Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase

 ,
, 
Abstract
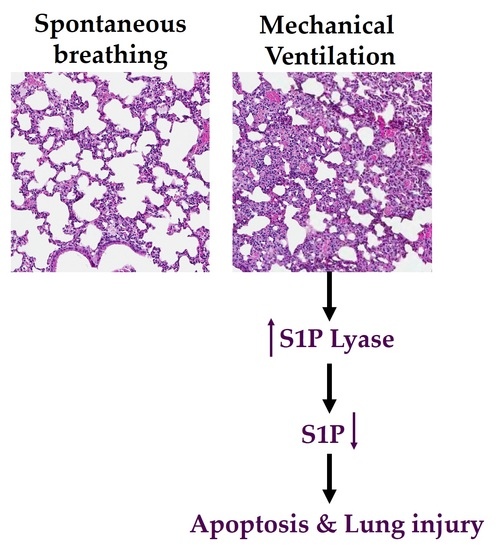
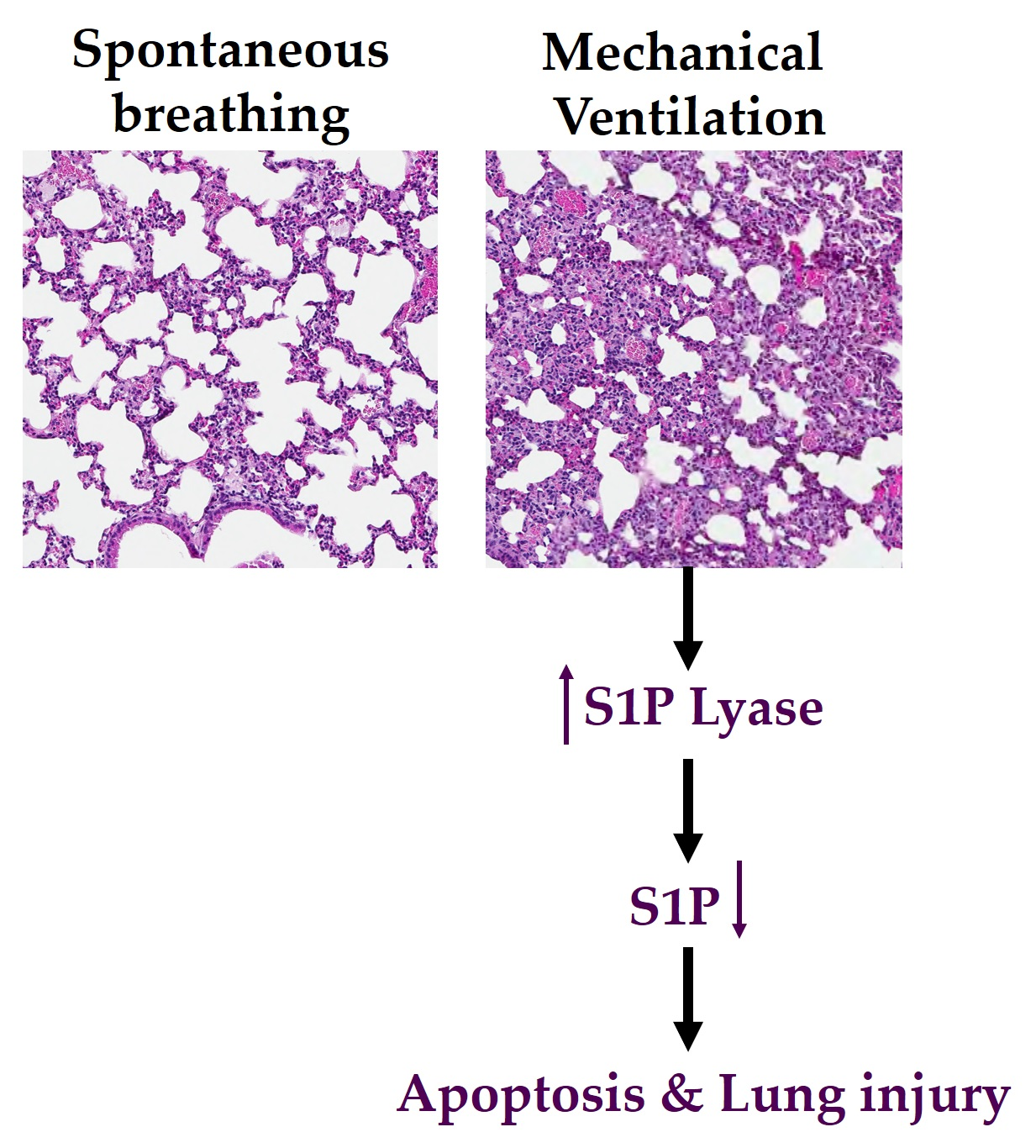
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
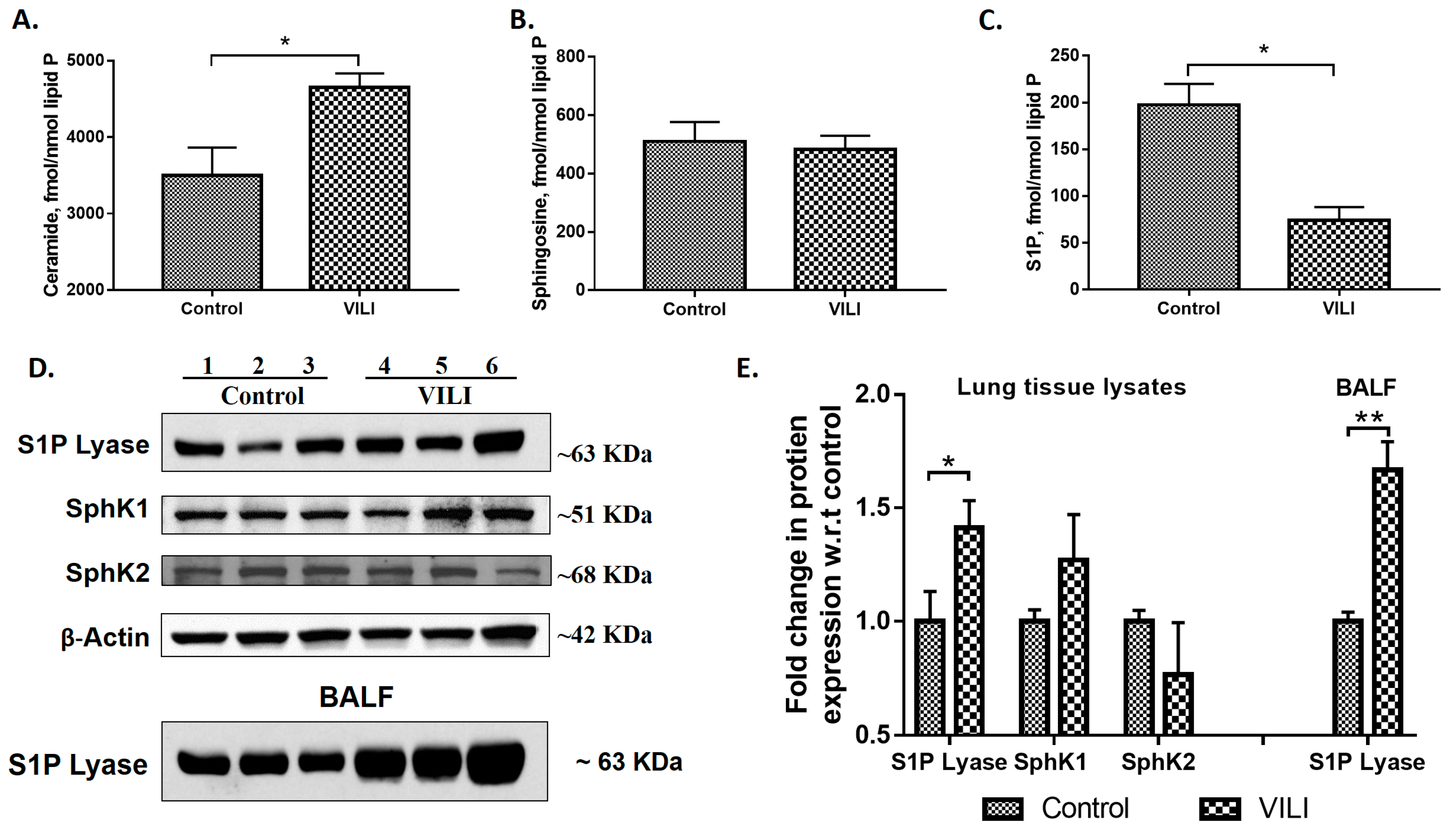
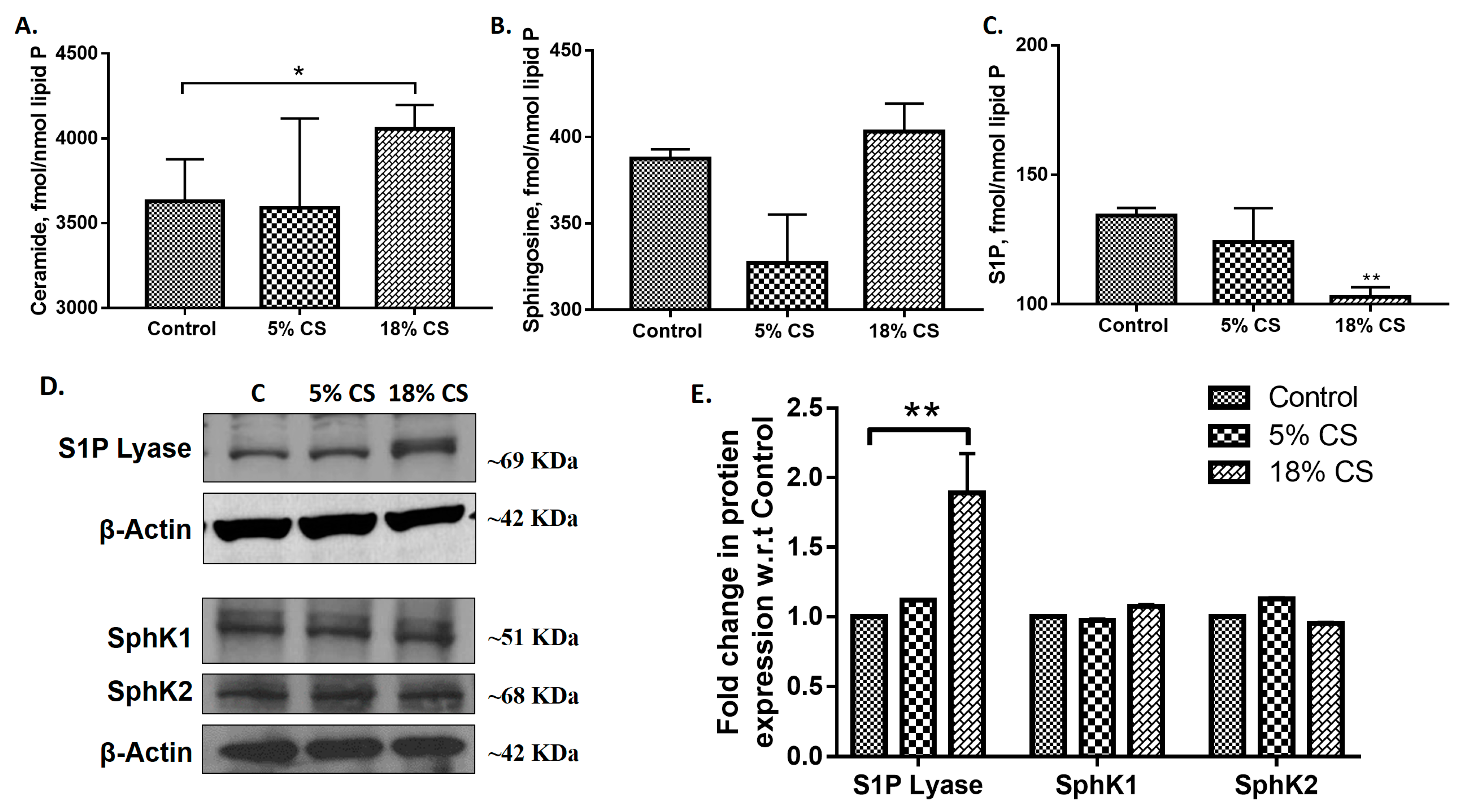
2.1. Mechanical Ventilation Modulates Sphingoid Bases Levels and Expression of S1P Lyase in Mouse Lung
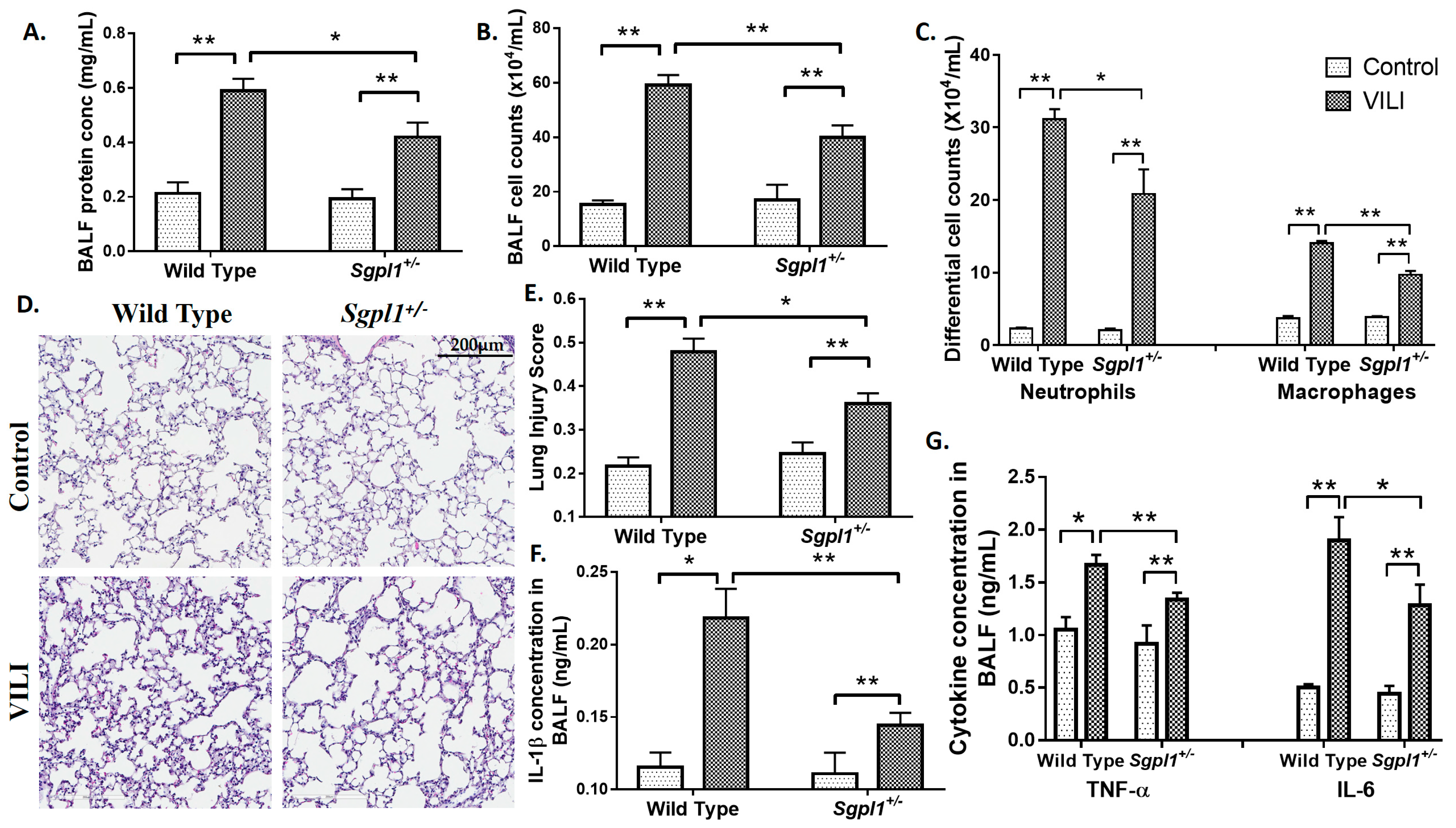
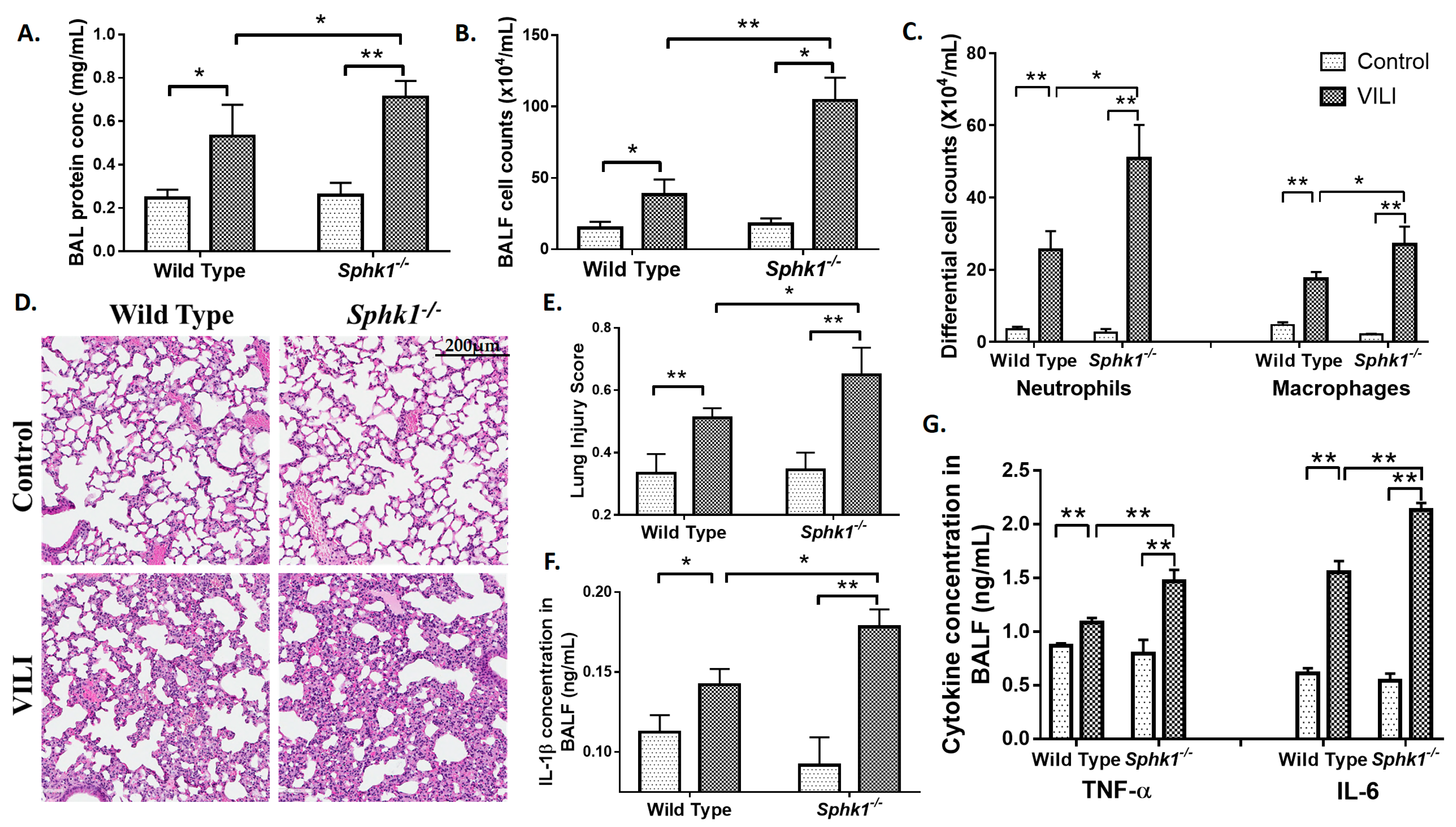
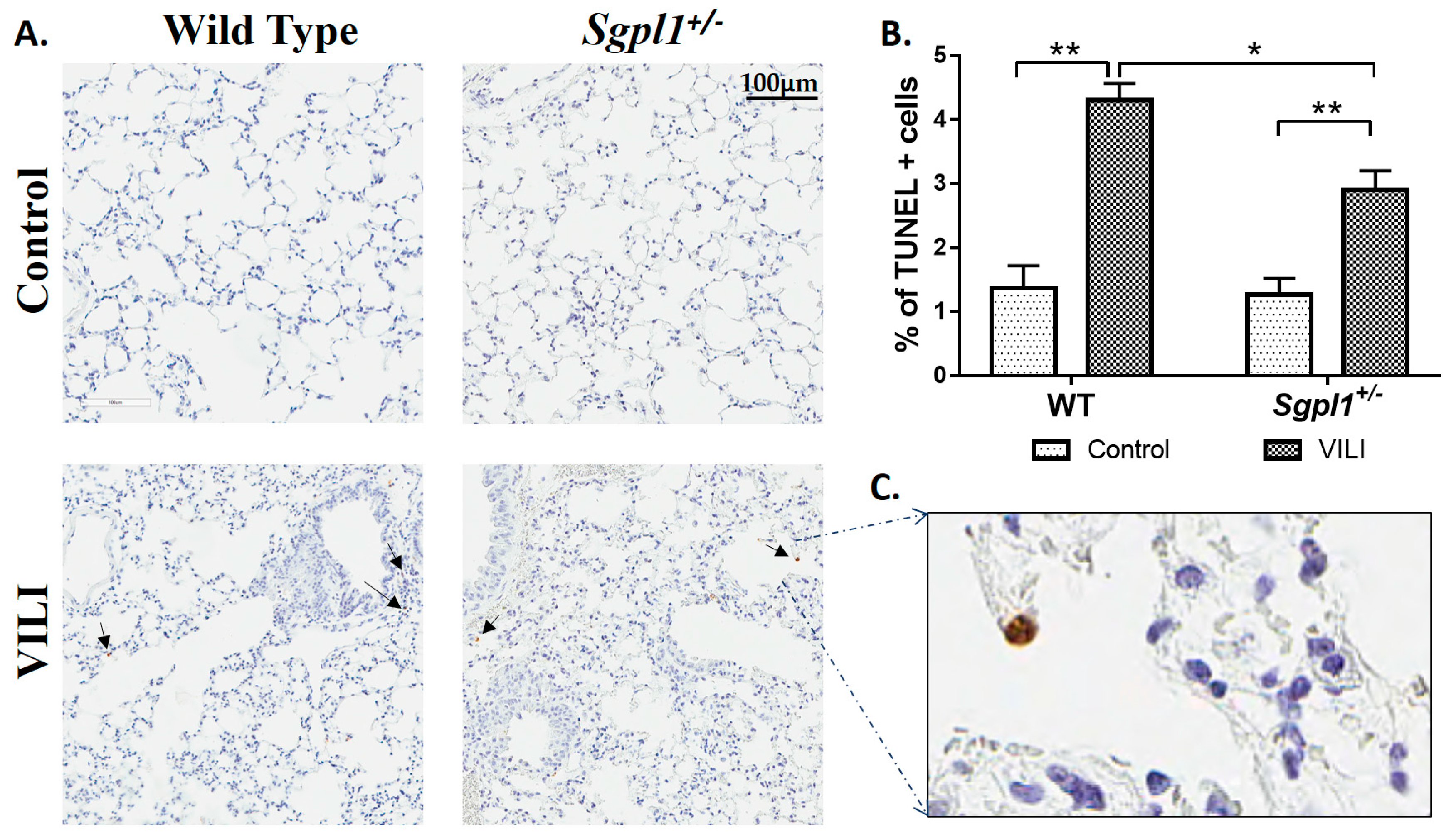
2.2. Genetic Deletion of S1PL, but Not Sphk1, Attenuates Ventilator-Induced Lung Injury and Inflammation
2.3. Cyclic Stretch Modulates Sphingoid Bases Levels and S1P Lyase Expression in Lung Epithelial Cells
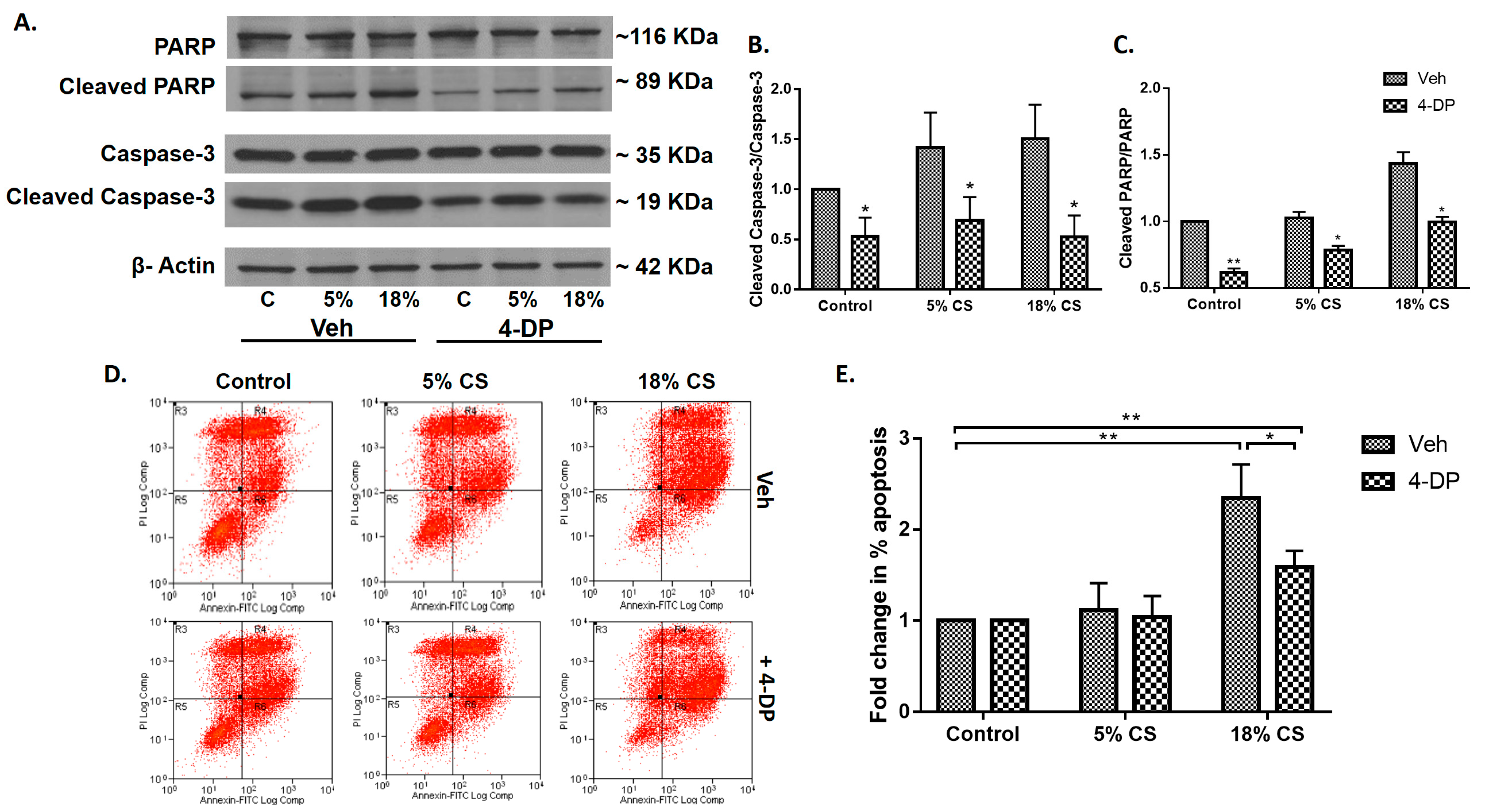
2.4. Inhibition of S1P Lyase by 4-Deoxypyridoxine Attenuates Cyclic Stretch-Induced Epithelial Cell Apoptosis
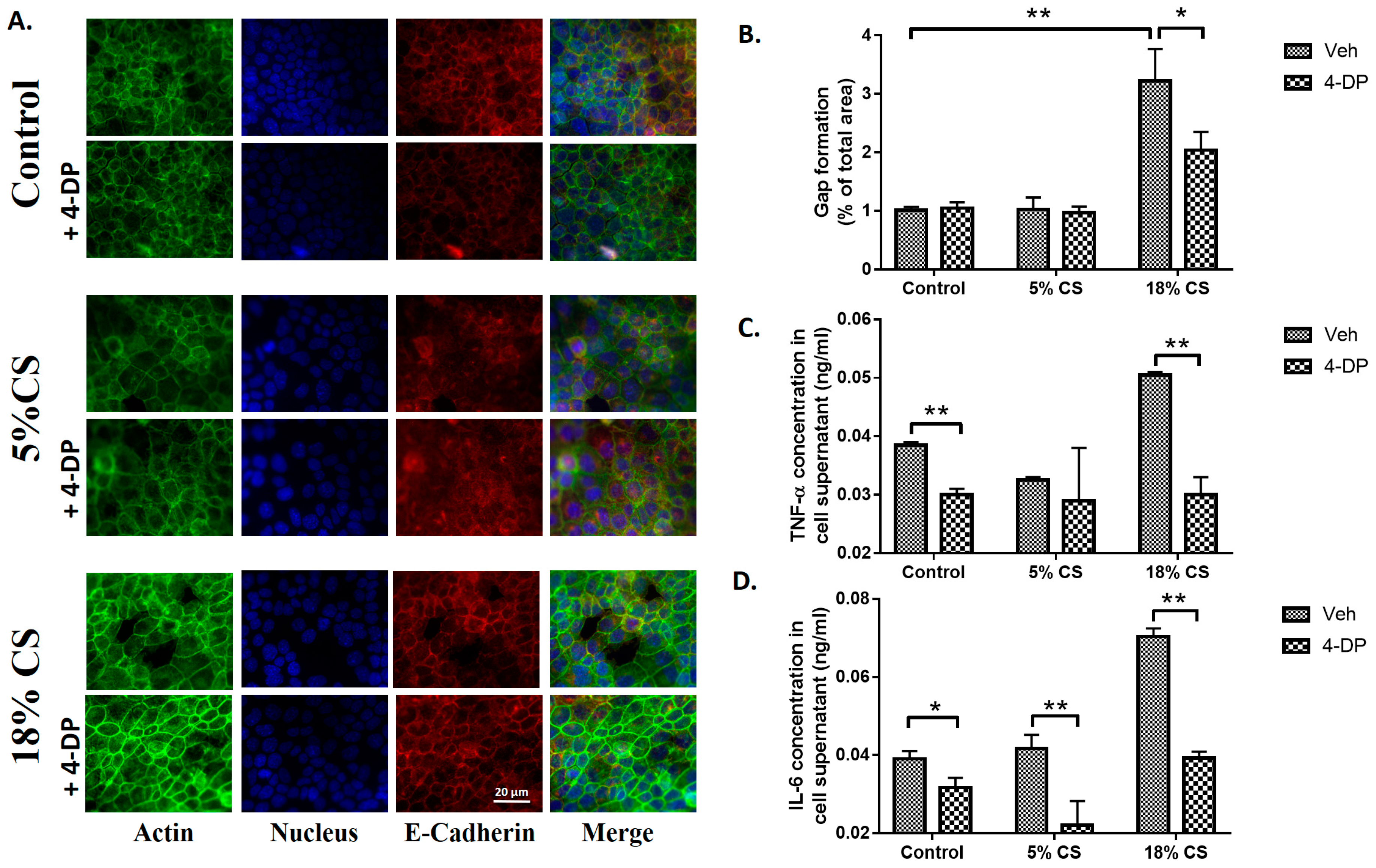
2.5. Inhibition of S1P Lyase Reduces Cyclic Stretch Induced Paracellular Gap Formation and Cytokine Secretion
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Murine Model of VILI
4.3. Analyses of Sphingoid Base-1-Phosphates
4.4. Cell Culture and Cyclic Stretch Experiments
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
References
- Matthay, M.A.; Uchida, T.; Fang, X. Clinical acute lung injury and acute respiratory distress syndrome. Curr. Treat. Options Cardiovasc. Med. 2002, 4, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Drake, R.E.; Doursout, M.F. Pulmonary edema and elevated left atrial pressure: Four hours and beyond. Physiology 2002, 17, 223–226. [Google Scholar] [CrossRef]
- Eddy, F.; Jesus, V.; Arthur, S.S. Novel approaches to minimize ventilator-induced lung injury. BMC Med. 2013, 11, 85. [Google Scholar] [CrossRef]
- Dreyfuss, D.; Saumon, G.; Hubmayr, R. Ventilator-Induced Lung Injury; CRC Press: Boca Raton, FL, USA, 2006; ISBN 9780849337161. [Google Scholar]
- Spieth, P.M.; Bluth, T.; Gama De Abreu, M.; Bacelis, A.; Goetz, A.E.; Kiefmann, R. Mechanotransduction in the lungs. Minerva Anestesiol. 2014, 80, 933–941. [Google Scholar] [PubMed]
- James, A.F.; Michael, A.M. Science review: Mechanisms of ventilator-induced injury. Crit. Care 2003, 7, 233–241. [Google Scholar] [CrossRef]
