%), and MMI, 1 of 3 genes (6.7%). Gene changes in one liver occurred with APAP (11 genes, 10.6%).
Evidence for effects on glutathione regulation was apparent by altered expression of glutathione transferase (GSTA3) for APAP (−3.1-fold), DCF (−34.4-fold), TBF (−2.3 to 2.0-fold), CBZ (3.4-fold), ETM (−2.0 to −4.4-fold). In particular, the mu class of GST enzymes (GSTM4) was altered by DCF (−3.1 to −7.5-fold) in all three livers. MMI and CSA had no effect on either GSTA3 or GSTM4. Various gene isoforms of glutathione peroxidase, the intracellular GPX1 and GPX2 and extracellular GPX3 are responsible for the majority of the glutathione-dependent hydrogen peroxide-reducing activity, were altered particularly by TBF (1.8 to 3.1-fold) and CBZ (1.5 to 2.6-fold), followed by DTL (−2.0-fold), CSA (−3.4-fold), APAP (−1.6 to 1.5-fold), DCF (−15.9-fold), and ETM, while MMI had no effect. Genes indicative of reactive intermediate formation, microsomal epoxide hydrolase 1 (EPHX1) which converts epoxides to diols was altered by DCF (−2.2 to −8.9-fold) in all three livers, followed by CBZ (1.6 to 1.8-fold), TBF (2.5-fold) and APAP (1.6-fold). Reactive oxygen formation is indicative of dual oxidase (DUOX1-2) changes, which was altered by TBF (1.8–2.3-fold), CBZ (−2.1 to 4-fold), CSA (2.5 to 4.3-fold) and APAP (−2.3 to −3.1-fold). Up-regulation of the NQO1 gene which encodes for NAD(P)H dehydrogenase (quinone 1), and is involved in the reduction of quinones to hydroquinones, was evident with MMI (1.9 to 2.9-fold) and TBF (2.6-fold). Additionally, MMI up-regulated the PRDX1 gene (3.7-fold), which encodes for a member of the peroxiredoxin family of antioxidant enzymes to reduce hydrogen peroxide and alkyl hydroperoxides. The gene PPP1R15B encodes for a phosphatase, that regulates a translation factor, was affected by DCF (2.0–26.4-fold) in all three livers, followed by TBF (−2.0 to 9.3-fold). The liver (HL871) that exhibited the greatest up-regulation for DCF (26.4-fold) was affected by several drugs, yet to a lesser extent, APAP 7.3-fold, MMI 8.2-fold, ETM 7.5-fold, DTL 8.2-fold and CSA 8.6-fold.
2.7. Mitochondrial Energy
An important category that gene expression can provide insight into is the effects of drugs on mitochondrial pathways. This category includes genes encoding for enzymes involved with the TCA cycle and mitochondrial energy. DCF affected only 1 gene in all three livers (2%). APAP altered 2 genes of 5 in two livers (6.7%), TBF 1 of 3 genes (4.5%) and 1 of 4 genes ETM (6%) in two livers, followed by changes in one liver by CBZ (6 genes, 7.8%), MMI (4 genes, 6.7%), DTL (4 genes, 3.3%) and CSA (3 genes, 2.8%).
The aconitase genes, ACO1 and ACO2, encode for proteins catalyzing steps in the TCA cycle. ACO1 aids to control iron levels inside cells and was affected in the same liver by MMI (−1.7-fold), TBF (−2.6-fold), CBZ (−3.7-fold), ETM (−2.5-fold), DTL (−2.1-fold), and CSA (−2.9-fold), while ACO2 was altered in a different liver by APAP (−1.5-fold). Genes which encode for enzymes that utilize NAD(+) and NADP(+) as electron acceptors the IDH 1-3 (isocitrate dehydrogenases) were altered. MMI (−1.7-fold) down-regulated IDH1, while APAP (−1.7-fold) and CBZ (−1.7-fold) down-regulated IDH2. APAP (1.3-fold) up-regulated IDH3B, while ETM (−1.8-fold) down-regulated IDH3B. The MDH1 gene encodes for a cytosolic enzyme malate dehydrogenase, which utilizes the NAD/NADH cofactor system in the citric acid cycle. The MDH1 gene was altered by APAP (−1.5 to 3.1-fold), MMI (3.1-fold), TBF (−1.9 to 3.6-fold), CBZ (−2.9-fold), ETM (−4.2 to 3.6-fold) and CSA (−1.9-fold). Furthermore, the SDHC gene, succinate dehydrogenase complex subunit C also known as mitochondrial complex II, encodes for a key enzyme complex of the TCA cycle and aerobic respiratory chains of the mitochondria, was down-regulated by CBZ (−5.7-fold) and DTL (−1.7-fold). SUCLA2 and SUCLG1 are genes that encode for mitochondrial matrix succinyl-CoA ligases which are accompanied by the substrate-level phosphorylation of ADP to ATP or GDP to GTP. Exposure to CBZ (−10.3-fold) down-regulated SUCLA2, and APAP (−1.4 to 3.3-fold) altered SUCLG1 gene expression levels.
The ACYL gene, which encodes for the enzyme ATP citrate lyase, is responsible for the synthesis of cytosolic acetyl-CoA was up-regulated by CSA (2.3-fold). The adenosine kinase (ADK) gene, which encodes for an enzyme that catalyzes the transfer of the gamma-phosphate from ATP to adenosine, as well as COX8A, which encodes a subunit of the cytochrome c oxidase complex of the mitochondrial respiratory chain were down-regulated by DTL (−5.8-fold, ADK and −6.1-fold, COX8A) in one liver. The mitochondrial uncoupling proteins (UCPs) separate oxidative phosphorylation from ATP synthesis with energy dissipated as heat, and referred to as the mitochondrial proton leak. DCF exposure caused a down-regulation of UCP2 (−2.4 to −5.1 fold) in all three livers. The CYC1 gene, cytochrome c1, encodes for a mitochondrial heme protein, which was up-regulated by MMI (1.4-fold) and TBF (1.3-fold) in the same liver. The DLD gene, dihydrolipoamide dehydrogenase, encodes for a mitochondrial enzyme that is part of several multi-enzyme complexes involved with energy metabolism. CBZ exposure caused a down-regulation of DLD gene expression levels (−27.3-fold).
2.8. Fatty Acid Metabolism
Drugs affecting the pathways of fatty acid metabolism have the potential to alter the energy status of the tissue. In this study, the drugs which affected genes in all three livers included: DCF altered 3 of 6 genes (8.2%), APAP altered 1 of 7 genes (9.6%), CBZ altered 1 of 9 genes (14.3%), TBF altered 1 of 5 genes (10.1%), and ETM altered 1 of 8 genes (14.5%), followed by CSA (3.8%) and DTL (24.6%) in which two livers responded, and MMI (3.3%) whereby one liver responded.
