Proteostasis of Huntingtin in Health and Disease

Abstract
:
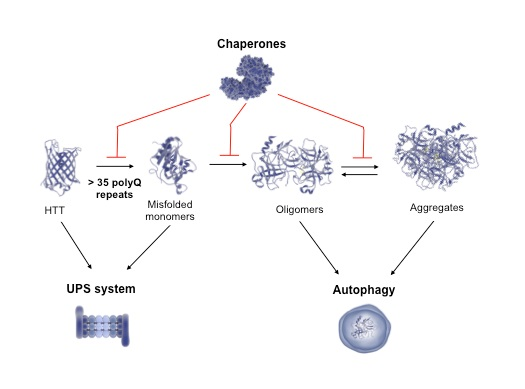{kind=link}
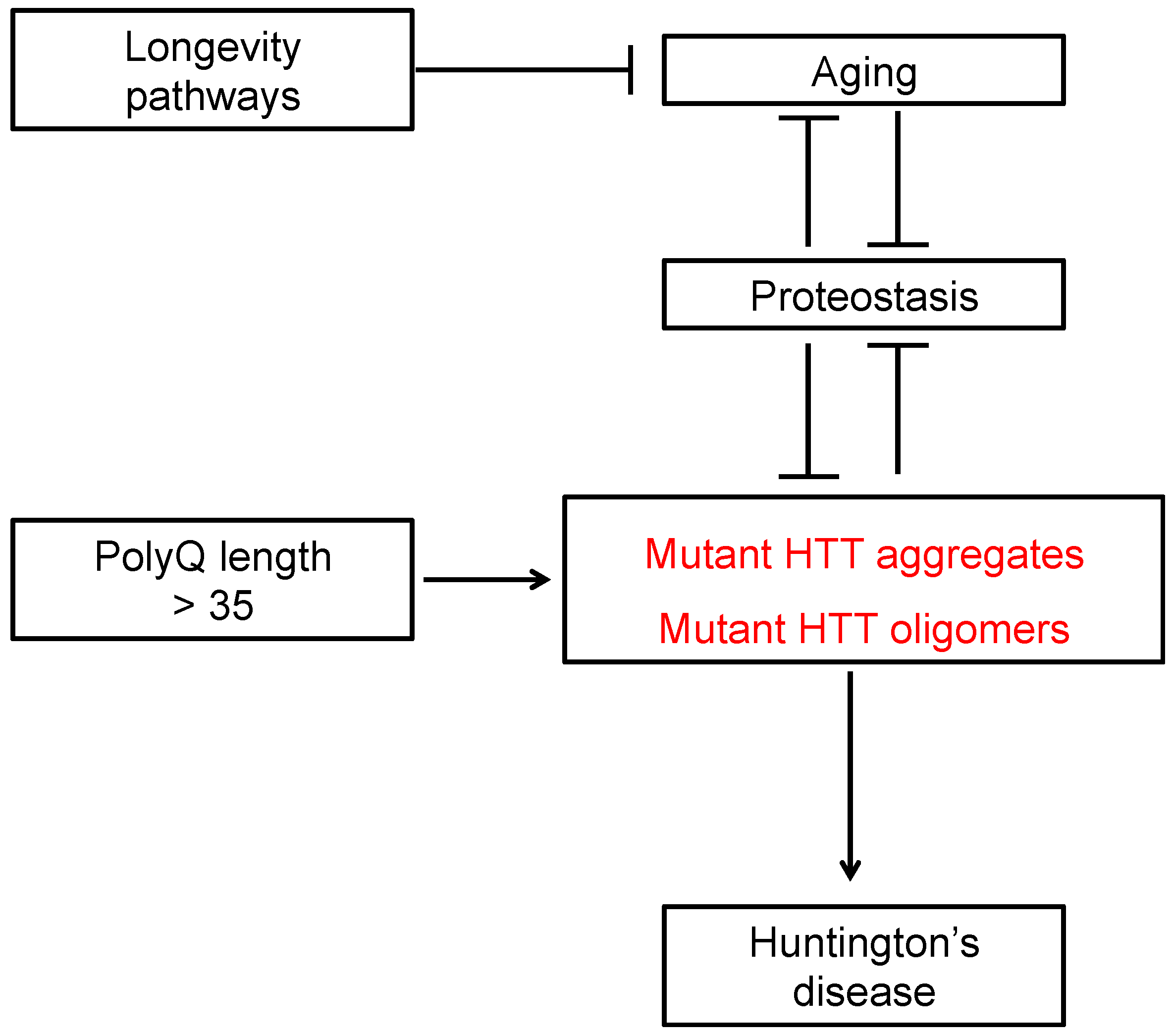{kind=link}
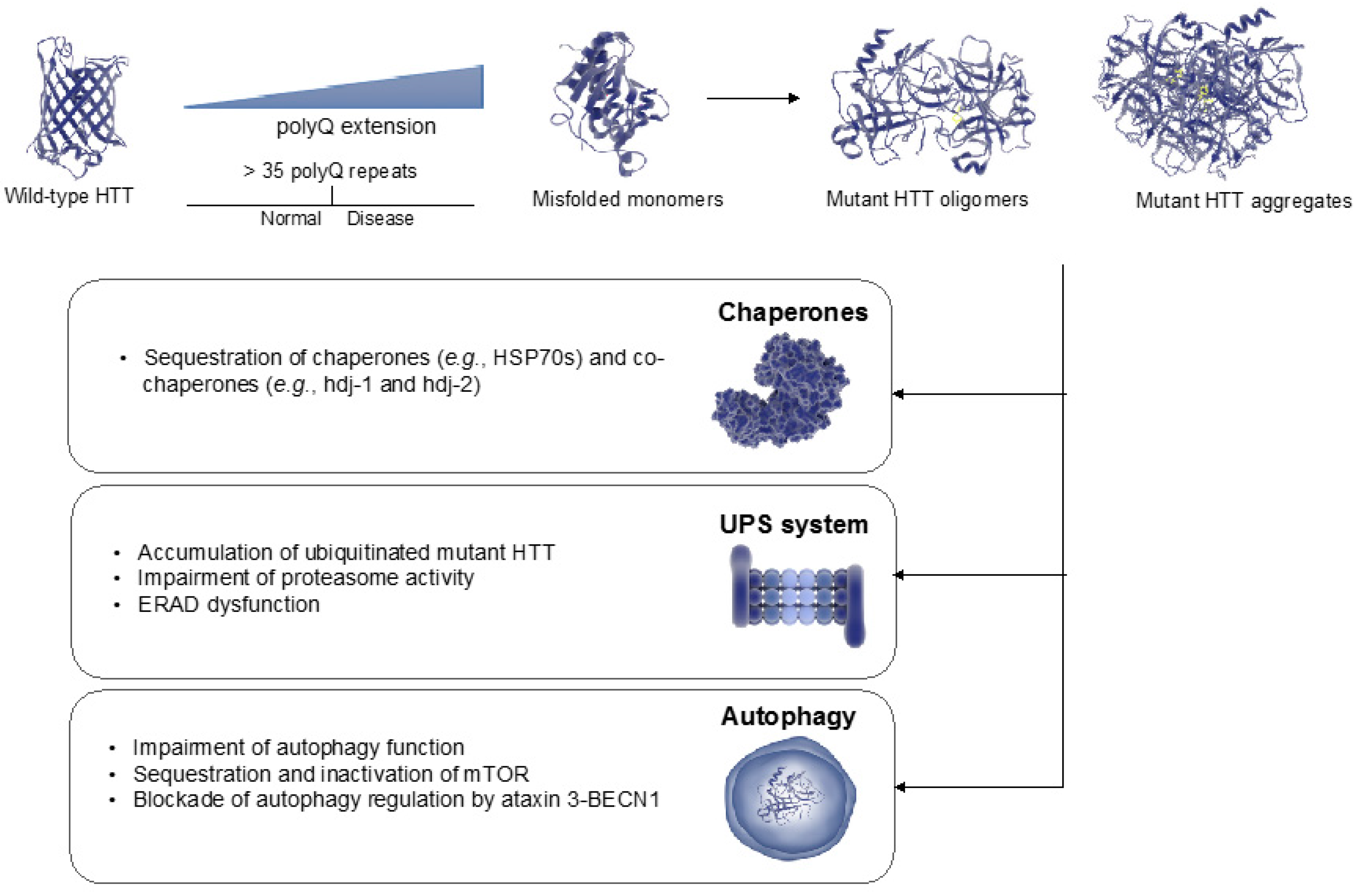{kind=link}
{kind=link}
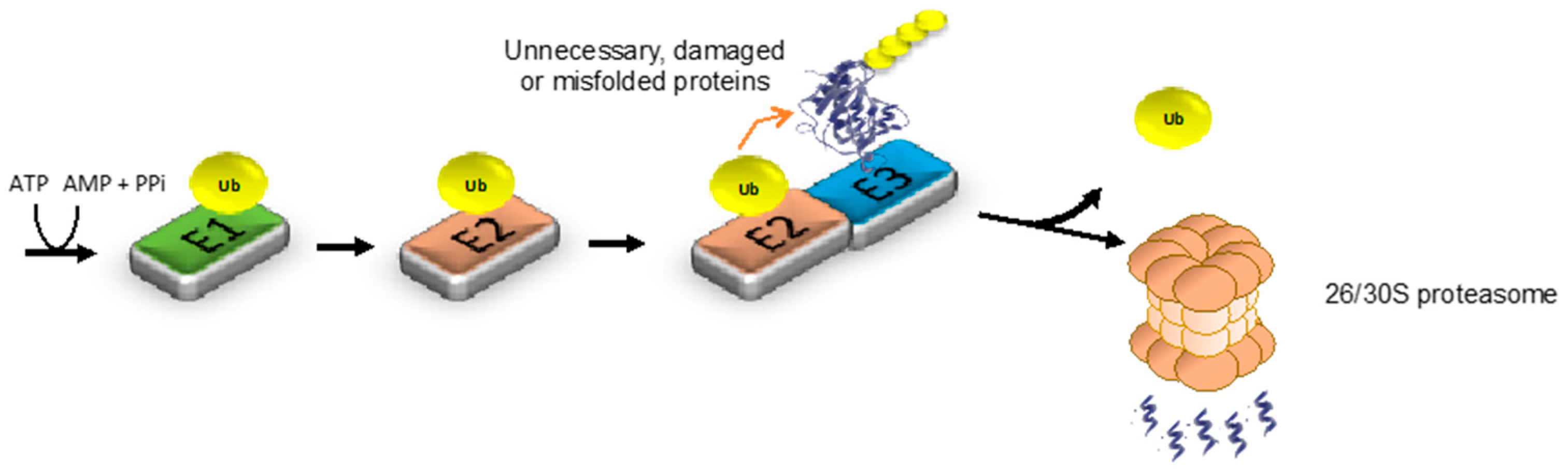{kind=link}
{kind=link}
1. Introduction
2. Regulation of Mutant HTT Aggregation by the Chaperome Network
3. The Mechanistic Links between the Ubiquitin Proteasome-System (UPS) and HD
4. The Dual Role of PolyQ-Expanded HTT in the Autophagy-Lysosome System
5. The Melding Fields of Proteostasis of Aging, Pluripotency and HD
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| CMA | Chaperon-Mediated Autophagy |
| HD | Huntington’s disease |
| HTT | Huntingtin |
| HSR | Heat-shock response |
| ER | Endoplasmic reticulum |
| ERAD | Endoplasmic reticulum-associated degradation |
| iPSCs | Induced pluripotent stem cells |
| TRiC/CCT | T-complex protein-1 ring complex/Chaperonin containing TCP1 complex |
| UPS | Ubiquitin-proteasome system |
References
- Finkbeiner, S. Huntington’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 2012, 27, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F. Huntington’s disease: Update and review of neuropsychiatric aspects. Int. J. Psychiatry Med. 1994, 24, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Graveland, G.A.; Williams, R.S.; DiFiglia, M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 1985, 227, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The biology of huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Bates, G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 2003, 361, 1642–1644. [Google Scholar] [CrossRef]
- Wang, C.E.; Tydlacka, S.; Orr, A.L.; Yang, S.H.; Graham, R.K.; Hayden, M.R.; Li, S.; Chan, A.W.; Li, X.J. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington’s disease. Hum. Mol. Genet. 2008, 17, 2738–2751. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.E.; Huang, S.; Xu, X.; Li, X.J.; Li, H.; Li, S. Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum. Mol. Genet. 2010, 19, 2445–2455. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vincente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK phosphorylates huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Carmichael, J.; Swartz, J.; Furlong, R.A.; Narain, Y.; Rankin, J.; Rubinsztein, D.C. Effects of heat shock, heat shock protein 40 (hdj-2), and proteasome inhibition on protein aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2898–2903. [Google Scholar] [CrossRef] [PubMed]