- Waters, C.M.; Ridge, K.M.; Sunio, G.; Venetsanou, K.; Sznajder, J.I. Mechanical stretching of alveolar epithelial cells increases Na-K-ATPase activity. J. Appl. Physiol. 1999, 87, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Nakos, G.; Kitsiouli, E.; Hatzidaki, E.; Koulouras, V.; Touqui, L.; Lekka, M.E. Phospholipases A2 and platelet-activating-factor acetylhydrolase in patients with acute respiratory distress syndrome. Crit. Care Med. 2005, 33, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gu, C.; Wang, Y. Upregulation of the tight junction protein occludin: Effects on ventilation-induced lung injury and mechanisms of action. BMC Pulm. Med. 2014, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, U.; Haitsma, J.J.; Goldmann, T.; Poelma, D.L.; Lachmann, B.; Uhlig, S. Ventilation-induced activation of the mitogen-activated protein kinase pathway. Eur. Respir. J. 2002, 20, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.P.; Dean, D.A. Cyclic stretch-induced nuclear localization of transcription factors results in increased nuclear targeting of plasmids in alveolar epithelial cells. J. Gene Med. 2008, 10, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 93, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Airola, M.V.; Hannun, Y.A. Sphingolipid metabolism and neutral sphingomyelinases. In Handbook of Experimental Pharmacology; Springer: Vienna, Austria, 2013; Volume 215, pp. 57–76. [Google Scholar]
- Lina, M.O.; Yusuf, A.H. Ceramide: A stress signal and mediator of growth suppression and apoptosis. J. Cell. Biochem. 1995, 58, 191–198. [Google Scholar] [CrossRef]
- Babak, O.; Julie, D.S. Cancer Treatment Strategies Targeting Sphingolipid Metabolism. In Sphingolipids as Signaling and Regulatory Molecules; Chalfant, C., Poeta, M.D., Eds.; Springer: New York, NY, USA, 2010; Volume 688, pp. 185–205. [Google Scholar]
- Natarajan, V.; Dudek, S.M.; Jacobson, J.R.; Moreno-Vinasco, L.; Huang, L.S.; Abassi, T.; Mathew, B.; Zhao, Y.; Wang, L.; Bittman, R.; et al. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Kihara, A.; Igarashi, Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem. Biophys. Res. Commun. 2004, 325, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.S.; Nierhaus, A.; Holzmann, M.; Mudersbach, E.; Bauer, A.; Robbe, L.; Zahrte, C.; Geffken, M.; Peine, S.; Schwedhelm, E.; et al. Decreased serum concentrations of sphingosine-1-phosphate in sepsis. Crit. Care 2015, 19, 372. [Google Scholar] [CrossRef] [PubMed]