Several genes affecting mitochondrial fatty acid oxidation were altered. The CPT1A gene encodes for carnitine palmitoyltransferse 1, which is located on the outer mitochondrial membrane, is a rate-limiting step involved in fatty acid metabolism. CPT1A was up-regulated in all three livers by ETM (2.3 to 16.1-fold), and up-regulated by TBF (1.5 to 8.1-fold) in two livers, and by APAP (2.1-fold), CBZ (2.1-fold) and CSA (2.5-fold) in the same one liver. The ACADM (acyl-coenzyme A dehydrogenase) gene encodes for a mitochondrial enzyme that catalyzes the initial step of medium-chain fatty acid beta-oxidation. ACADM gene expression was altered by TBF (−2.1 to 3.4-fold), APAP (−1.9 to 3.2-fold), ETM (4.1-fold), CSA (2.5-fold), and DTL (−5.2-fold). The ACADVL (very long-chain specific acyl-CoA dehydrogenase) gene encodes for an inner mitochondrial protein to catalyze the first step of fatty acid beta-oxidation of long chain and very long chain fatty acids, typically C16-acylCoA and longer. An up-regulation of ACADVL was triggered by TBF (1.7–2.7-fold), CBZ (3.3-fold), ETM (1.8-fold) and CSA (1.9-fold). The ECHS1 gene, enoly-coA hydratase short chain 1, encodes for a protein that catalyzes the second step of the mitochondrial fatty acid beta-oxidation pathway. DTL down-regulated ECHS1 (−2.4 to −4.9-fold) in two livers. The HADHA gene, the α subunit, and HADHB, the beta subunit gene encodes for 3-ketonacyl-CoA thiolase, the trifunctional mitochondrial enzyme that catalyzes the last steps of mitochondrial beta-oxidation of long chain fatty acids. The HADHA and HADHB genes were up-regulated by TBF (2.1 to 3.2-fold) in one liver and were down-regulated by DTL (−1.5 to −3.1-fold) in two livers. The ACAA1 gene encodes for acetyl-Coenzyme A acyltransferase 1, which is operative in the beta oxidation system of peroxisomes; whereas the ACAA2 gene encodes for acetyl-Coenzyme A acyltransferase 2, to catalyze the last step of mitochondrial fatty acid beta oxidation. ETM up-regulated ACAA1 (1.8 to 4.7-fold) and ACAA2 (2.4-fold), while DTL down-regulated ACAA1 (−2.8 to −34-fold) and ACAA2 (−2.5 to −5.9-fold) gene expression.
Additionally, genes affecting peroxisomal fatty acid oxidation were altered. Three ACOT genes were altered. The ACOT1 gene, acyl-coenyzme A thioesterase 1, encodes for an enzyme that regulates intracellular levels of CoA esters, Coenzyme A, and free fatty acids to modulate lipid metabolism. The ACOT8 gene encodes for a peroxisomal thioesterase, which is more involved with the oxidation of fatty acids rather than in their formation. In addition, the ACOT12 gene encodes for a lipid transfer protein. APAP up-regulated ACOT1 (2.3-fold) and ACOT 8 (1.6-fold); whereas, ACOT12 was down-regulated by APAP (−1.4-fold), DCF (−3.9-fold) and DTL (−11.2-fold), and up-regulated by CBZ (1.6-fold). The ACOX2 gene, encodes for the branched-chain acyl-CoA oxidase, which is involved in the degradation of long branched fatty acids and bile acid intermediates in peroxisomes. DCF down-regulated this gene in the three livers (−4.4 to −12.5-fold), while CBZ (1.5-fold) up-regulated the gene in one liver. The EHHADH gene, enoyl-CoA hydratase, encodes for a bifunctional enzyme and is one of the four enzymes of the peroxisomal beta-oxidation pathway. The EHHADH gene was down-regulated by DTL (−1.8 to −5.4-fold) in two livers, and by CBZ (−14.7-fold) in one liver, and up-regulated by ETM (1.6-fold) in one liver.
The ACAT2 gene encodes for a cytosolic enzyme involved in lipid metabolism, cytosolic acetyl-CoA acetyltransferase. ACAT2 gene expression was down-regulated by DCF (−1.9 to −3.5-fold) in the three livers, and by DTL (−2.2 to −4.4-fold) in two livers. The APOE gene encodes for the apolipoprotein E protein, produced primarily in the liver, and mediates cholesterol metabolism. APOE is polymorphic, which alters its structure and function, and has physiological consequences. APAP (−1.7 to 3.3-fold) altered APOE gene expression in all three livers, while CSA (3.4-fold) altered it in one liver. The APOF gene encodes for apolipoprotein F, which is synthesized in the liver and found in plasma to form complexes with lipoproteins involved with the transport of cholesterol. DCF (−5.2 to −8.7) caused down-regulation of the APOF gene in all three livers, while DTL (−1.7 to −4.9-fold) down-regulated the gene in two livers and APAP (−2.2-fold) in one liver. Furthermore, the PON1 (paraoxonase 1) gene is activated by PPAR-γ in the liver and encodes for an enzyme which is a component of high-density lipoprotein. PON-1 gene expression was down-regulated by DCF (−7.0-fold) and DTL (−2.5-fold), and up-regulated by CBZ (1.8-fold), each in different livers. Moreover, the ALB gene encodes for the protein albumin is synthesized in the liver and transports hormones, fatty acids, and other compounds in the serum. ALB gene expression was altered by CBZ (−3.8 to 1.4-fold) in all three livers, while DTL (−7.6-fold) exposure down-regulated ALB gene expression in one liver.
2.9. Heat Shock and ER Stress
Heat shock and ER stress genes were affected across the livers by each drug. In particular, DCF altered 11 of 14 genes (24.5%) in all three livers. The other drugs affected genes in two or one liver, CSA (24 genes, 25.5%) exposure, followed by APAP (15 genes, 16.3%), ETM (12 genes, 18.1%), TBF (9 genes, 13.5%), CBZ (9 genes, 13.5%), DTL (11 genes, 12.3%) and MMI (8 genes, 15%).
Genes encoding for mitochondrial heat shock proteins were altered by DCF, APAP, TBF and CBZ. The HSPA9 gene encodes for a mitochondrial heat shock protein that plays a role in the control of cell proliferation. Up-regulation of HSP9A gene expression occurred with DCF (2.1 to 3.6-fold) in all three livers, while APAP up-regulated HSP9A in one liver (1.3-fold). The HSPD1 gene encodes for a mitochondrial protein essential for the folding and assembly of newly imported proteins in the mitochondria, and may function as a signaling molecule in the innate immune system. HSPD1 gene expression was altered by TBF (1.8-fold) and by CBZ (−2.1 to 1.9-fold). The HSPE1 gene encodes for a mitochondrial protein essential in biogenesis, and DTL (−1.8 to −1.6-fold) down-regulated HSPE1 expression in two livers.
