
Cross-Kingdom Regulation of Putative miRNAs Derived from Happy Tree in Cancer Pathway: A Systems Biology Approach

 ,
,
Abstract
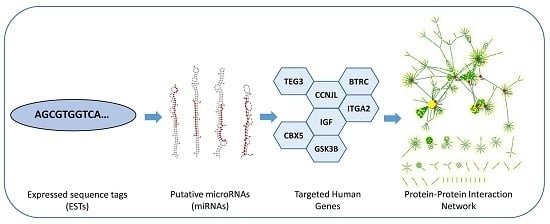
:
1. Introduction
2. Results
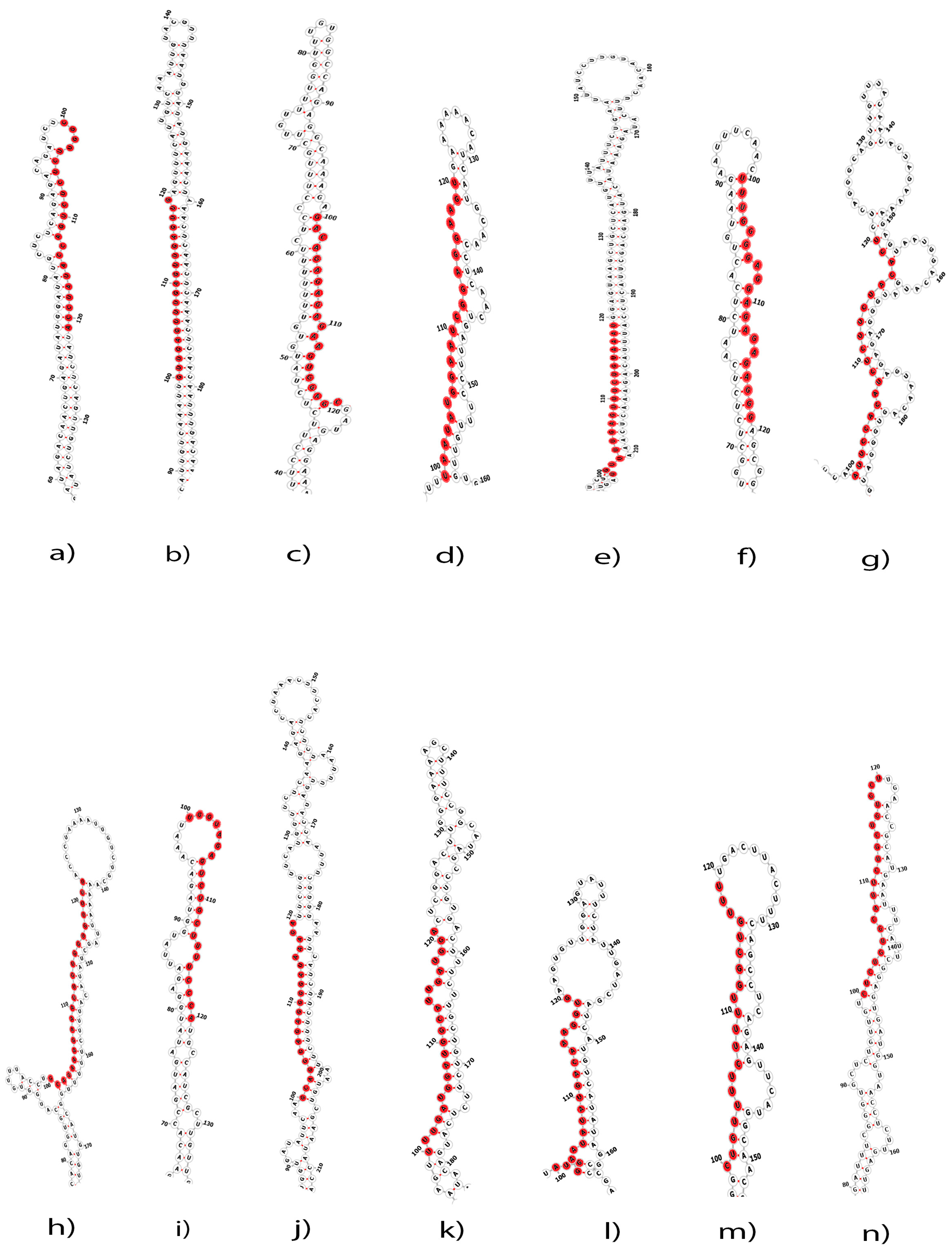
2.1. Putative C. acuminata miRNAs
2.2. Target Gene Identification and Their Functional Enrichment Analysis
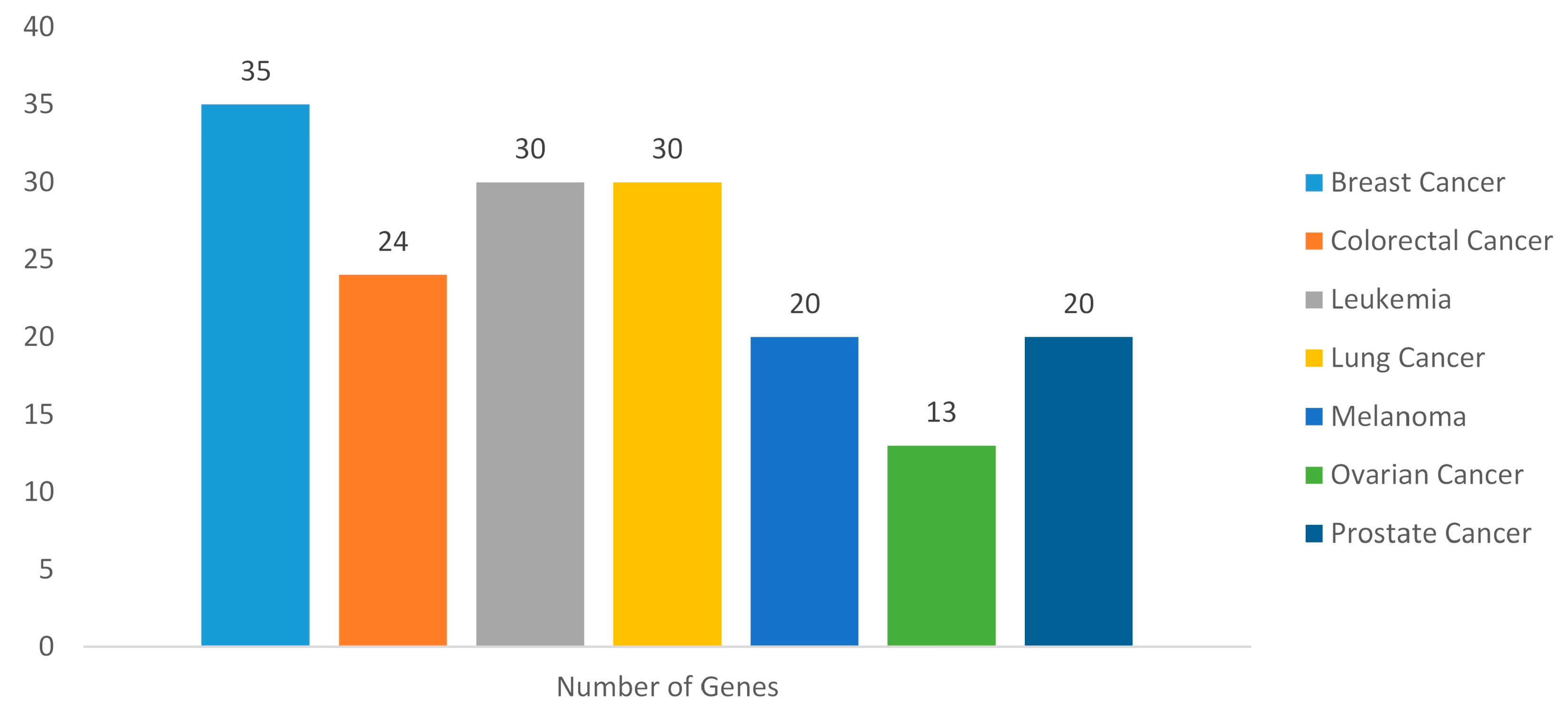
2.3. Gene Disease Associations
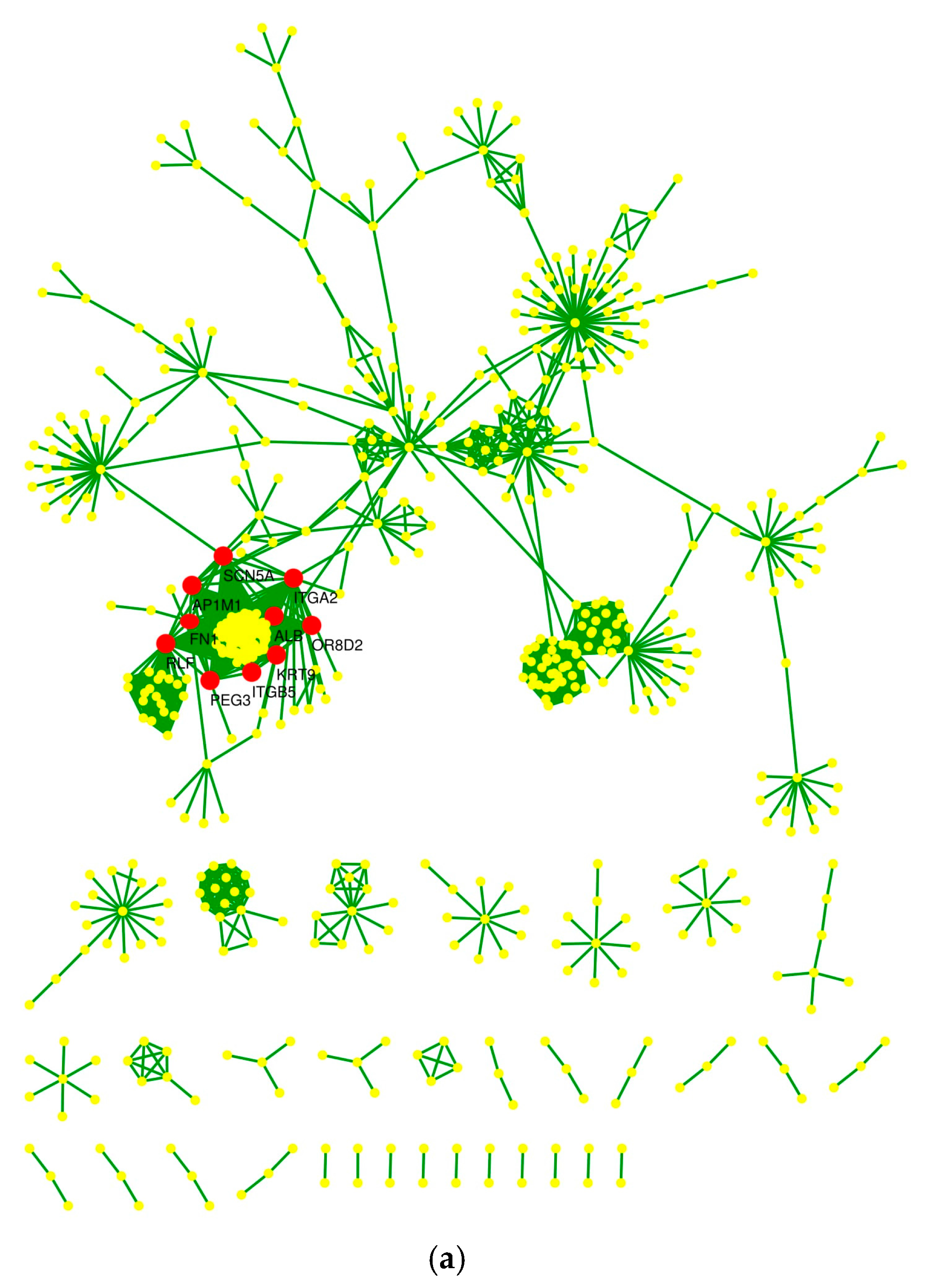
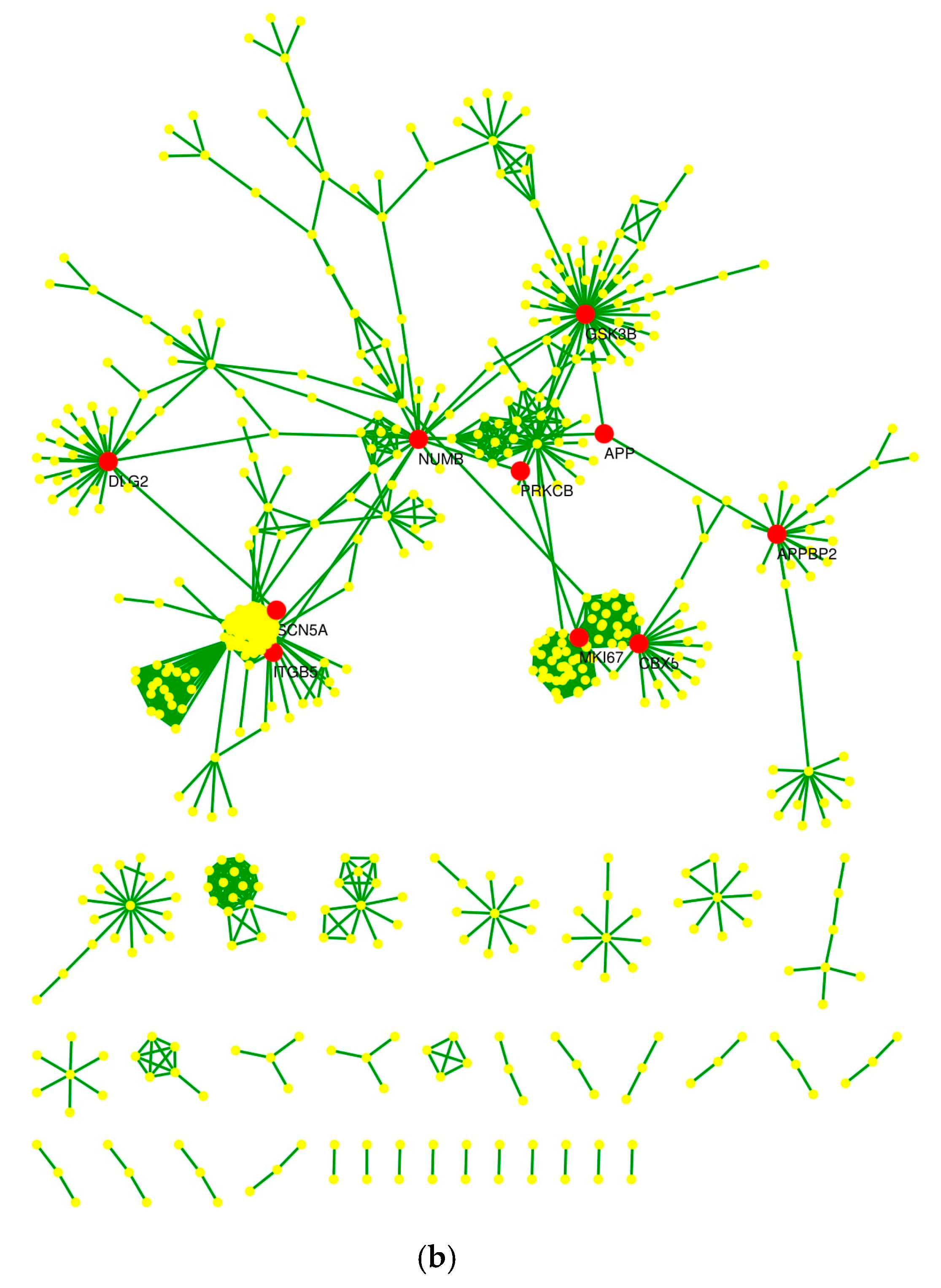
2.4. Protein–Protein Interaction Network and Statistical Validation
2.5. Hub Proteins and Centrality Parameters
3. Discussion
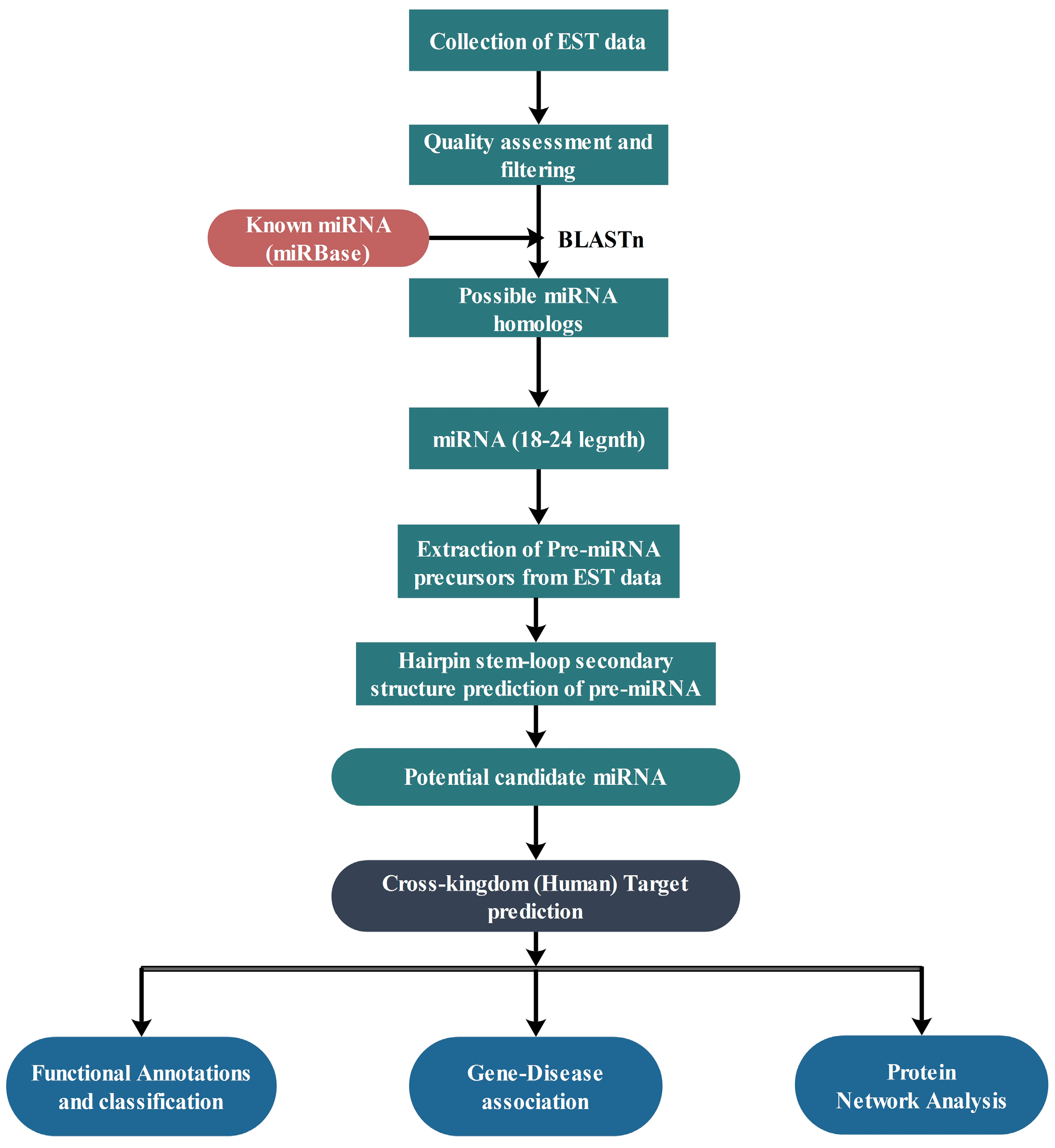
4. Materials and Methods
4.1. Data Retrieval
4.2. Prediction of Putative Novel miRNAs
4.3. Prediction of Secondary Structure
4.4. Nomenclature of miRNAs
4.5. Prediction of Potential Targets of miRNAs
4.6. Functional Analysis of Target Genes
4.7. Gene-Disease Associations
4.8. Protein-Protein Interaction Data and Network Construction
4.9. Statistical Validation Using Network Randomization
4.10. Regulatory Network Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Carrington, J.C.; Ambros, V. Role of MicroRNAs in Plant and Animal Development. Science 2003, 301, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Aukerman, M.J.; Sakai, H. Regulation of Flowering Time and Floral Organ Identity by a MicroRNA and Its APETALA2-Like Target Genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Khraiwesh, B.; Zhu, J.; Zhu, J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 2012, 1819, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Kragler, F.; Varkonyi-Gasic, E.; Haywood, V.; Archer-Evans, S.; Lee, Y.M.; Lough, T.J.; Lucas, W.J. A Systemic Small RNA Signaling System in Plants. Plant Cell 2004, 16, 1979–2000. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam Encodes a Developmentally Regulated microRNA that Controls Cell Proliferation and Regulates the Proapoptotic Gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef]
- Hwang, H.; Mendell, J.T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 2006, 94, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Stappert, L.; Roese-Koerner, B.; Brastle, O. The role of microRNAs in human neural stem cells, neuronal differentiation and subtype specification. Cell Tissue Res. 2014, 359, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Ruohola-Baker, H. In Regulation of Stem Cell Populations by microRNAs. Adv. Exp. Med. Biol. 2013, 786, 329–351. [Google Scholar] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, S.; Fu, Z.; Wang, Y.; Wang, N.; Liu, Y.; Zhao, C.; Wu, J.; Hu, Y.; Zhang, J.; et al. Effective detection and quantification of dietetically absorbed plant microRNAs in human plasma. J. Nutr. Biochem. 2015, 26, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Sang, X.; Hong, Z. Beyond nutrients: Food-derived microRNAs provide cross-kingdom regulation. Bioessays 2012, 34, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yang, Z.; Li, J.; Minakhina, S.; Yang, M.; Padgett, R.W.; Stewart, R.; Chen, X. Methylation as a crucial step in plant microRNA biogenesis. Science 2005, 307, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hou, D.; Chen, X.; Li, D.; Zhu, L.; Zhang, Y.; Li, J.; Bian, Z.; Liang, X.; Cai, X.; et al. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: Evidence of cross-kingdom regulation by microRNA. Cell Res. 2012, 22, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhu, Y.; Sun, B.; Shao, Y.; Jing, A.; Wang, J.; Xiao, Z. Assessing the survival of exogenous plant microRNA in mice. Food Sci. Nutr. 2014, 2, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Farmer, L.M.; Agyekum, A.A.; Hirschi, K.D. Detection of dietary plant-based small RNAs in animals. Cell Res. 2015, 25, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Farmer, L.M.; Agyekum, A.A.; Elbaz-Younes, I.; Hirschi, K.D. Detection of an abundant plant-based small RNA in healthy consumers. PLoS ONE 2015, 10, e0137516. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Li, Y.; Ahmad, A.; Azmi, A.S.; Bao, G.; Ali, S.; Banerjee, S.; Kong, D.; Sarkar, F.H. Targeting CSC-Related miRNAs for Cancer Therapy by Natural Agents. Curr. Drug Targets 2012, 13, 1858–1868. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Mao, Z.; Shi, Y.; Chen, Y.; Sun, Y.; Zhang, Q.; Song, L.; Peng, L. MicroRNA-7 inhibits cell proliferation, migration and invasion in human non-small cell lung cancer cells by targeting FAK through ERK/MAPK signaling pathway. Oncotarget 2016, 7, 77468–77481. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Chen, F.; Shen, S.; Chen, W.; Chen, L.; Su, Q.; Zhang, L.; Bi, J.; Zeng, W.; Li, W.; et al. MicroRNA-129–5p inhibits hepatocellular carcinoma cell metastasis and invasion via targeting ETS1. Biochem. Biophys. Res. Commun. 2015, 461, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, M.; Li, X.; Tang, H. miR-490-3p Modulates Cell Growth and Epithelial to Mesenchymal Transition of Hepatocellular Carcinoma Cells by Targeting Endoplasmic Reticulum-Golgi Intermediate Compartment Protein 3 (ERGIC3). J. Biol. Chem. 2012, 288, 4035–4047. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, S.; Bae, H.; Kang, H.; Kim, S.J. Genome-wide identification of target genes for miR-204 and miR-211 identifies their proliferation stimulatory role in breast cancer cells. Sci. Rep. 2016, 6, 25287. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, X.; Liu, J.; Dong, L.; Chen, Q.; Liu, J.; Kong, H.; Zhang, Q.; Qi, X.; Hou, D.; et al. Honeysuckle-encoded atypical microRNA2911 directly targets influenza A viruses. Cell Res. 2015, 25, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Pastrello, C.; Tsay, M.; McQuaid, R.; Abovsky, M.; Pasini, E.; Shirdel, E.; Angeli, M.; Tokar, T.; Jamnik, J.; Kotlyar, M.; et al. Circulating plant miRNAs can regulate human gene expression in vitro. Sci. Rep. 2016, 6, 32773. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.R.; Fong, M.Y.; Somlo, G.; Wu, J.; Swiderski, P.; Wu, X.; Wang, S.E. Cross-kingdom inhibition of breast cancer growth by plant miR159. Cell Res. 2016, 26, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Thapa, S.; Pradhan, D.; Thorat, S.S.; Talukdar, N.C. Intra-specific genetic diversity, phytochemical analysis and antioxidant activities of a potential Himalayan Swertia (Swertia bimaculata Hook. F. & Thomas.). Ind. Crops Prod. 2013, 49, 341–347. [Google Scholar]
- Elsayed, W.; El-Shafie, L.; Hassan, M.K.; Farag, M.A.; El-Khamisy, S. Isoeugenol is a selective potentiator of camptothecin cytotoxicity in vertebrate cells lacking TDP1. Sci. Rep. 2016, 6, 26626. [Google Scholar] [CrossRef] [PubMed]