- Gairhe, S.; Joshi, S.R.; Bastola, M.M.; McLendon, J.M.; Oka, M.; Fagan, K.A.; McMurtry, I.F. Sphingosine-1-phosphate is involved in the occlusive arteriopathy of pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Navarro, R.; Juan, G.; Peiro, T.; Serrano, A.; Ramon, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Natarajan, V. Sphingolipids in pulmonary fibrosis. Adv. Biol. Regul. 2015, 57, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Berdyshev, E.; Mathew, B.; Fu, P.; Gorshkova, I.A.; He, D.; Ma, W.; Noth, I.; Ma, S.F.; Pendyala, S.; et al. Targeting sphingosine kinase 1 attenuates bleomycin-induced pulmonary fibrosis. FASEB J. 2013, 27, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Berdyshev, E.V.; Tran, J.T.; Xie, L.; Chen, J.; Ebenezer, D.L.; Mathew, B.; Gorshkova, I.; Zhang, W.; Reddy, S.P.; et al. Sphingosine-1-phosphate lyase is an endogenous suppressor of pulmonary fibrosis: Role of S1P signaling and autophagy. Thorax 2015, 70, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Jolly, P.S.; Rosenfeldt, H.M.; Milstien, S.; Spiegel, S. The roles of sphingosine-1-phosphate in asthma. Mol. Immunol. 2002, 38, 1239–1245. [Google Scholar] [CrossRef]
- Harijith, A.; Pendyala, S.; Reddy, N.M.; Bai, T.; Usatyuk, P.V.; Berdyshev, E.; Gorshkova, I.; Huang, L.S.; Mohan, V.; Garzon, S.; et al. Sphingosine Kinase 1 Deficiency Confers Protection against Hyperoxia-Induced Bronchopulmonary Dysplasia in a Murine Model: Role of S1P Signaling and Nox Proteins. Am. J. Pathol. 2013, 18, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Harikumar, K.B. Sphingosine 1-Phosphate: A Novel Target for Lung Disorders. Front Immunol. 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Snoek, K.G.; Reiss, I.K.M.; Tibboel, J.; van Rosmalen, J.; Capolupo, I.; van Heijst, A.; Schaible, T.; Post, M.; Tibboel, D. Sphingolipids in congenital diaphragmatic hernia; results from an international multicenter study. PLoS ONE 2016, 11, e0155136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-García, J.L.; Naz, S.; Nin, N.; Rojas, Y.; Erazo, M.; Martínez-Caro, L.; García, A.; de