- Tipping, K.W.; van Oosten-Hawle, P.; Hewitt, E.W.; Radford, S.E. Amyloid fibres: Inert end-stage aggregates or key players in disease? Trends Biochem. Sci. 2015, 40, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Ardley, H.C.; Hung, C.C.; Robinson, P.A. The aggravating role of the ubiquitin-proteasome system in neurodegeneration. FEBS Lett. 2005, 579, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Hernandez, M.; Valera, A.G.; Moran, M.A.; Gomez-Ramos, P.; Alvarez-Castelao, B.; Castano, J.G.; Hernandez, F.; Lucas, J.J. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J. Neurochem. 2006, 98, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, C.I.; Staniszewski, K.E.; Mensah, K.N.; Matouschek, A.; Morimoto, R.I. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 2004, 23, 4307–4318. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kukushkin, Y.; Gupta, R.; Chen, T.; Konagai, A.; Hipp, M.S.; Hayer-Hartl, M.; Hartl, F.U. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 2013, 154, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Suhr, S.T.; Senut, M.C.; Whitelegge, J.P.; Faull, K.F.; Cuizon, D.B.; Gage, F.H. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J. Cell Biol. 2001, 153, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef]
- Lajoie, P.; Snapp, E.L. Formation and toxicity of soluble polyglutamine oligomers in living cells. PLoS ONE 2010, 5, e15245. [Google Scholar] [CrossRef] [PubMed]
- Leitman, J.; Ulrich Hartl, F.; Lederkremer, G.Z. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 2013, 4, 2753. [Google Scholar] [CrossRef] [PubMed]
- Schaffar, G.; Breuer, P.; Boteva, R.; Behrends, C.; Tzvetkov, N.; Strippel, N.; Sakahira, H.; Siegers, K.; Hayer-Hartl, M.; Hartl, F.U. Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol. Cell 2004, 15, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kikuchi, S.; Katada, S.; Nagai, Y.; Nishizawa, M.; Onodera, O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 2008, 17, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Mahlke, C.; Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003, 421, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Snell, R.G.; MacMillan, J.C.; Cheadle, J.P.; Fenton, I.; Lazarou, L.P.; Davies, P.; MacDonald, M.E.; Gusella, J.F.; Harper, P.S.; Shaw, D.J. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 1993, 4, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Brignull, H.R.; Moore, F.E.; Tang, S.J.; Morimoto, R.I. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J. Neurosci. 2006, 26, 7597–7606. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Cellular homeostasis and aging. Ann. Rev. Biochem. 2016, 85, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, R.A.; Ellis, R.J. Chaperones: Needed for both the good times and the bad times. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20130091. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Ann. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Gidalevitz, T.; Prahlad, V.; Morimoto, R.I. The stress of protein misfolding: From single cells to multicellular organisms. Cold Spring Harb. Perspect. Biol. 2011, 3, a009704. [Google Scholar] [CrossRef] [PubMed]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Rudiger, S.; Buchberger, A.; Bukau, B. Interaction of Hsp70 chaperones with substrates. Nat. Struct. Biol. 1997, 4, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Haslbeck, M.; Vierling, E. A first line of stress defense: Small heat shock proteins and their function in protein homeostasis. J. Mol. Biol. 2015, 427, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Albanese, V.; Yam, A.Y.; Baughman, J.; Parnot, C.; Frydman, J. Systems analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell 2006, 124, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, V.; Meister-Broekema, M.; Minoia, M.; Carra, S.; Kampinga, H.H. Barcoding heat shock proteins to human diseases: Looking beyond the heat shock response. Dis. Model. Mech. 2014, 7, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.Y.; Warrick, J.M.; Gray-Board, G.L.; Paulson, H.L.; Bonini, N.M. Mechanisms of chaperone suppression of polyglutamine disease: Selectivity, synergy and modulation of protein solubility in Drosophila. Hum. Mol. Genet. 2000, 9, 2811–2820. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Kubota, H.; Pack, C.G.; Matsumoto, G.; Hirayama, S.; Takahashi, Y.; Kimura, H.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 2006, 8, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Nollen, E.A.; Garcia, S.M.; van Haaften, G.; Kim, S.; Chavez, A.; Morimoto, R.I.; Plasterk, R.H. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2004, 101, 6403–6408. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.; Geller, R.; Spiess, C.; Frydman, J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006, 8, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Tanaka, M.; Wang, G.; Nukina, N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: Their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 2000, 9, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Sun, Y.; Opal, P.; Antalffy, B.; Mestril, R.; Orr, H.T.; Dillmann, W.H.; Zoghbi, H.Y. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum. Mol. Genet. 2001, 10, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Funez, P.; Nino-Rosales, M.L.; de Gouyon, B.; She, W.C.; Luchak, J.M.; Martinez, P.; Turiegano, E.; Benito, J.; Capovilla, M.; Skinner, P.J.; et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 2000, 408, 101–106. [Google Scholar] [PubMed]
- Kazemi-Esfarjani, P.; Benzer, S. Genetic suppression of polyglutamine toxicity in Drosophila. Science 2000, 287, 1837–1840. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Cummings, C.J.; Zoghbi, H.Y. Expanding our understanding of polyglutamine diseases through mouse models. Neuron 1999, 24, 499–502. [Google Scholar] [CrossRef]
- Warrick, J.M.; Chan, H.Y.; Gray-Board, G.L.; Chai, Y.; Paulson, H.L.; Bonini, N.M. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 1999, 23, 425–428. [Google Scholar] [PubMed]