- Lukasik, A.; Zielenkiewicz, P. Plant MicroRNAs—Novel Players in Natural Medicine? Int. J. Mol. Sci. 2016, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L.; Vaishnav, P. Natural products for cancer chemotherapy. Microb. Biotechnol. 2010, 4, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, D.; Rizzetto, L.; Tocci, N.; Rivero, D.; Asquini, E.; Si-Ammour, A.; Bonechi, E.; Ballerini, C.; Viola, R. Plant microRNAs as novel immunomodulatory agents. Sci. Rep. 2016, 6, 25761. [Google Scholar] [CrossRef] [PubMed]
- Hirschi, K.D.; Pruss, G.J.; Vance, V. Dietary delivery: A new avenue for microRNA therapeutics? Trends Biotechnol. 2015, 33, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Richard, T.S.; Kamdje, A.H.N.; Mukhtar, F. Medicinal plants in breast cancer therapy. J. Dis. Med. Plants 2015, 1, 19–23. [Google Scholar]
- Hong, M.; Wang, N.; Tan, H.Y.; Tsao, S.; Feng, Y. MicroRNAs and Chinese Medicinal Herbs: New Possibilities in Cancer Therapy. Cancers 2015, 7, 1643–1657. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.C.; Whang-Peng, J.; Adamson, R.H. Studies on the Antitumor Activity, Mechanism of Action, and Cell Cycle Effects of Camptothecin. J. Natl. Cancer Inst. 1971, 46, 789–795. [Google Scholar] [PubMed]
- Li, L.H.; Fraser, T.J.; Olin, E.J.; Bhuyan, B.K. Action of Camptothecin on Mammalian Cells in Culture. Cancer Res. 1972, 32, 2643. [Google Scholar] [PubMed]
- Drewinko, B.; Freireich, E.J.; Gottlieb, J.A. Lethal activity of camptothecin sodium on human lymphoma cells. Cancer Res. 1974, 34, 747–750. [Google Scholar] [PubMed]
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar] [CrossRef]
- Fukuoka, M.; Niitani, H.; Suzuki, A.; Motomiya, M.; Hasegawa, K.; Nishiwaki, Y.; Kuriyama, T.; Ariyoshi, Y.; Negoro, S.; Masuda, N. A phase II study of CPT-11, a new derivative of camptothecin, for previously untreated non-small-cell lung cancer. JCO 1992, 10, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Sirikantaramas, S.; Asano, T.; Sudo, H.; Yamazaki, M.; Saito, K. Camptothecin: Therapeutic Potential and Biotechnology. Curr. Pharm. Biotechnol. 2007, 8, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Baker, T.L.; Burnett, J.P.; Theisen, R.L.; Lazarus, D.; Peters, C.G.; Clouthier, S.G.; Eliasof, S.; Wicha, M.S. CRLX101, an investigational camptothecin-containing nanoparticle-drug conjugate, targets cancer stem cells and impedes resistance to antiangiogenic therapy in mouse models of breast cancer. Breast Cancer Res. Treat. 2015, 150, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chen, P.C.; Wang, C.K.; Wang, C.W.; Chang, Y.J.; Tai, C.J. Antitumor effects and biological mechanism of action of the aqueous extract of the Camptotheca acuminata fruit in human endometrial Carcinoma cells. Evid. Based Complement. Altern. Med. 2014, 2014, 564810. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; Wuyts, J.; RouzÃ, P.; Van, D.P. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 2004, 20, 2911–2917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhu, M.; Wang, X.; Tan, H.; Tsao, S.; Feng, Y. Berberine-induced tumor suppressor p53 up-regulation gets involved in the regulatory network of MIR-23a in hepatocellular carcinoma. Biochim. Biophys. Acta 2014, 1839, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Zhang, X.; Lin, K.; Ye, H.; Feng, S.; Zhang, H.; Chen, Y. Camptothecin Induces Apoptosis in Cancer Cells via MicroRNA-125b-Mediated Mitochondrial Pathways. Mol. Pharmacol. 2012, 81, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Colaiacovo, M.; Subacchi, A.; Bagnaresi, P.; Lamontanara, A.; Cattivelli, L.; Faccioli, P. A computational-based update on microRNAs and their targets in barley (Hordeum vulgare L.). BMC Genom. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Monavar Feshani, A.; Mohammadi, S.; Frazier, T.P.; Abbasi, A.; Abedini, R.; Karimi Farsad, L.; Ehya, F.; Hosseini Salekdeh, G.; Mardi, M. Identification and validation of Asteraceae miRNAs by the expressed sequence tag analysis. Gene 2012, 493, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Wang, W.; Liu, P. Identification and functional analysis of novel and conserved microRNAs in tomato. Mol. Biol. Rep. 2014, 41, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Nithin, C.; Patwa, N.; Thomas, A.; Bahadur, R.P.; Basak, J. Computational prediction of miRNAs and their targets in Phaseolus vulgaris using simple sequence repeat signatures. BMC Plant Biol. 2015, 15, 140. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Saini, S.; Fukuhara, S.; Majid, S.; Shahryari, V.; Yamamura, S.; Chiyomaru, T.; Deng, G.; Tanaka, Y.; Dahiya, R. MicroRNA-4723 Inhibits Prostate Cancer Growth through Inactivation of the Abelson Family of Nonreceptor Protein Tyrosine Kinases. PLoS ONE 2013, 8, e78023. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Guan, J. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Populo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Labagnara, M.; Friedman, M.; Kwasnicki, A.; Murali, R. Glioblastoma: Molecular Pathways, Stem Cells and Therapeutic Targets. Cancers 2015, 7, 538–555. [Google Scholar] [CrossRef] [PubMed]
- Notarnicola, M.; Miccolis, A.; Tutino, V.; Lorusso, D.; Caruso, M.G. Low Levels of Lipogenic Enzymes in Peritumoral Adipose Tissue of Colorectal Cancer Patients. Lipids 2012, 47, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Baier, S.R.; Zempleni, J. The Intestinal Transport of Bovine Milk Exosomes Is Mediated by Endocytosis in Human Colon Carcinoma Caco-2 Cells and Rat Small Intestinal IEC-6 Cells. J. Nutr. 2015, 145, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.K.; Mooney, P.; Jia, L.; Eves, R.; Raptis, L.; Mak, A.S. Doubles Game: Src-Stat3 versus p53-PTEN in Cellular Migration and Invasion. Mol. Cell. Biol. 2010, 30, 4980–4995. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Fan, X.; Xu, X.; Huang, J.; Xu, S.; Geng, Q.; Li, R.; Chen, D.; Yan, G. Epigenetic Silencing of ITGA2 by MiR-373 Promotes Cell Migration in Breast Cancer. PLoS ONE 2015, 10, e0135128. [Google Scholar] [CrossRef] [PubMed]
- Bianchi-Smiraglia, A.; Paesante, S.; Bakin, A.V. Integrin β-5 contributes to the tumorigenic potential of breast cancer cells through the Src-FAK and MEK-ERK signaling pathways. Oncogene 2013, 32, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Bao, R.; Jiang, L.; Wang, Z.; Wang, X.; Zhang, F.; Liang, H.; Li, H.; Ye, Y.; Xiang, S.; et al. MicroRNA-29c-5p suppresses gallbladder carcinoma progression by directly targeting CPEB4 and inhibiting the MAPK pathway. Cell Death Differ. 2017, 24, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Lu, Z.; Liu, G.; Fang, Y.; Liu, J.; Huang, Z.; Xu, X. Numb downregulation suppresses cell growth and is associated with a poor prognosis of human hepatocellular carcinoma. Int. J. Mol. Med. 2015, 36, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Tammam, J.; Ware, C.; Efferson, C.; O’Neil, J.; Rao, S.; Qu, X.; Gorenstein, J.; Angagaw, M.; Kim, H.; Kenific, C.; et al. Down-regulation of the Notch pathway mediated by a γ-secretase inhibitor induces anti-tumour effects in mouse models of T-cell leukaemia. Br. J. Pharmacol. 