Paula, M.; Fernández-Segoviano, P.; Casals, C.; et al. A Metabolomic Approach to the Pathogenesis of Ventilator-induced Lung Injury. Anesthesiology 2014, 120, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Jaecklin, T.; Otulakowski, G.; Kavanagh, P.B. Do soluble mediators cause ventilator-induced lung injury and multi-organ failure? Intensive Care Med. 2010, 36, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Ikeda, M.; Kariya, Y.; Lee, E.Y.; Lee, Y.M.; Igarashi, Y. Sphingosine-1-phosphate lyase is involved in the differentiation of F9 embryonal carcinoma cells to primitive endoderm. J. Biol. Chem. 2003, 278, 14578–14585. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.; Gorshkova, I.; Usatyuk, P.V.; Kalari, S.; Zhao, Y.; Pyne, N.J.; Pyne, S.; Garcia, J.G.N.; Natarajan, V. Intracellular Sphhingosine-1-Phosphate is essential for Exogenous Sphingosine-1-Phosphate-induced Endothelial Cell Motility. Role of Sphingosine Kinase 1 and Sphingosine-1-Phospate Lyase. PLoS ONE. 2011, 6, e16571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Gorshkova, A.I.; Berdyshev, E.; He, D.; Fu, P.; Ma, W.; Su, Y.; Usatyuk, V.P.; Pendyala, S.; Oskouian, B.; et al. Protection of LPS-Induced Murine Acute Lung Injury by Sphingosine-1-Phosphate Lyase Suppression. Am. J. Respir. Cell Mol. Biol. 2011, 45, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Kim, H.P.; Choi, A.M.K. Apoptosis in Lung Injury and Disease. In Essentials of Apoptosis; Dong, Z., Yin, X.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 523–545. ISBN 978-1-60327-381-7. [Google Scholar]
- Wrigge, H.; Stüber, F.; Putensen, C. Ventilator-Associated Systemic Inflammation. In Yearbook of Intensive Care and Emergency Medicine 2001; Vincent, J.L., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; Volume 2001, pp. 35–43. ISBN 978-3-642-59467-0. [Google Scholar]
- Dos Santos, C.C.; Slutsky, A.S. Invited Review: Mechanisms of ventilator-induced lung injury: A perspective. J. Appl. Physiol. 2000, 89, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Yabu, T.; Shiba, H.; Shibasaki, Y.; Nakanishi, T.; Imamura, S.; Touhata, K.; Yamashita, M. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ. 