- Krobitsch, S.; Lindquist, S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.; Outeiro, T.F.; DeVit, M.J.; Lindquist, S.L.; Muchowski, P.J. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or α-synuclein. Science 2003, 302, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Kotliarova, S.; Murata, S.; Tanaka, K.; Nukina, N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 2005, 280, 11635–11640. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Nukina, N. BAG-1 associates with the polyglutamine-expanded huntingtin aggregates. Neurosci. Lett. 2005, 378, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Sang, C.; Adachi, H.; Minamiyama, M.; Waza, M.; Tanaka, F.; Doyu, M.; Sobue, G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc. Natl. Acad. Sci. USA 2005, 102, 16801–16806. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, K.; Marubuchi, S.; Qi, M.L.; Enokido, Y.; Tamura, T.; Inagaki, R.; Murata, M.; Kanazawa, I.; Wanker, E.E.; Okazawa, H. The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J. Neurosci. 2007, 27, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Herbst, M.; Wanker, E.E. Small molecule inducers of heat-shock response reduce polyQ-mediated huntingtin aggregation. A possible therapeutic strategy. Neurodegener. Dis. 2007, 4, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. The heat shock response: Systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Fujikake, N.; Nagai, Y.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef] [PubMed]
- Neef, D.W.; Turski, M.L.; Thiele, D.J. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010, 8, e1000291. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Cunliffe, H.; Weiss, A.; Katsyuba, E.; Sathasivam, K.; Seredenina, T.; Woodman, B.; Moussaoui, S.; Frentzel, S.; Luthi-Carter, R.; et al. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Investig. 2011, 121, 3306–3319. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Nylandsted, J.; Castilho, R.F.; Leist, M.; Jaattela, M.; Brundin, P. Overexpression of heat shock protein 70 in R6/2 Huntington’s disease mice has only modest effects on disease progression. Brain Res. 2003, 970, 47–57. [Google Scholar] [CrossRef]
- Hay, D.G.; Sathasivam, K.; Tobaben, S.; Stahl, B.; Marber, M.; Mestril, R.; Mahal, A.; Smith, D.L.; Woodman, B.; Bates, G.P. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum. Mol. Genet. 2004, 13, 1389–1405. [Google Scholar] [CrossRef] [PubMed]
- Zourlidou, A.; Gidalevitz, T.; Kristiansen, M.; Landles, C.; Woodman, B.; Wells, D.J.; Latchman, D.S.; de Belleroche, J.; Tabrizi, S.J.; Morimoto, R.I.; et al. Hsp27 overexpression in the R6/2 mouse model of Huntington’s disease: Chronic neurodegeneration does not induce Hsp27 activation. Hum. Mol. Genet. 2007, 16, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Lam, W.; Hannus, M.; Sonnichsen, B.; Imarisio, S.; Fleming, A.; Tarditi, A.; Menzies, F.; Ed Dami, T.; Xu, C.; et al. siRNA screen identifies QPCT as a druggable target for Huntington’s disease. Nat. Chem. Biol. 2015, 11, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Seidel, K.; Meister, M.; Dugbartey, G.J.; Zijlstra, M.P.; Vinet, J.; Brunt, E.R.; van Leeuwen, F.W.; Rub, U.; Kampinga, H.H.; den Dunnen, W.F. Cellular protein quality control and the evolution of aggregates in spinocerebellar ataxia type 3 (SCA3). Neuropathol. Appl. Neurobiol. 