The HSP90B1 gene encodes for a protein in the endoplasmic reticulum, which plays a critical role in folding proteins, and is an essential immune chaperone to regulate both the innate and adaptive immunity. DCF up-regulated HSP90B1 (2.0 to 10.9-fold) in all three livers, while CSA caused up-regulation in two livers (1.9 to 6.4-fold), and APAP caused up-regulation in one liver (4.3-fold). ETM (−2.1 to 3.9-fold) altered HSP90B expression in two livers. The HSPB8 gene encodes for a protein superfamily of small heat-shock proteins and functions as a chaperone, involved with macroautophagy, the regulation of cell proliferation and apoptosis. ETM (−2.9 to 3.3-fold) caused changes in HSPB8 expression in two livers, and CSA (2.6-fold) in one liver. The CRYAB gene encodes for a member of the small heat shock proteins (HSP20), α-crystallin B chain protein which functions as a molecular chaperone that binds to misfolded proteins to prevent protein aggregation, as well as inhibit apoptosis and contribute to intracellular architecture. CRYAB gene expression was altered by TBF (1.9 to 2.8-fold) and DTL (−1.8 to −3.4-fold) in two livers and by CBZ (−2.9-fold), ETM (3.2-fold) and CSA (−3.8-fold) in one liver. Several genes that encode for members of the DNAJ/HSP40 protein family, and function in a wide range of cellular events, such as protein folding and oligomeric protein complex assembly, were altered. In particular, DCF exposure up-regulated DNAJC6 (2.9 to 8.6-fold) gene expression in all three livers, and APAP (2.3-fold) in one liver, while DTL down-regulated DNAJC6 (−2.3 to −3.1-fold) in two livers. APAP also up-regulated DNAJB6 (1.4 to 2.3-fold) in two livers, and DNAJA2 (1.4-fold) in one liver, and DTL down-regulated DNAJB1 (−1.5 to −3.9-fold) in two livers. CSA up-regulated DNAJC3 (2.4-fold) and DNAJC5 (2-fold) in one liver.
Both the SEL1L and SERP1 genes encode for proteins in the endoplasmic reticulum that have a role in unfolded protein response. The SEL1L protein aids the transport of unfolded proteins from the ER to the cytosol, to be degraded by the proteasome in an ubiquitin-dependent manner. SERP1 encodes for a stress protein that interacts and protects unfolded target proteins against degradation during ER stress. DCF up-regulated both SEL1L (2.5 to 12.8-fold) and SERP1 (2.4 to 7.1-fold) in all three livers. Additionally, both SEL1L and SERP1 expression were altered in a liver by APAP, MMI, TBF, ETM, and CSA. The SEC62 gene encodes for an integral membrane protein located in the endoplasmic reticulum, which is part of the SEC61 complex involved with protein translocation apparatus to aid the transport of ER proteins subject to the ubiquitin-proteasome dependent degradation pathway. SEC62 gene expression was modulated by APAP (−2.6 to 6.4-fold), TBF (−3.4 to 7.9-fold), and CSA (−2.7 to 9.6-fold) in the same two livers, and by CBZ (−4.6-fold), MMI (7.2-fold), and ETM (5.7-fold) in one liver.
Several genes activated encoded for heat shock transcription factors, which activate heat-shock response genes under conditions of stress. Both ATF4 and ATF6 genes encode for transcription factors following ER stress. ATF4 belongs to a family of DNA-binding proteins that includes the AP-1 family of transcription factors, the cAMP-response element binding proteins (CREBS), and is involved in protein–protein interactions. ATF6 is embedded in the ER and functions as a stress sensor and transducer of the unfolded protein response. DCF exposure up-regulated ATF4 (3.3 to 5.1-fold) and ATF6 (2.3 to 2.8-fold) in all three livers. TBF (2.1-fold) and CSA (1.5-fold) up-regulated ATF4 in one liver. The DDIT3 gene encodes for a multifunctional transcription factor, the DNA damage-inducible transcript 3 protein, which induces cell cycle arrest and apoptosis in response to ER stress. DCF up-regulated DDIT3 (4.9 to 24.8-fold) gene expression in all three livers, while APAP (−1.9-fold) and ETM (−1.8-fold) down-regulated this gene in one liver. The MBTPS2 gene, (membrane-bound transcription factor site-2 protease) encodes for a member of the intramembrane proteases that cleave several transcription factors involved in the ER stress response and the sterol control of transcription. MBTPS2 gene expression was up-regulated by APAP (2.4-fold), altered by TBF (−2.4 to 2.4-fold), and down-regulated by CSA (−2.5-fold) and CBZ (−1.5-fold).
Several genes induced by misfolded proteins in the ER (DERL1, EIF2AK3, SYVN1, HSF2, HERPUD1) were up-regulated by DCF only in this study. The DERL1 gene (degradation in endoplasmic reticulum protein-1) encodes for a protein that targets misfolded ER proteins for destruction. The EIF2AK3 gene (eukaryotic translation initiation factor 2-α kinase 3) encodes for an enzyme, located in the ER and induced by misfolded proteins, that phosphorylates and inactivates EIF2 (eukaryotic translation-initiation factor 2), to reduce translational initiation and hence repress protein synthesis. DCF up-regulated the DERL1 gene (2.8 to 3.8-fold) and the EIF2AK3 gene (3.4 to 11.5-fold) in all three livers. Furthermore, DCF up-regulated the SYVN1 (E3 ubiquitin-protein ligase synoviolin) gene, which encodes for an enzyme to remove unfolded proteins accumulated during ER stress by retrograde transport to the cytosol from the ER and via the ubiquitin-proteasome system. DCF up-regulated the SYVN1 gene (2.2 to 2.5-fold) expression in all three livers. Additionally, HSF2 encodes for heat shock factor protein 2, which binds to the heat-shock element to activate heat-shock response genes under conditions of stress. The HERPUD1 gene (homocysteine-responsive endoplasmic reticulum-resident ubiquitin-like domain member 1 protein) encodes for a protein involved in polypeptide folding and in the destruction of misfolded proteins by the ER-associated degradation system. DCF exposure caused an up-regulation of HSF2 (8-fold) and HERPUD1 (7-fold) gene expression in the same liver.
2.10. Steatosis, Cholestasis, Phospholipidosis
Perturbations in genes associated with steatosis, cholestasis and phospholipidosis were particularly evident across the three livers with APAP (18 genes, 25%), DCF (15 genes, 22.4%), DTL (21 genes, 20.5%) and CSA (17 genes, 19.8%), followed by CBZ (13 genes, 19.5%), TBF (14 genes, 19.1%), ETM (14 genes, 18.1%), and MMI (11 genes, 23.3%).