2009, 158, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Shan, G.; Zhang, P.; Li, P.; Du, F.; Yang, Y. Numb Gene Enhances Radiation Sensitivity of Nonsmall Cell Lung Cancer Stem Cells. Cancer Biother. Radiopharm. 2016, 31, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Carey, K.A.; Farnfield, M.M.; Tarquinio, S.D.; Cameron-Smith, D. Impaired Expression of Notch Signaling Genes in Aged Human Skeletal Muscle. J. Gerontol. Ser. A 2007, 62, 9–17. [Google Scholar] [CrossRef]
- Giebel, B.; Wodarz, A. Notch Signaling: Numb Makes the Difference. Curr. Biol. 2012, 22, R133–R135. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.P.; Gazdar, A.F.; Virmani, A.K.; Sepetavec, T.; Hande, K.R.; Minna, J.D.; Roberts, J.R.; Carbone, D.P. Chromosome 19 Translocation, Overexpression of Notch3, and Human Lung Cancer. J. Natl. Cancer Inst. 2000, 92, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, A.R.; Pritchard, J.I.; Hansen, S.; Benson, M.; Stoeck, J.; Hatton, B.A.; Russell, T.L.; Ellenbogen, R.G.; Bernstein, I.D.; Beachy, P.A.; et al. The SmoA1 Mouse Model Reveals That Notch Signaling Is Critical for the Growth and Survival of Sonic Hedgehog-Induced Medulloblastomas. Cancer Res. 2004, 64, 7794. [Google Scholar] [CrossRef] [PubMed]
- Curry, C.L.; Reed, L.L.; Golde, T.E.; Miele, L.; Nickoloff, B.J.; Foreman, K.E. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi’s sarcoma tumor cells. Oncogene 2005, 24, 6333–6344. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Li, M.; Nakayama, K.; Mao, T.; Davidson, B.; Zhang, Z.; Kurman, R.J.; Eberhart, C.G.; Shih, I.; Wang, T. Notch3 Gene Amplification in Ovarian Cancer. Cancer Res. 2006, 66, 6312–6318. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, B.; Warneke, J.A.; Nickoloff, B.J.; Giaccia, A.J.; Powell, M.B. Notch1 is an effector of Akt and hypoxia in melanoma development. J. Clin. Investig. 2008, 118, 3660–3670. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.Y.; Liu, S.; Yam, J.; Ngan, H.; Chan, D.W.; Liu, X. Epigenetic silencing of microRNA-199b-5p is associated with acquired chemoresistance via activation of JAG1-Notch1 signaling in ovarian cancer. Oncotarget 2014, 5, 944. [Google Scholar]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Frey, W.D.; He, H.; Kim, H.; Ekram, M.B.; Bakshi, A.; Faisal, M.; Perera, B.P.U.; Ye, A.; Teruyama, R. Peg3 Mutational Effects on Reproduction and Placenta-Specific Gene Families. PLoS ONE 2014, 8, e83359. [Google Scholar] [CrossRef]
- Feng, W.; Marquez, R.T.; Lu, Z.; Liu, J.; Lu, K.H.; Issa, J.J.; Fishman, D.M.; Yu, Y.; Bast, R.C. Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down-regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer 2008, 112, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.C.; Gostout, B.S.; Shridhar, V.; Wu, X.; Smith, D.I.; Podratz, K.C.; Jiang, S. Biallelic methylation and silencing of paternally expressed gene 3 (PEG3) in gynecologic cancer cell lines. Gynecol. Oncol. 2005, 99, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.; Preiss, F.; Brauns-Schubert, P.; Schlicher, L.; Charvet, C. GSK-3 at the crossroads of cell death and survival. J. Cell. Sci. 2014, 127, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Ishioka, C. Inhibition of glycogen synthase kinase-3 β induces apoptosis and mitotic catastrophe by disrupting centrosome regulation in cancer cells. Sci. Rep. 2015, 5, 13249. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Cao, W.; Li, L.; Li, S.; Liu, T.; Wan, H.; Liu, M.; Li, X.; Tang, H. MicroRNA-519d targets MKi67 and suppresses cell growth in the hepatocellular carcinoma cell line QGY-7703. Cancer Lett. 2011, 307, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Zubakov, D.; Stupar, Z.; Kovacs, G. Differential expression of a new isoform of DLG2 in renal oncocytoma. BMC Cancer 2006, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Goulart, M.A.; Wurcel, A.G. Shigellosis in men who have sex with men: An overlooked opportunity to counsel with pre-exposure prophylaxis for HIV. Int. J. STD AIDS 2016, 27, 1236–1238. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Chiou, G.; Huang, P.; Lo, W.; Wang, C.; Lu, K.; Yu, C.; Alterovitz, G.; Huang, W.; Lo, J.; et al. Network Biology of Tumor Stem-like Cells Identified a Regulatory Role of CBX5 in Lung Cancer. Sci. Rep. 2012, 2, 584. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, A.; Saito-Ohara, F.; Inoue, J.; Aoki, D.; Susumu, N.; Yokoyama, T.; Nozawa, S.; Inazawa, J.; Imoto, I. Association of 17q21-q24 Gain in Ovarian Clear Cell Adenocarcinomas with Poor Prognosis and Identification of PPM1D and APPBP2 as Likely Amplification Targets. Clin. Cancer Res. 2003, 9, 1995–2004. [Google Scholar] [PubMed]
- Gangora-Castillo, E.; Childs, K.L.; Fedewa, G.; Hamilton, J.P.; Liscombe, D.K.; Magallanes-Lundback, M.; Mandadi, K.K.; Nims, E.; Runguphan, W.; Vaillancourt, B.; et al. Development of Transcriptomic Resources for Interrogating the Biosynthesis of Monoterpene Indole Alkaloids in Medicinal Plant Species. PLoS ONE 2012, 7, e52506. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Honer, Z.S.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Wang, Q.; Cobb, G.P.; Anderson, T.A. Computational identification of microRNAs and their targets. Comput. Biol. Chem. 2006, 30, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.L.S.; Mishra, S.K. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics 2007, 23, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Pinero, J.; Bravo, Ã.; Queralt-Rosinach, N.; Gutiarrez-Sacristan, A.; Deu-Pons, J.; Centeno, E.; Garcia-Garcia, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database 2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Inter J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Chin, C.; Chen, S.; Wu, H.; Ho, C.; Ko, M.; Lin, C. Cytohubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Scardoni, G.; Tosadori, G.; Faizan, M.; Spoto, F.; Fabbri, F.; Laudanna, C. Biological network analysis with CentiScaPe: Centralities and experimental dataset integration. F1000Res 2015, 3, 139. [Google Scholar] [CrossRef] [PubMed]
- Valente, T.W.; Foreman, R.K. Integration and radiality: Measuring the extent of an individual’s connectedness and reachability in a network. Soc. Netw. 1998, 20, 89–105. [Google Scholar] [CrossRef]
- Freeman, L.C. A Set of Measures of Centrality Based on Betweenness. Sociometry 1977, 40, 35–41. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabasi, A.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Shimbel, A. Structural parameters of communication networks. Bull. Math. Biophys. 1953, 15, 501–507. [Google Scholar] [CrossRef]
- Bonacich, P. Some unique properties of eigenvector centrality. Soc. Netw. 2007, 29, 555–564. [Google Scholar] [CrossRef]