2012, 38, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Hageman, J.; Rujano, M.A.; van Waarde, M.A.; Kakkar, V.; Dirks, R.P.; Govorukhina, N.; Oosterveld-Hut, H.M.; Lubsen, N.H.; Kampinga, H.H. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 2010, 37, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, V.; Mansson, C.; de Mattos, E.P.; Bergink, S.; van der Zwaag, M.; van Waarde, M.A.; Kloosterhuis, N.J.; Melki, R.; van Cruchten, R.T.; Al-Karadaghi, S.; et al. The S/T-rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol. Cell 2016, S1097–S2765. [Google Scholar] [CrossRef] [PubMed]
- Leitner, A.; Joachimiak, L.A.; Bracher, A.; Monkemeyer, L.; Walzthoeni, T.; Chen, B.; Pechmann, S.; Holmes, S.; Cong, Y.; Ma, B.; et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 2012, 20, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Meyer, A.S.; Reissmann, S.; Frydman, J. Mechanism of the eukaryotic chaperonin: Protein folding in the chamber of secrets. Trends Cell Biol. 2004, 14, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Noormohammadi, A.; Khodakarami, A.; Gutierrez-Garcia, R.; Lee, H.J.; Koyuncu, S.; Konig, T.; Schindler, C.; Saez, I.; Fatima, A.; Dieterich, C.; et al. Somatic increase of CCT8 mimics proteostasis of human pluripotent stem cells and extends C. elegans lifespan. Nat. Commun. 2016, 7, 13649. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Kazantsev, A.; Preisinger, E.; Dranovsky, A.; Goldgaber, D.; Housman, D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. USA 1999, 96, 11404–11409. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.K.; Paulson, H.L.; Pendse, S.J.; Saionz, S.J.; Bonini, N.M.; Pittman, R.N. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J. Cell Biol. 1998, 143, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Integration of clearance mechanisms: The proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2010, 2, a006734. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Morantte, I.; Liu, Z.; Douglas, P.M.; Merkwirth, C.; Rodrigues, A.P.; Manning, G.; Dillin, A. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 2012, 489, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Zabel, C.; Nguyen, H.P.; Hin, S.C.; Hartl, D.; Mao, L.; Klose, J. Proteasome and oxidative phoshorylation changes may explain why aging is a risk factor for neurodegenerative disorders. J. Proteom. 2010, 73, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Lucas, J.J.; Hen, R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000, 101, 57–66. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Mechanisms underlying ubiquitination. Ann. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Ann. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Kish-Trier, E.; Hill, C.P. Structural biology of the proteasome. Ann. Rev. Biophys. 2013, 42, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 2005, 398, 364–378. [Google Scholar] [PubMed]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.P. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: From basic mechanisms to emerging roles. Keio J. Med. 2013, 62, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stadtmueller, B.M.; Hill, C.P. Proteasome activators. Mol. Cell 2011, 41, 8–19. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S.; Mitra, S. The ubiquitin-proteasome pathway in Huntington’s disease. ScientificWorldJournal 2008, 8, 421–433. [Google Scholar] [PubMed]
- Jana, N.R.; Zemskov, E.A.; Wang, G.; Nukina, N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 2001, 10, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, K.A.; Mechan, A.O.; Jones, L.; Wanker, E.E.; Nukina, N.; Mann, D.M. Huntington’s disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp. Neurol. 1999, 156, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Stenoien, D.L.; Cummings, C.J.; Adams, H.P.; Mancini, M.G.; Patel, K.; DeMartino, G.N.; Marcelli, M.; Weigel, N.L.; Mancini, M.A. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the hdj-2 chaperone. Hum. Mol. Genet. 1999, 8, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Waelter, S.; Boeddrich, A.; Lurz, R.; Scherzinger, E.; Lueder, G.; Lehrach, H.; Wanker, E.E. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 2001, 12, 1393–1407. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Perl, D.P.; Brownell, A.L.; Olanow, C.W. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 2004, 56, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Tonoki, A.; Kuranaga, E.; Tomioka, T.; Hamazaki, J.; Murata, S.; Tanaka, K.; Miura, M. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol. Cell Biol. 2009, 29, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Sonntag, K.C.; Kim, W.; Cattaneo, E.; Isacson, O. Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS ONE 2007, 2, e238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safren, N.; El Ayadi, A.; Chang, L.; Terrillion, C.E.; Gould, T.D.; Boehning, D.F.; Monteiro, M.J. Ubiquilin-1 overexpression increases the lifespan and delays accumulation of huntingtin aggregates in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2014, 9, e87513. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Fishman, P.S.; Thakor, N.V.; Oyler, G.A. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 2003, 278, 22044–22055. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhong, X.; Ballar, P.; Luo, S.; Shen, Y.; Rubinsztein, D.C.; Monteiro, M.J.; Fang, S. Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 2007, 313, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef]