Several genes for liver transporters were altered in this study, including the ATP-binding sub-families ABCB1, ABCB4, ABCC2, and ABCC3, and the solute carriers including OSTb, SLCO1A2, and SLC10A1. The ABCB1 gene, which encodes for the ATP-dependent membrane efflux pump P-glycoprotein 1 that pumps harmful substances out of the liver cells into the bile ducts, was up-regulated by in all three livers by DCF (2.8 to 4.1-fold), and in one liver by APAP (1.5-fold), TBF (2.2-fold) and CBZ (1.5-fold), and down-regulated by DTL (21-fold). The ABCB4 gene was down-regulated in all three livers by APAP (−1.5 to −2.2-fold). ABCC2, the canalicular multispecific organic anion transporter 1 gene, and ABCC3, the canalicular multispecific organic anion transporter 2 gene were both induced by APAP, MMI, TBF, CBZ, and ETM, as a hepatoprotective response. The OSTΒ gene, which encodes for the organic solute transporter beta to transport endogenous compounds like taurocholate, prostaglandin E2 across cell membranes, was up-regulated by DCF (5.2-fold) and down-regulated by APAP (−2.7-fold) and ETM (−2.4-fold). The SLCO1A2 gene that encodes for a sodium independent organic anion transporter of bile acids was down-regulated in three livers by DCF (−7.9 to −28.9-fold), and up-regulated in one liver by MMI (2.1-fold) and CBZ (1.8-fold). The sodium-bile acid cotransporter gene SLC10A1 was down-regulated in one liver by APAP (−18.3-fold) and TBF (−2.6-fold).
Several of the drugs altered the expression of genes involved with the cholesterol pathway. The SC4MOL gene, which encodes for a sterol-C4-methyl oxidase that is involved in the cholesterol synthesis pathway, was altered by APAP (−1.6 to 3.3-fold), TBF (−1.9 to 6.8-fold) and CSA (−2.3 to 6.6-fold) in two livers, and by CBZ (6.4-fold) and ETM (6-fold) in one liver. The cytochrome P450 genes associated with conversion of cholesterol to bile acids, CYP7A1 and CYP7B1 were both affected by APAP (−2.8 to 6-fold), ETM (−3.3 to 4.6-fold), DTL (−19.4 to 4.9-fold), CSA (−14.3 to 6.6-fold), TBF (−4.5 to 6.4-fold), and MMI (4.7 to 4.8-fold). DCF affected only CYP7A1 (−51.1-fold) in one liver. Involved in the conjugation and elimination of bile acids is the enzyme encoded by UGT2B4 (UDP glucuronosyltransferase 2 family, polypeptide B4), which was up-regulated in two livers by APAP (2.1 to 11.4-fold), MMI (2.2 to 4.5-fold), TBF (2.7 to 6.0-fold), and in one liver by ETM (4.1-fold), DTL (5.8-fold) and CSA (4.9-fold).
The alcohol dehydrogenase gene ADH1C, and the aldehyde dehydrogenase ALDH1A1 gene encode for enzymes that facilitate the inter-conversion between alcohols and aldehydes with the reduction of NAD+ to NADH. Both genes were down-regulated in all three livers by DCF, and in one liver by DTL. An up-regulation of the ALDH1A1 gene was detected in two livers with CBZ (1.6 to 2.5-fold) and one liver with APAP (1.9-fold) and MMI (2.2-fold). The ASNS gene, asparagine synthetase, encodes for a protein involved in the synthesis of asparagine, a non-essential amino acid produced in the liver. DCF caused up-regulation of the ASNS gene expression in all three livers (4.4 to 7.4-fold), while TBF up-regulated it in one liver (2.9-fold).
The FABP1 (fatty acid-binding protein 1) gene which encodes for a cytoplasmic protein expressed in the liver that binds long-chain fatty acids, fatty acyl CoA, bilirubin, and heme to limit cytotoxicity, was down-regulated in all three livers by APAP (−1.6 to −2.3-fold), in two livers by MMI (−3.5 to −1.7-fold) and DTL (−1.5 to −4.3-fold), and in one liver by DCF (−22-fold) and CBZ (−3.2-fold), while up-regulated in one liver by TBF (7.2-fold) and ETM (5.6-fold). The LPL (lipoprotein lipase) gene encodes for a member of the hepatic lipase and endothelial lipase family that hydrolyzes triglycerides in lipoproteins. LPL was up-regulated in all three livers by DCF (2.9 to 4.4-fold), and in one liver by APAP (1.9-fold) and ETM (4.5-fold). The WIPI1 gene encodes for the WD repeat domain phosphoinositide-interacting protein 1, which regulates the assembly of protein and phospholipid interactions, was up-regulated in all three livers by DCF (2.9 to 6.7-fold), and in one liver by CSA (1.8-fold).
DCF was the only drug that altered FXC1, mitochondrial import inner membrane translocase, and MTTP (microsomal triglyceride transfer protein) gene expression. FXC1 encodes for a protein that mediates the import and insertion of hydrophobic membrane proteins into the mitochondrial inner membrane, and was up-regulated in all three livers (2.5 to 4.0-fold). MTTP encodes for a protein involved with lipoprotein assembly and was down-regulated by DCF in all three livers (−2.8 to −11.5-fold).
DTL was the only drug that altered the aquaporin genes AQP (−10.9-fold) and AQP4 (−2.6-fold). These genes encode for integral membrane pore proteins that selectively allow water molecules to go in and out of the cell, while preventing the passage of ions and other solutes.
2.11. DNA Damage and Repair
The drugs which altered genes in this category in all three livers included CSA (9 genes, 10.4%), DTL (7 genes, 8.2%), TBF (6 genes, 7.9%), DCF (2 genes, 4.1%), followed by two livers MMI (7 genes, 13.3%), ETM (4 genes, 4.8%), CBZ (2 genes, 2.6%), and in one liver APAP (2 genes, 1.9%). Specifically, two genes CDKN1 and ERCC1 were altered by DCF in all three livers. The CDKN1 and CDKN1A genes encode for cyclin-dependent kinase inhibitors, in particular p21 (CDKN1A). DCF up-regulated CDKN1 in all three livers (2.1 to 3.8-fold) while DTL down-regulated it in two livers. CDKN1A was affected only by CSA (−3.2-fold) in one liver. The ERCC1 gene encodes for the DNA excision repair protein ERCC-1, which is part of an enzyme complex that participates in DNA repair and DNA recombination. DCF up-regulated ERCC1 in all three livers (2.5 to 24.6-fold), and in one liver by APAP (4.6-fold), MMI (6.7-fold), TBF (6.7-fold), DTL (7.8-fold), ETM (2.5-fold). CSA altered ERCC1 gene expression levels in two livers (−2.1 to 8.4-fold). The MSH2 gene encodes for the DNA mismatch repair involved with many types of DNA repair, which was affected in two livers by TBF (−2.6 to 3.5-fold), DTL (−1.7 to 3.2-fold), and CSA (−4.4 to 3.6-fold), and up-regulated in one liver by MMI (3.9-fold) and ETM (3.4-fold).