- Hwang, W.; Kim, T.; Ramanathan, M.; Zhang, A. Bridging centrality: Graph mining from element level to group level. In Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Las Vegas, NV, USA, 24–27 August 2008; pp. 336–344. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No | EST ID | miRNA Name | Homolog miRNA | Mature Sequence | MSL | PSL | MFE (ΔG) | MFE in Kcal/mol | (G + C) % | MFEI |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | medp_camac_20101112|9453 | cac-miR-5653 | ath-miR5653 | GTTGAGTTTGAGTTGAGTTG | 20 | 205 | −100 | −102.7 | 35.12 | −1.389 |
| 2 | medp_camac_20101112|7558 | cac-miR-5780d | gma-miR5780d | TGTTTTGAGTTTCTG-TAAAT | 21 | 210 | −80.8 | −83.96 | 32.86 | −1.171 |
| 3 | medp_camac_20101112|10526 | cac-miR-3440-3p | aly-miR3440-3p | CGGTTCTCTCTGACCATATCCA | 22 | 141 | −74.1 | −75.88 | 45.39 | −1.158 |
| 4 | medp_camac_20101112|33501 | cac-miR-8157-3p | ssa-miR-8157-3p | CTCTGTGCATTCTGCTGTGCT | 21 | 220 | −67.7 | −72.58 | 52.27 | −0.589 |
| 5 | medp_camac_20101112|6119 | cac-miR-6903-5p | mmu-miR-6903-5p | TGGTAGAGT-CTGCTTTTCCCA | 22 | 220 | −61.3 | −64.88 | 40.45 | −0.689 |
| 6 | medp_camac_20101112|44664 | cac-miR-7398f-5p | mdo-miR-7398f-5p | ATT-CCACATCTCTTCTACACT | 22 | 220 | −60.4 | −64.34 | 44.09 | −0.623 |
| 7 | medp_camac_20101112|9293 | cac-miR-156e-3p | bdi-miR156e-3p | GACAGAGAGAGAAGTGGAGC | 20 | 183 | −58.5 | −61.48 | 42.08 | −0.760 |
| 8 | medp_camac_20101112|29 | cac-miR-5532 | osa-miR5532 | ATGGAATATATGACAAAGGTG | 21 | 220 | −57.4 | −61.99 | 39.09 | −0.667 |
| 9 | medp_camac_20101112|6065 | cac-miR-4723-3p | hsa-miR-4723-3p | TTTGGGGAGGAG--AGAGAGGG | 22 | 217 | −56.3 | −61.33 | 45.16 | −0.574 |
| 10 | medp_camac_20101112|447 | cac-miR-5049-3p | bdi-miR5049-3p | TAATATGGAATCGGAGGAAGT | 21 | 220 | −53.3 | −57.51 | 39.55 | −0.613 |
| 11 | medp_camac_20101112|3541 | cac-miR-5291c | mtr-miR5291c | TTTGATGGATGGCATTG-ATGGA | 23 | 221 | −53.2 | −57.84 | 41.63 | −0.578 |
| 12 | medp_camac_20101112|4893 | cac-miR-548d-3p | mml-miR-548d-3p | GCAGAAAGAAATTGTGGTGTTTT | 23 | 222 | −53.1 | −58.01 | 37.39 | −0.640 |
| 13 | medp_camac_20101112|18253 | cac-miR-29c-5p | ssa-miR-29c-5p | CTGTTTTCTTTTGGCTGTTT | 20 | 219 | −52.2 | −56.62 | 42.47 | −0.561 |
| 14 | medp_camac_20101112|10789 | cac-miR-7009-3p | mmu-miR-7009-3p | GCAGGGAGAGGGGATAAAGA | 20 | 219 | −51.4 | −55.09 | 36.99 | −0.635 |
| Sr. No | miRNA Name | miRNA_Acc. | Target Gene |
|---|---|---|---|
| 1 | cac-miR-3440-3p | medp_camac_20101112|10526 | CCNJL |
| 2 | cac-miR-7009-3p | medp_camac_20101112|10789 | GPATCH8, POM121C, TMEM14E, ZDHHC3, IYD, IYD, FCRLA, CLCN6, PIP4K2B, ARPP19, CBX5, MGAT4A, HS2ST1, CEP350, ZNF609, DSTYK, BNC2, KANSL2, BCL11A, HAUS3, HMBOX1, SOX7, DIDO1, MUM1L1, APCDD1, POM121, ANKRD52, COX18, TDRD1, IYD, PPAPDC2 |
| 3 | cac-miR-29c-5p | medp_camac_20101112|18253 | ZNF37A, SOGA3, ARGFX, CELF2, CELF2, SPATA6L, PIK3R3, METTL20, RPS6KC1, TTC26, POLR3B, FBXL17, MEF2C, CD44, CCR1, ITGA2, KCNJ10, AKAP5, PEG3, CDC42EP3, SCN8A, RNF144A, ABHD5, TFB1M, RHOU, TMEM106C, GINS4, MRPL11, MRPL11, GPATCH4, ZNF169 |
| 4 | cac-miR-5532 | medp_camac_20101112|29 | EXOSC3, KIF13A, EXOSC3, C11orf87 |
| 5 | cac-miR-8157-3p | medp_camac_20101112|33501 | C3orf18, RBM33 |
| 6 | cac-miR-5291c | medp_camac_20101112|3541 | GLIS3, CNOT11, C11orf87 |
| 7 | cac-miR-7398f-5p | medp_camac_20101112|44664 | NCOA4, NCOA4, STAMBP |
| 8 | cac-miR-5049-3p | medp_camac_20101112|447 | CLHC1, CACUL1, MMAA |