- Venkatraman, P.; Wetzel, R.; Tanaka, M.; Nukina, N.; Goldberg, A.L. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 2004, 14, 95–104. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.J.; Bence, N.F.; Jayakumar, R.; Kopito, R.R. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 2005, 17, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Patel, C.N.; Bersuker, K.; Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Brandeis, M.; Kopito, R.R. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 2012, 196, 573–587. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Ann. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Rubinsztein, D.C. Huntington’s disease: Degradation of mutant huntingtin by autophagy. FEBS J. 2008, 275, 4263–4270. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Chaperone-mediated autophagy. Autophagy 2007, 3, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Chaperone-mediated autophagy: Selectivity pays off. Trends Endocrinol. Metab. 2010, 21, 142–450. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T.; Johansen, T.; Dikic, I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009, 5, 732–733. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Qin, Z.H.; Wang, Y.; Kegel, K.B.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Yoder, J.; Aronin, N.; DiFiglia, M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 2003, 12, 3231–3244. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. P62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Perlstein, E.O.; Imarisio, S.; Pineau, S.; Cordenier, A.; Maglathlin, R.L.; Webster, J.A.; Lewis, T.A.; O’Kane, C.J.; Schreiber, S.L.; et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 2007, 3, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; La Spada, A.R. The many faces of autophagy dysfunction in Huntington’s disease: From mechanism to therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sun, X.; Hu, D.; Wang, Y.J.; Fujioka, H.; Vyas, R.; Chakrapani, S.; Joshi, A.U.; Luo, Y.; Mochly-Rosen, D.; et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 2016, 7, 12646. [Google Scholar] [CrossRef] [PubMed]
- Wellington, C.L.; Ellerby, L.M.; Gutekunst, C.A.; Rogers, D.; Warby, S.; Graham, R.K.; Loubser, O.; van Raamsdonk, J.; Singaraja, R.; Yang, Y.Z.; et al. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J. Neurosci. 2002, 22, 7862–7872. [Google Scholar] [PubMed]
- Martin, D.D.; Heit, R.J.; Yap, M.C.; Davidson, M.W.; Hayden, M.R.; Berthiaume, L.G. Identification of a post-translationally myristoylated autophagy-inducing domain released by caspase cleavage of huntingtin. Hum. Mol. Genet. 2014, 23, 3166–3179. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Salecker, I.; Dong, X.; Yao, X.; Arnheim, N.; Faber, P.W.; MacDonald, M.E.; Zipursky, S.L. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 1998, 21, 633–642. [Google Scholar] [CrossRef]