2.12. Apoptosis
Several genes indicative of apoptosis were affected in all three livers by DCF, 6 of 9 genes (14.3%); while drugs which altered the same gene in two livers, included CSA, 4 of 8 genes (11.3%), DTL, 3 of 7 genes (8.2%), TBF, 3 of 4 genes (7.9%), and CBZ, 1 of 6 genes (9.1%). Various apoptotic genes were altered in the three individual livers by APAP (7 genes, 6.7%), and in only two of the livers by ETM, (7 genes, 8.4%) and MMI (4 genes, 6.7%).
The BID gene encodes for the BH3 interacting-domain death agonist, which is a pro-apoptotic member of the Bcl-2 family. The BID gene expression was up-regulated by APAP (2.8-fold) ETM (2.3-fold) and CBZ (1.4-fold). BCL2L1 encodes for a BCL-2 member located at the outer mitochondrial membrane that is an apoptosis regulator. CSA altered BCL2L1 gene expression in two livers. CSA also up-regulated the APAF1 gene (2.2-fold), apoptotic protease activating factor 1, which encodes a cytoplasmic protein that forms one of the central hubs in the apoptosis regulatory network.
The FAS gene encodes for the FAS receptor, which resides on the cell surface to form a death-inducing signaling complex that leads to apoptosis. FAS gene expression was up-regulated in the same liver by APAP (5.4-fold), DCF (11-fold), MMI (8.3-fold), TBF (8.0-fold), ETM (5.5-fold), DTL (6.9-fold), and CSA (7.6-fold). The FADD gene encodes for the Fas-associated protein with Death Domain, which forms a death inducing signaling complex during apoptosis. FADD gene expression was up-regulated in all three livers by DCF (2.1 to 6.8-fold), in two livers by TBF (1.8 to 2.9-fold) and CSA (2.2 to 2.9-fold), and in one liver by APAP (2.5-fold), MMI (2.7-fold), ETM (2.2-fold), DTL (2.7-fold).
The TNFRSF10A and TNFRSF10B genes encode for members of the TNF-receptor superfamily involved with apoptosis. DCF up-regulated both these genes in all three livers, while CSA up-regulated TNFRSF10A in two livers and CBZ in one liver. The TNFSF10 gene encodes for a cytokine that is a member of the tumor necrosis factor TNF ligand family, which preferentially induces apoptosis in transformed cells. DCF down-regulated TNFSF10 gene expression levels in all three livers (−5.0 to −24.8-fold).
Altered GADD45 gene expression is indicative of growth arrest conditions. DCF up-regulated GADD45 gene expression levels in all three livers (2.5 to 30.7-fold). The XIAP gene encodes for an inhibitor of apoptosis. This gene was up-regulated in all three livers by DCF (2.0 to 5.5-fold), and in the same liver by ETM (3.3-fold), DTL (2.8-fold) and CSA (3.3-fold).
2.13. Necrosis
Genes indicative of necrosis were most evident following exposure to APAP (8 genes, 8.7%) and ETM (5 genes, 7.2%), followed by CSA (5 genes, 4.7%), TBF (3 genes, 3.4%), DTL (3 genes, 2.5%), CBZ (1 gene, 1.3%), MMI (1 gene, 1.7%), and DCF (1 gene, 0.7%).
BMF encodes for a Bcl-2 protein, which has been associated with apoptosis and necrosis, was up-regulated by ETM (2.2 to 2.8-fold) and CSA (2.2-fold). NUDT13 (nudix hydrolase 13) gene expression was up-regulated in the same liver by APAP (3.9-fold), MMI (2.6-fold), TBF (3.0-fold), ETM (2.8-fold), DTL (2.9-fold), and CSA (4.8-fold). Furthermore, in this same liver APAP exposure caused the up-regulation of several other genes associated with necrosis, FOXI1 (2.3-fold), GALNT5 (2.3-fold), HOXA3 (2.2-fold), JPH3 (2.3-fold), KCNIP1 (2.3-fold), S100A7A (2.3-fold).
2.14. Immune Response, Inflammation
Genes indicative of inflammation and an immune response exhibited altered expression by most drugs in this study. DCF affected 3 of 4 genes (7.5%) in the three livers. Genes altered in two livers occurred following CSA, 2 of 10 genes (11.3%), ETM, 2 of 7 genes (10.8%), DTL, 1 of 6 genes (5.7%), CBZ, 1 of 7 genes (10.4%), TBF, 1 of 4 genes (5.6%), and MMI 1 of 5 genes (10%). APAP exposure altered 6 genes (5.8%) in two different livers.
AHSG (α2-HS glycoprotein) encodes for a plasma binding protein synthesized by hepatocytes. Gene expression of AHSG was down-regulated by DCF (−4.2 to −2.0-fold) in all three livers and by MMI (−1.7 to −2.4-fold) in two livers. The interleukin genes exhibited a varied response to the drugs in this study. IL1Β gene expression was down-regulated by ETM (−2.5 to −10.2-fold), DTL (−3.5 to −2.4-fold), and CSA (−8.2 to −3.9-fold) in the same two livers, and by MMI (−2.9-fold) in one liver; whereas APAP induced the expression of IL1A (2.2-fold) and IL2 (2.4-fold), CBZ increased IL4 (56-fold), and CSA increased IL10 (2.8-fold) gene expression levels. The C9 gene, which encodes for a member of the complement system, was down-regulated by DCF (−3.8 to −7.6-fold) in the three livers, while up-regulated by CSA (2-fold) in one liver.
EP300 encodes for a protein that is involved in regulating cell growth, differentiation and division. This gene was up-regulated in the same liver by TBF (2.2-fold), ETM (2.5-fold), DTL (2.2-fold) and CSA (2.5-fold). HRG (histidine-rich glycoprotein) encodes for a glycoprotein produced in the liver and located in the plasma and platelets. HRG gene expression was up-regulated by APAP (4.1-fold) in one liver. PTGS2 (prostaglandin-endoperoxide synthase 2) encodes for the cyclooxygenase 2 (COX 2), and the gene expression was up-regulated by DCF (16.9 to 34.8-fold) in two livers, which is tied to the pharmacological mechanism of DCF.