2015, 22, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Uhlig, S. The role of sphingolipids in respiratory disease. Ther. Adv. Respir. Dis. 2011, 5, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Brodlie, M.; McKean, M.C.; Johnson, G.E.; Gray, J.; Fisher, A.J.; Corris, P.A.; Lordan, J.L.; Ward, C. Ceramide Is Increased in the Lower Airway Epithelium of People with Advanced Cystic Fibrosis Lung Disease. Am. J. Respir. Crit. Care Med. 2010, 182, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.J. Lung-protective ventilation strategies and adjunctive treatments for the emergency medicine patient with acute respiratory failure. Emerg. Med. Clin. 2014, 32, 871–887. [Google Scholar] [CrossRef] [PubMed]
- Petrusca, D.N.; Van Demark, M.; Gu, Y.; Justice, M.J.; Rogozea, A.; Hubbard, W.C.; Petrache, I. Smoking Exposure Induces Human Lung Endothelial Cell Adaptation to Apoptotic Stress. Am. J. Respir. Cell Mol. Biol. 2014, 50, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and Intracellular Actions of Sphingosine-1-Phosphate. In Sphingolipids as Signaling and Regulatory Molecules; Chalfant, C., Poeta, M.D., Eds.; Springer: New York, NY, USA, 2010; Volume 688, pp. 141–155. [Google Scholar]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Saba, J.D. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzym. Regul. 2010, 50, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Nishiuma, T.; Nishimura, Y.; Okada, T.; Kuramoto, E.; Kotani, Y.; Jahangeer, S.; Nakamura, S. Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1085–L1093. [Google Scholar] [CrossRef] [PubMed]
- Price, M.M.; Oskeritzian, C.A.; Falanga, Y.T.; Price, M.M.; Oskeritzian, C.A.; Falanga, Y.T.; Harikumar, K.B.; Allegood, J.C.; Alvarez, S.E.; Conard, D.; et al. A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent mouse model of allergic asthma. J. Allergy Clin. Immunol. 2013, 131, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The Sphingosine Kinase 1/Sphingosine-1-Phosphate Pathway in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase in immunity and cancer: Silencing the siren. Trends Mol. Med. 2007, 13, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Bagdanoff, J.T.; Donoviel, M.S.; Nouraldeen, A.; Carlsen, M.; Jessop, T.C.; Tarver, J.; Aleem, S.; Dong, L.; Zhang, H.; Boteju, L.; et