| 9 | cac-miR-548d-3p | medp_camac_20101112|4893 | ATP2B4, ATP2B4, GSK3B, MTUS2 |
| 10 | cac-miR-4723-3p | medp_camac_20101112|6065 | IGF1, GRINA, GPNMB, CREBZF, FAM3C, C1orf226, PHF21A, IGF1, SERPINA1, DLG2, SYTL4, DCTN5, TGOLN2, TGOLN2, UNC5B, B4GALNT1 CPM, MKI67, PRELP, RGS7, SLC1A2, CBFA2T3, CRTAP, TGOLN2, MYO9A, EID1, RPS6KA6, ABLIM3, ZBTB7A, PACS1, PARVA, PCDHA1, PLEKHH1, PRX, RAP2C, EBF1, RNF39, C6orf25, MARVELD1, ANKRD27, DCTN5, EAF1, ITPRIP, BTRC, STK35, C6orf25, C6orf25, DBF4B, RNF39, SLC41A1, GALNT10, FMNL3, BACH1, PATE2 |
| 11 | cac-miR-6903-5p | medp_camac_20101112|6119 | SLC30A4, TRIM62 |
| 12 | cac-miR-5780d | medp_camac_20101112|7558 | NPY1R, NUMB, GLIS3, DCTN4, C11orf93, ZNF540, TIPARP, STARD13, STARD13, PROX1, MIP, UBXN4, SERTAD2, CLIP3, HPCAL4, CCDC93, FAM49A |
| 13 | cac-miR-156e-3p | medp_camac_20101112|9293 | DDX17, KAZN, ASB4, PGLYRP4, JPH3, EPG5, TPCN2 |
| 14 | cac-miR-5653 | medp_camac_20101112|9453 | LOC100996485, GYPA, APPBP2, EFR3B, RAP2A, TRUB1, NAP1L5 |
| Rank | Protein | Score |
|---|---|---|
| 1 | ALB | 2056 |
| 1 | KRT9 | 2056 |
| 1 | OR8D2 | 2056 |
| 4 | ITGA2 | 1047 |
| 4 | RLF | 1047 |
| 6 | AP1M1 | 1032 |
| 7 | PEG3 | 1030 |
| 8 | ITGB5 | 1029 |
| 8 | SCN5A | 1029 |
| 8 | FN1 | 1029 |
| Rank | Protein | Score |
|---|---|---|
| 1 | NUMB | 428 |
| 2 | ITGB5 | 394 |
| 3 | GSK3B | 122 |
| 4 | APP | 93 |
| 5 | PRKCB | 76 |
| 6 | MKI67 | 75 |
| 7 | SCN5A | 74 |
| 8 | DLG2 | 73 |
| 9 | CBX5 | 31 |
| 9 | APPBP2 | 31 |
| Rank | Radiality | Betweenness | Degree | Stress | Eigen Vector | Bridging |
|---|---|---|---|---|---|---|
| 1 | NUMB | NUMB | RLF | NUMB | RLF | PRKCB |
| 2 | ITGB5 | ITGB5 | ITGA2 | GSK3B | ITGA2 | APP |
| 3 | PRKCB | GSK3B | AP1M1 | ITGB5 | AP1M1 | DPYSL2 |
| 4 | SCN5A | PRKCB | PEG3 | PRKCB | PEG3 | NOTCH1 |
| 5 | APP | MKI67 | ATM | MKI67 | ITGB5 | CREB3 |
| 6 | ITGB3 | DLG2 | FN1 | APP | FN1 | MDM2 |
| 7 | RLF | APP | ITGB5 | DLG2 | SCN5A | PRKCA |
| 8 | AP1M1 | SCN5A | SCN5A | BTRC | ATM | PRKAR2A |
| 9 | ITGA2 | APPBP2 | ADRA1B | SCN5A | ADRA1B | DLG4 |
| 10 | FN1 | BTRC | AGA | CBX5 | AGA | ATP2B4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, D.; Kumar, S.; Ayachit, G.; Bhairappanavar, S.B.; Ansari, A.; Sharma, P.; Soni, S.; Das, J. Cross-Kingdom Regulation of Putative miRNAs Derived from Happy Tree in Cancer Pathway: A Systems Biology Approach. Int. J. Mol. Sci. 2017, 18, 1191. https://doi.org/10.3390/ijms18061191
Kumar D, Kumar S, Ayachit G, Bhairappanavar SB, Ansari A, Sharma P, Soni S, Das J. Cross-Kingdom Regulation of Putative miRNAs Derived from Happy Tree in Cancer Pathway: A Systems Biology Approach. International Journal of Molecular Sciences. 2017; 18(6):1191. https://doi.org/10.3390/ijms18061191
Chicago/Turabian StyleKumar, Dinesh, Swapnil Kumar, Garima Ayachit, Shivarudrappa B. Bhairappanavar, Afzal Ansari, Priyanka Sharma, Subhash Soni, and Jayashankar Das. 2017. "Cross-Kingdom Regulation of Putative miRNAs Derived from Happy Tree in Cancer Pathway: A Systems Biology Approach" International Journal of Molecular Sciences 18, no. 6: 1191. https://doi.org/10.3390/ijms18061191
APA StyleKumar, D., Kumar, S., Ayachit, G., Bhairappanavar, S. B., Ansari, A., Sharma, P., Soni, S., & Das, J. (2017). Cross-Kingdom Regulation of Putative miRNAs Derived from Happy Tree in Cancer Pathway: A Systems Biology Approach. International Journal of Molecular Sciences, 18(6), 1191. https://doi.org/10.3390/ijms18061191