3. Discussion
The goal of this study was to characterize human response ex vivo to drugs associated with liver side-effects clinically. In this study, functional markers of oxidative stress plus gene expression profiles indicative of organelle and tissue dysfunction differentiated the drug induced tissue stresses across drugs in the same human liver tissue. These drugs demonstrated diverse effects and can serve as reference drugs to evaluate the safety risks of drugs in development. The ex vivo model used in this study, organotypic human liver slices, has the leverage as a relevant model since cell-cell and cell-matrix interactions are represented in their normal architecture to mimic in vivo dynamics to forecast drug safety risks. The doses of the drugs were selected either from the literature citing plasma concentrations associated with clinical side-effects or from previous in vitro human liver preparations or from human liver slice studies in which the concentration exhibited a change in tissue function [
8,
30,
32]. For example, high doses of the pain medication APAP (4 g dose or blood levels of 1 mM) are associated with altered liver function in some healthy males [
28]. All drugs were tested side-by-side in each human liver, and the drugs were dosed daily to mimic initial dosing in clinical studies.
Several drugs in this study are known to perturb tissue anti-oxidant status, and the metabolism of the drug contributes to oxidative stress, mitochondrial injury, and ER stress in some individuals. For example, the acetaminophen metabolite
N-acetyl-p-benzoquinone imine (NAPQI) is conjugated with GSH, and other metabolites formed by peroxidase-like enzymes can reduce the liver anti-oxidant status [
33]. APAP metabolite profiles have been shown to distinguish responders from non-responders toward APAP hepatotoxicity [
34]. Mitochondria are a key target of APAP due to a reduction of mitochondrial GSH levels by ROS, leading to necrotic cell death [
35,
36,
37,
38]. Diclofenac, a non-steroidal anti-inflammatory agent, generates reactive metabolites that bind to cellular macromolecules and proteins to alter tissue anti-oxidant status. Mitochondrial injury is proving to be a key factor in DCF hepatotoxicity, and an immune mediated hypersensitivity is seen in some individuals [
18,
39,
40]. Methimazole, an agent to decrease thyroid size, and linked with hematologic effects and hepatoxicity, co-oxidizes GSH to GSSG via the metabolism of the thione moiety [
41]. The reduced GSH tissue contributes to subsequent toxic effects [
42,
43,
44,
45]. Clinically, a reduction of the MMI dose from 30 to 15 mg/day reduces the incidence of side-effects [
46]. Two additional compounds that could affect the tissue anti-oxidant status as a consequence of metabolism are terbinafine, an allylamine derivative used as an antifungal agent, and carbamazepine, an iminostilbene used for seizures. Both drugs likely generate reactive metabolites because hepatotoxicity, detectable by increases in serum transaminases, is associated with hypersensitivity or an immunological response in some individuals, yet evidence for adduct formation has not been reported [
29,
30,
47,
48].
Pharmaceutical agents that modulate mitochondria function raise concern about the potential for mitochondrial injury. Drugs in this study associated with a direct mitochondrial interaction include etomoxir considered for diabetes and heart failure, the immunosuppressant cyclosporine, and dantrolene a smooth muscle relaxant. ETM irreversibly inhibits the rate-limiting enzyme of mitochondrial β-oxidation, carnitine palmitoyltransferase-1 (CPT-1), to decrease the use of free fatty acids as a source of energy so as to increase the utilization of glucose for ATP production [
49]. CSA inhibits the mitochondrial permeability transition pore through binding to a regulator of the pore cyclophilin D. High CSA concentrations are associated with mitochondrial injury [
1,
50,
51,
52,
53,
54]. Dantrolene, a potential muscle relaxant, affects Ca
+2 homeostasis via ryanodine receptors. The hepatic injury may in part be due to a perturbation of Ca
+2 homeostasis or the formation of reactive oxygen species [
55]. The RyR1 receptor is located on the inner mitochondrial membrane of excitable cells and is postulated to exist within liver mitochondria [
1]. Hepatocytes also possess a truncated type 1 ryanodine (RYR1) receptor in the endoplasmic reticulum [
56]. Both CSA and DTL at low doses can influence calcium homeostasis to minimize apoptosis or necrosis during ischemia-reperfusion injury [
57,
58].
Functional tissue biomarkers of oxidative stress used in this study were liver slice ATP and GSH levels following drug exposure. ATP is a sensitive marker of oxidative stress and an indicator of mitochondrial function. GSH, the major anti-oxidant synthesized in the liver, is an indicator of overall tissue anti-oxidant status. About 15% of the cellular GSH resides within the mitochondria, and mitochondria are a major source of ROS production, which drugs can disrupt to impact mitochondrial function and cause injury [
1,
40,
59,
60]. Both APAP and DCF caused significant time-dependent decreases of ATP levels, and DTL caused significant decreases at 72 h. GSH levels were significantly increased with APAP, MMI and CBZ followed by TBF, DTL, and ETM. Since the metabolism of these drugs consumes GSH (APAP, MMI, CBZ and TBF) or affects the mitochondria directly (DTL, ETM), the tissue likely responds by synthesizing GSH. Previous human liver slice studies using several concentrations per drug revealed that 1 mM APAP, 500 μM MMI, 10 μM CSA, and 10 μM DTL altered ATP or GSH levels in most but not all human liver slice studies [
8,
27].
3.1. Metabolism Genes
The induction of cytochrome P450s by xenobiotics can have a protective effect, and yet may trigger deleterious effects. For example, POR, the cytochrome p450 oxidoreductase, facilitates electron transfer from NADPH to all microsomal P450 enzymes. The gene expression of
POR was altered by DCF and TBF. If this were to result in an increased respiration rate, it could further increase the formation of ROS within mitochondria to perturb Ca
+2 homeostasis and cell signaling [
1,
35,
36]. Furthermore, mitochondrial DNA, lipids and proteins are important targets of ROS [
61]. The cytochromes CYP1A1 and CYP1A2, inducible by xenobiotics, interact with mitochondria and may be linked with trans-membrane potential and apoptosis [
62]. In this study, TBF, MMI, DTL, APAP, CBZ, and DCF caused changes in the
CYP1A gene expression. DCF and DTL also affected
FMO gene expression levels. Induction of FMO enzymes in mice is protective toward APAP hepatotoxicity [
63], whereas inhibition of FMO suppressed MMI hepatotoxicity [
42].
3.2. Oxidative Stress, Mitochondrial Energy, Heat Shock and ER Stress, Apoptosis, Necrosis, DNA Damage and Repair
The APAP and DCF induced decreases of ATP levels and changes in GSH levels were paralleled by gene expression changes of oxidative stress (10.6% APAP, 7.5% DCF) and mitochondrial energy (6.7% APAP, 2.0% DCF). In particular, the oxidative stress genes affected by both drugs included glutathione regulation (
GSTA3,
GSTA4), reactive intermediate formation (
GPXs,
EPHX1), and a regulator of translation (PPP1R15B). Mitochondrial energy genes altered by APAP included
MDH1 (malate dehydrogenase), which is part of the TCA cycle, and the mitochondrial matrix succinyl-CoA ligase (
SUCLG1), while DCF altered a mitochondrial uncoupling protein (
UCP2), which can separate oxidative phosphorylation from ATP synthesis. Other studies report that mitochondrial uptake of DCF via the anion carrier results in uncoupling of respiration and opening of the permeability transition pore to initiate cell death [
39]. Additionally, the binding of DCF reactive electrophiles to mitochondrial proteins triggers apoptosis [
64] and possibly a hypersensitivity reaction [
65].