- Kikis, E.A.; Gidalevitz, T.; Morimoto, R.I. Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 2010, 694, 138–159. [Google Scholar] [PubMed]
- Gidalevitz, T.; Ben-Zvi, A.; Ho, K.H.; Brignull, H.R.; Morimoto, R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006, 311, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Alavez, S.; Lithgow, G.J. Pharmacological maintenance of protein homeostasis could postpone age-related disease. Aging Cell 2012, 11, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, O.; Arpalahti, L.; Rantanen, V.; Hautaniemi, S.; Holmberg, C.I. Insulin/IGF-1 signaling regulates proteasome activity through the deubiquitinating enzyme UBH-4. Cell Rep. 2013, 3, 1980–1995. [Google Scholar] [CrossRef] [PubMed]
- Melendez, A.; Talloczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Consortium, H.D.I. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar]
- Jeon, I.; Lee, N.; Li, J.Y.; Park, I.H.; Park, K.S.; Moon, J.; Shim, S.H.; Choi, C.; Chang, D.J.; Kwon, J.; et al. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells 2012, 30, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prat, L.; Sousa-Victor, P.; Munoz-Canoves, P. Proteostatic and metabolic control of stemness. Cell Stem Cell 2017, 20, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Gutierrez-Garcia, R.; Vilchez, D. Embryonic stem cells: A novel paradigm to study proteostasis? FEBS J. 2017, 284, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Simic, M.S.; Dillin, A. Proteostasis and aging of stem cells. Trends Cell Biol. 2014, 24, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Boyer, L.; Morantte, I.; Lutz, M.; Merkwirth, C.; Joyce, D.; Spencer, B.; Page, L.; Masliah, E.; Berggren, T.W.; et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 2012, 489, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Wang, Y.; Lalli, M.A.; Guzman, E.; Godshalk, S.E.; Zhou, H.; Kosik, K.S. Primary cilium-autophagy-Nrf2 (PAN) axis activation commits human embryonic stem cells to a neuroectoderm fate. Cell 2016, 165, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koyuncu, S.; Fatima, A.; Gutierrez-Garcia, R.; Vilchez, D. Proteostasis of Huntingtin in Health and Disease. Int. J. Mol. Sci. 2017, 18, 1568. https://doi.org/10.3390/ijms18071568
Koyuncu S, Fatima A, Gutierrez-Garcia R, Vilchez D. Proteostasis of Huntingtin in Health and Disease. International Journal of Molecular Sciences. 2017; 18(7):1568. https://doi.org/10.3390/ijms18071568
Chicago/Turabian StyleKoyuncu, Seda, Azra Fatima, Ricardo Gutierrez-Garcia, and David Vilchez. 2017. "Proteostasis of Huntingtin in Health and Disease" International Journal of Molecular Sciences 18, no. 7: 1568. https://doi.org/10.3390/ijms18071568
APA StyleKoyuncu, S., Fatima, A., Gutierrez-Garcia, R., & Vilchez, D. (2017). Proteostasis of Huntingtin in Health and Disease. International Journal of Molecular Sciences, 18(7), 1568. https://doi.org/10.3390/ijms18071568