al. Inhibition of sphingosine 1-phosphate lyase for the treatment of rheumatoid arthritis: Discovery of (E)-1-(4-((1R,2S,3R)-1,2,3,4-tetrahydroxybutyl)-1H-imidazol-2-yl) ethanone oxime (LX2931) and (1R,2S,3R)-1-(2-(isoxazol-3-yl)-1H-imidazol-4-yl)butane-1,2,3,4-tetraol (LX2932). J. Med. Chem. 2010, 53, 8650–8662. [Google Scholar] [CrossRef] [PubMed]
- Hemdan, N.Y.; Weigel, C.; Reimann, C.M.; Gräler, M.H. Modulating sphingosine 1-phosphate signaling with DOP or FTY720 alleviates vascular and immune defects in mouse sepsis. Eur. J. Immunol. 2016, 46, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Fyrst, H.; Saba, J.D. Sphingosine-1-phosphate lyase in development and disease: Sphingolipid metabolism takes flight. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2008, 1781, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Japtok, L.; Bäumer, W.; Kleuser, B. Sphingosine-1-phosphate as signaling molecule in the skin: Relevance in atopic dermatitis. Allergo J. Int. 2014, 23, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Pugin, J. Molecular mechanisms of lung cell activation induced by cyclic stretch. Crit. Care Med. 2003, 31, S200–S206. [Google Scholar] [CrossRef] [PubMed]
- Desai, L.P.; White, S.R.; Waters, C.M. Cyclic mechanical stretch decreases cell migration by inhibiting phosphatidylinositol 3-kinase- and focal adhesion kinase-mediated JNK1 activation. J. Biol. Chem. 2010, 285, 4511–4519. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, N.; DiPaolo, B.C.; Lawrence, G.G.; Chhour, P.; Yehya, N.; Margulies, S.S. Cyclic Stretch–Induced Oxidative Stress Increases Pulmonary Alveolar Epithelial Permeability. Am. J. Respir. Cell Mol. Biol. 2013, 49, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Adyshev, D.M.; Elangovan, V.R.; Moldobaeva, N.; Mapes, B.; Sun, X.; Garcia, J.G.N. Mechanical Stress Induces Pre–B-cell Colony-Enhancing Factor/NAMPT Expression via Epigenetic Regulation by miR-374a and miR-568 in Human Lung Endothelium. Am. J. Respir. Cell Mol. Biol. 2014, 50, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Meliton, A.Y.; Munoz, N.M.; Meliton, L.N.; Birukova, A.A.; Leff, A.R.; Birukov, K.G. Mechanical induction of group V phospholipase A2 causes lung inflammation and acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L689–L700. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bao, H.; Wang, K.X.; Zhang, P.; Yao, Q.P.; Chen, X.H.; Huang, K.; Qi, Y.X.; Jiang, Z.L. Secreted miR-27a Induced by Cyclic Stretch Modulates the Proliferation of Endothelial Cells in Hypertension via GRK6. Sci. Rep. 2017, 7, 41058. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gross, C.; Desai, A.A.; Zemskov, E.; Wu, X.; Garcia, A.N.; Jacobson, J.R.; Yuan, J.X.J.; Garcia, J.G.N.; Black, S.M. Endothelial cell signaling and ventilator-induced lung injury: Molecular mechanisms, genomic analyses, and therapeutic targets. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L452–L476. [Google Scholar] [CrossRef] [PubMed]
- Vlahakis, N.E.; Schroeder, M.A.; Limper, A.H.; Hubmayr, R.D. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 277, L167–L173. [Google Scholar] [CrossRef]
- Ohtoyo, M.; Tamura, M.; Machinaga, N.; Muro, F.; Hashimoto, R. Sphingosine 1-phosphate lyase inhibition by 2-acetyl-4-(tetrahydroxybutyl) imidazole (THI) under conditions of vitamin B6 deficiency. Mol. Cell. Biochem. 2015, 400, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Wadgaonkar, R.; Patel, V.; Grinkina, N.; Romano, C.; Liu, J.; Zhao, Y.; Sammani, S.; Garcia, J.G.; Natarajan, V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L603–L613. [Google Scholar] [CrossRef] [PubMed]
- Makena, P.S.; Gorantla, V.K.; Ghosh, M.C.; Bezawada, L.; Kandasamy, K.; Balazs, L.; Luellen, C.L.; Thompson, K.E.; Parthasarathi, K.; Ichijo, H.; et al. Deletion of Apoptosis Signal–Regulating Kinase–1 Prevents Ventilator-Induced Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2012, 46, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Damico, R.; Damarla, M.; Boueiz, A.; Pae, H.H.; Skirball, J.; Hasan, E.; Peng, X.; Chesley, A.; Crow, M.T.; et al. Alveolar cell apoptosis is dependent on p38 MAP kinase-mediated activation of xanthine oxidoreductase in ventilator-induced lung injury. J. Appl. Physiol. 2008, 105, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Jacobson, J.R.; Berdyshev, E.; Huang, Y.; Sun, X.; Zhao, Y.; Gerhold, L.M.; Siegler, J.; Evenoski, C.; Wang, T.; et al. Role of sphingolipids in murine radiation-induced lung injury: Protection by sphingosine 1-phosphate analogs. FASEB J. 2011, 25, 3388–3400. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.V.; Gorshkova, I.A.; Garcia, J.G.; Natarajan, V.; Hubbard, W.C. Quantitative analysis of sphingoid base-1-phosphates as bisacetylated derivatives by liquid chromatography-tandem mass spectrometry. Anal. Biochem. 2005, 339, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Sasaki, T.; Kawai, H.; Olivera, A.; Mi, Y.; van Echten-Deckert, G.; Hajdu, R.; Rosenbach, M.; Keohane, C.A.; Mandala, S.; et al. Mice Deficient in Sphingosine Kinase 1 Are Rendered Lymphopenic by FTY720. J. Biol. Chem. 2004, 279, 52487–52492. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Fu, P.; Xing, J.; Yakubov, B.; Cokic, I.; Birukov, K.G. Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L837–L848. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M. An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Vidyani Suryadevara, Sphingolipids in Ventilator-Induced Lung Injury. Master’s Thesis, University of Illinois, Chicago, IL, USA, 15 December 2014.







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suryadevara, V.; Fu, P.; Ebenezer, D.L.; Berdyshev, E.; Bronova, I.A.; Huang, L.S.; Harijith, A.; Natarajan, V. Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase. Int. J. Mol. Sci. 2018, 19, 114. https://doi.org/10.3390/ijms19010114
Suryadevara V, Fu P, Ebenezer DL, Berdyshev E, Bronova IA, Huang LS, Harijith A, Natarajan V. Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase. International Journal of Molecular Sciences. 2018; 19(1):114. https://doi.org/10.3390/ijms19010114
Chicago/Turabian StyleSuryadevara, Vidyani, Panfeng Fu, David Lenin Ebenezer, Evgeny Berdyshev, Irina A. Bronova, Long Shuang Huang, Anantha Harijith, and Viswanathan Natarajan. 2018. "Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase" International Journal of Molecular Sciences 19, no. 1: 114. https://doi.org/10.3390/ijms19010114
APA StyleSuryadevara, V., Fu, P., Ebenezer, D. L., Berdyshev, E., Bronova, I. A., Huang, L. S., Harijith, A., & Natarajan, V. (2018). Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase. International Journal of Molecular Sciences, 19(1), 114. https://doi.org/10.3390/ijms19010114