An increase of unfolded proteins in the endoplasmic reticulum (ER) triggers a stress response, which includes the increased expression of proteins involved in polypeptide folding, the inhibition of translation to prevent further accumulation of unfolded proteins, the destruction of misfolded proteins, and an increased transcriptional regulation of protein synthesis leading to cell death [
3,
66]. The ER is the major intracellular storage for Ca
+2 and interacts with mitochondria to exchange metabolic signals via lipids, proteins, metabolites and Ca
+2. Mitochondrial Ca
+2 overload triggers the mitochondrial pore to release mediators including Ca
+2, cytochrome c, and pro-apoptotic proteins [
38,
61,
67,
68,
69,
70].
In this study, both APAP and DCF altered the expression of a mitochondrial heat shock gene (
HSP9A), the protein folding genes (
DNAJ family), and genes encoding for the transport of unfolded proteins to the proteasome for degradation (
SEL1L,
SERP1). DCF caused changes in several genes induced by misfolded proteins in the ER (
DERL1,
EIF2AK3,
STVN1,
HSF2,
HERPUD1), and several genes that encode for heat shock transcription factors (
ATF4,
ATF6,
DDIT3). Consequences of ER and mitochondria stress can trigger various cell types to release pro-inflammatory or cytotoxic mediators to activate cell death signaling pathways. APAP induced a greater proportion of genes linked with necrosis (8.7%), while DCF induced gene changes linked with apoptosis (14.3%). Genes for the death receptor FAS (
FAS,
FADD), associated with necrosis [
59], were up-regulated by APAP and DCF. Additionally, DCF up-regulated
GADD45, a gene associated with growth arrest, and genes of the TSF-receptor family (
TNFRS10A and B) involved with apoptosis. Marked decreases of liver GSH have been demonstrated to sensitize hepatocytes to the oxidative effects of cytokines such as tumor necrosis factor [
66,
71]. APAP caused the up-regulation of several genes linked with necrosis (
FOXl1,
GALNT5,
HOXA3,
JPH3,
KCNIP1,
NUDT13,
and S100A7A). The concentrations of APAP (1 mM) and DCF (1 mM), which induced these changes are considered high, however such APAP concentrations have been tested in humans. The FDA is requiring additional labeling for APAP reminding consumers to be mindful of the dose taken [
72]. Acetaminophen is the primary choice for pain in US hospitals, and is a concern for the elderly, which likely have a compromised liver GSH status.
Both TBF and CBZ (100 μM) increased liver slice GSH levels and had an impact on oxidative stress (18% TBF, 11.7% CBZ) and mitochondrial energy gene expression (4.5% TBF, 7.8% CBZ). In particular, both drugs affected the expression of glutathione transferase GSTA3, gene markers of reactive intermediate formation with glutathione peroxidase isoforms (GPX2 and 3), microsomal epoxide hydrolase 1 (EPHX1), and reactive oxygen formation (DUOX2). TBF additionally up-regulated the NQO1 gene expression, involved in the reduction of quinones to hyroquinones, and the translation factor PPP1R15B. Genes involved with the TCA cycle and cellular energy, aconitase (ACO1) and malate dehydrogenase (MDH1), were altered by TBF and CBZ. Additionally, CBZ affected the mitochondrial gene expression of succinate dehydrogenase (SDHC), succinyl CoA ligase, and dihydrolipoamide dehydrogenase (DLD) involved in multi-enzyme complexes associated with energy metabolism, while TBF affected the mitochondrial cytochrome C1 (CYC1) gene expression. Drug induced tissue stress reflected by changes of heat shock and ER stress genes represented about 13% for both drugs. In particular, a mitochondrial heat shock gene (HSPD1) indicative of protein folding of imported proteins, ER protein folding chaperone genes (HSP90B1, CRYAB), unfolded protein response genes (SEL1L, SERP1, SEC62), and the membrane transcription factor MBTPS2 gene. Genes indicative of apoptosis represented 7.9% for TBF and 9.1% for CBZ, and necrosis gene changes represented 3.4% with TBF and 1.3% for CBZ. The apoptosis genes induced by TBF included the FAS death pathway and caspase 8, while CBZ affected GADD45. Differences between the two drugs became evident with a greater change of genes linked with regulation for TBF (7.9%) than for CBZ (1.3%). In particular, TBF altered the mismatch repair gene (MSH2), and the excision repair genes (ERCC1 and 6).
MMI (500 μM) exposure altered the fewest oxidative stress (6.7%) genes, yet important gene indicators of oxidative stress. Two genes that combat oxidative stress were up-regulated by MMI,
NQO1 involved in the reduction quinones to hydroquinones, and
PRDX1 which reduces hydrogen peroxide and alkyl hydroperoxides. Of particular interest for MMI is that Kupffer cells contain myeloperoxidases, which contribute to its metabolism and co-oxidation of GSH. In this study, the utilization of GSH was compensated for by an increase of liver slice GSH levels, and hence no effect on GSH utilization genes in this study. In a previous human liver-blood co-culture study, liver slice GSH levels were decreased by MMI exposure at 72 h, which is likely due to the overall liver GSH status being lower than in this study. Blood cells revealed a decline of GSH levels due to MMI metabolism prior to a decline of liver GSH levels [
25]. MMI also affected mitochondrial energy genes (6.7%), including
MDH1 and
CYC1, that corresponded to decreased ATP levels at 72 h. MMI induced heat shock gene expression changes (15%) involved with the ubiquitin pathway for protein degradation (
UBE2G2,
SEC62,
SEL1L, and
SERP1). Genes linked with apoptosis (6.7%) and necrosis (1.7%) were altered the least in comparison to the other drugs. However, the proportion of genes indicative of DNA damage and repair (13.3%) represented the greatest change compared to the other drugs. In particular, the gene changes dealt with DNA repair,
BRCA2,
ERCC1 and
MSH2.
The drugs known to interact directly with mitochondria DTL, ETM, and CSA trigged oxidative stress (7.4% DTL, 10.8% ETM, 8.5% CSA) and mitochondrial energy (3.3% DTL, 6% ETM, 2.8% CSA) gene expression changes. Glutathione regulation genes were affected, such as GSTA3 (ETM), and GPX2 (DTL, CSA), as well as genes linked to reactive oxygen formation, DUOX1 and DUOX2 (CSA). Heat shock and ER stress related genes were greatly affected by CSA (25.5%), followed by ETM (18.1%) and DTL (12.3%). HSP90B1 gene expression, involved with protein folding and as an immune chaperone, was altered in two livers by CSA and ETM. Consequences for apoptosis gene expression changes represented 11.3% CSA, 8.4% ETM and 8.2% DTL, and for necrosis represented 4.7% CSA, 7.2% ETM, and 2.5% DTL. The FADD and FAS genes, linked with the apoptotic death pathway, were perturbed by CSA, DTL and ETM, as well as TNFRSF10A by CSA.
3.3. Fatty Acid Metabolism, Steatosis, Cholestasis, Phospholipidosis, Immune Response and Inflammation
The perturbation of fatty acid metabolism can have consequences on the genes associated with steatosis, cholestasis, and phospholipidosis. The combined categories were greatest for DTL (45%), followed by APAP (34.6%), DCF (30.6%), ETM (32.6%), CBZ (33.8%), TBF (29.2%), MMI (26.5% MMI), and CSA (25.6%) of the total gene expression changes. Gene changes in at least two livers were greater for DCF (9 of 12 genes) and APAP (6 of 8 genes) for the steatosis, cholestasis, phosopholipidosis category; whereas DTL altered more genes in two livers for fatty acid metabolism (7 of 12 genes). Several liver transporter genes exhibited altered expression, in particular the efflux p-glycoprotein transporter
ABCB1 (DCF, APAP, DTL, CBZ), the cannicular organic anion transporter 1 and 2,
ABCC2 and
ABCC3, (APAP, MMI, TBF, CBZ and ETM), the organic solute transporter gene
OSTβ (DCF, APAP, ETM), the organic anion transporter
SLCO1A2 (DCF, MMI, CBZ), and the sodium-bile acid co-transporter
SLC10A1 (APAP, TBF). The perturbation of several transporter genes indicates that the tissue is ramping up the export of bile acids and solutes to prevent cholestatic hepatotoxicity [
73].
Genes associated with the cholesterol synthesis,
SC4MOL (APAP, TBF, CSA, CBZ, ETM), the conversion of cholesterol to bile acids
CYP7A1 and
CYP7B1 (APAP, ETM, DTL, CSA, TBF, MMI and DCF), and the elimination of bile acids
UGT2B4 (APAP, MMI, TBF, ETM, DTL, CSA) exhibited altered expression. The
FABP1 gene, which encodes for a protein that binds long chain fatty acids, acyl CoA, bilirubin and heme to limit cytotoxicity was affected by all drugs except CSA. Cyclooxygenase 2 (
PTGS2) gene expression was affected by DCF, which is in line with the pharmacological mechanism of DCF inhibiting this enzyme. Various interleukin genes were altered including
IL1β (ETM, DTL, CSA, MMI),
IL1A and
IL2 (APAP),
IL4 (CBZ),
IL10 (CSA),
C9 (DCF, CSA), suggestive of an inflammation and possible immune response. Proinflammatory and cytotoxic mediators released from macrophages, may contribute to APAP hepatotoxicity in some individuals [
74].
Drugs that influence fatty acid metabolism can alter both mitochondrial function and peroxisome function to affect the overall energy levels of the tissue. Drug effects on mitochondrial genes are of particular concern since mitochondrial toxicity could become an issue. Several mitochondrial genes (
CPT1,
ACADM,
ACAA1,
ACADVL,
ACAA2,
HADHA,
ECHS1,
HADHB) involved in the β-oxidation pathway of fatty acids were altered by ETM (6 genes), DTL (6 genes), TBF (3 genes), CBZ (3 genes), CSA (3 genes) and APAP (2 genes). Neither DCF nor MMI influenced these mitochondrial genes. DCF affected only a mitochondrial inner translocase gene
FXC1. Severe or chronic impairment of mitochondrial β-oxidation free fatty acid (FFA) metabolism can lead to liver lipid deposits, microvesicular steatosis and inflammation, and necrosis in rats and man [
75,
76]. Hepatotoxicity is attributed to interference of the respiratory complexes, decreased ATP production, and oxidative stress [
75,
77,
78,
79]. Direct inhibitors of β-oxidation (e.g., tetracyclines, phenformin, troglitazone) or drugs that sequester CoA (e.g., valproic acid) to inhibit β-oxidation have caused dicarboxylic aciduria, and lactic acidosis [
78,
79]. In humans, drug induced lactic acidosis can lead to hepatic failure, and is correlated with enlarged mitochondria [
80]. In previous liver slice studies, compounds that inhibited CPT1 or which complexed with CoA to limit the capacity of the β-oxidation pathway led to mitochondrial injury at high doses. In rats the mitochondria were enlarged both in vivo and in vitro, and in human liver slices the mitochondria exhibited granular inclusions [
81,
82].
In this study, 8 drugs associated with liver effects clinically were compared side-by-side in individual human liver slice experiments. ATP and GSH levels following drug exposure were sensitive functional markers of oxidative stress and mitochondria dysfunction. Gene expression profiles indicative of organ dysfunction and injury further characterized and distinguished drug effects (
Table 5). Each drug had direct effects on the livers and caused multifactorial effects, for which consequences on mitochondrial energy, ER calcium regulation, cholestasis, and inflammation, will be underlying mechanisms of the clinical effects. Activation of oxidative stress, mitochondrial energy, heat shock and ER stress genes was particularly evident with CSA (37%), TBF (36%), ETM (35%), APAP (34%), DCF (34%) and CBZ (33%). Adding DNA damage and repair, apoptosis, necrosis, inflammation and immune response revealed CSA (75%) with the greatest effect followed by ETM (66%), TBF (61%), DCF (61%), MMI (60%), APAP (57%), CBZ (56%), and DTL (48%). Gene changes in fatty acid metabolism, steatosis, cholestasis plus immune response and inflammation were affected by DTL (51%), followed by CBZ and ETM (44%–43%), APAP and DCF (40%–38%), then MMI, TBF and CSA (37%–35%).
A comparison of drugs in development to reference drugs aids in drug candidate selection with the widest safety margin, and the identification of serum concentrations that would lead to organ injury, if such concentrations were achieved. Drug specific safety biomarkers can be identified for clinical studies, which may include formation of a metabolite, mitochondrial injury markers, or indicators of cholestasis, inflammation and immune response markers. Some of the drugs in this study are also linked with hypersensitivity reactions. As more studies are done early markers of hypersensitivity will be identified. To improve the objectivity and semiquantitation to assess drug induced liver injury (DILI) and clinical assessments the Roussel Uclaf Causality Assessment Method (RUCAM) is showing success particularly with hypersensitivity cases [
83,
84]. Further validation is needed, as well as reliable assessments for hepatitis or cholestasis [
84]. The multifactorial nature of drug induced liver injury and idiosyncratic-DILI requires continued approaches of multiparametric endpoints including drug metabolism enzymes, antioxidant enzymes, drug transporters, and inflammation plus modeling to identify predictive assays and endpoints for diagnosing individual drug-induced liver injury [
85,
86].







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}