Understanding the Role of Intrinsic Disorder of Viral Proteins in the Oncogenicity of Different Types of HPV

Abstract
:1. Introduction
2. Results
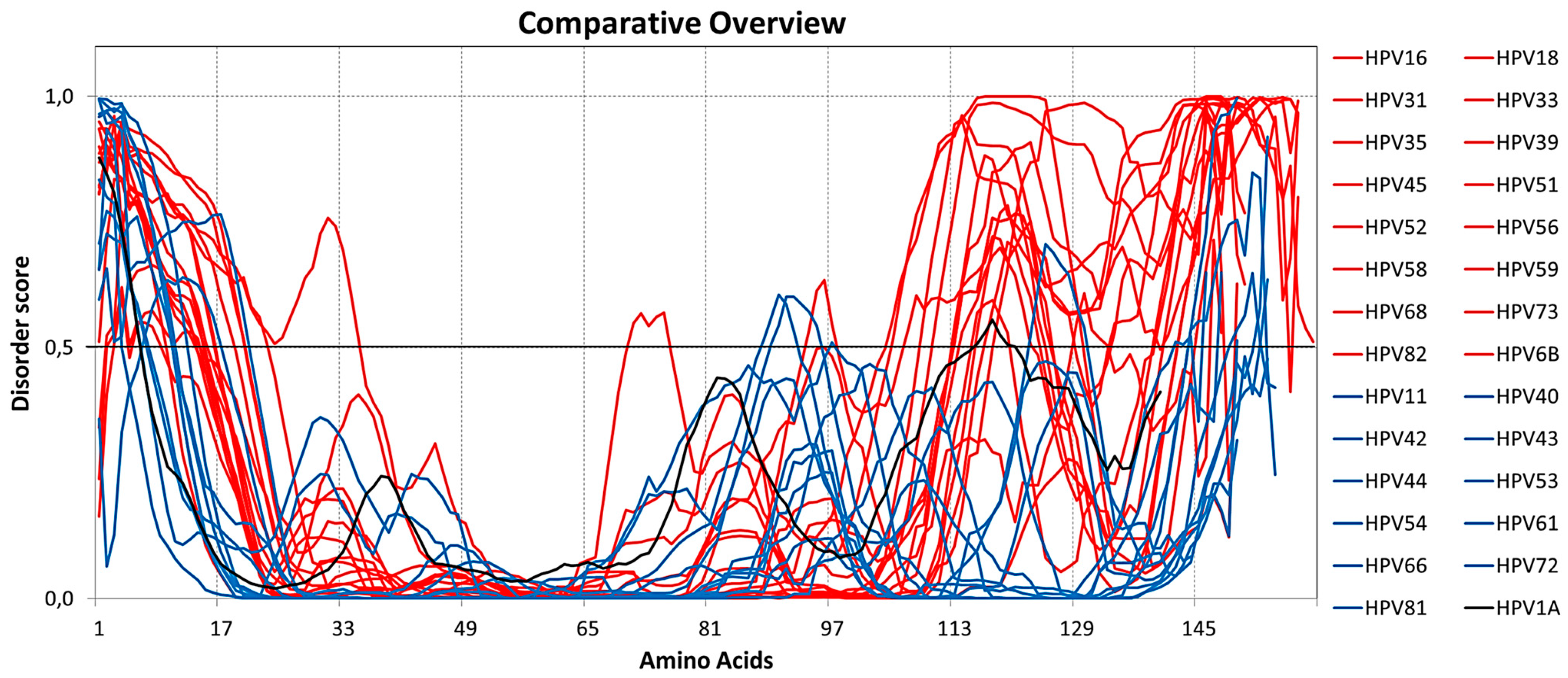
2.1. Analysis of Intrinsic Disorder
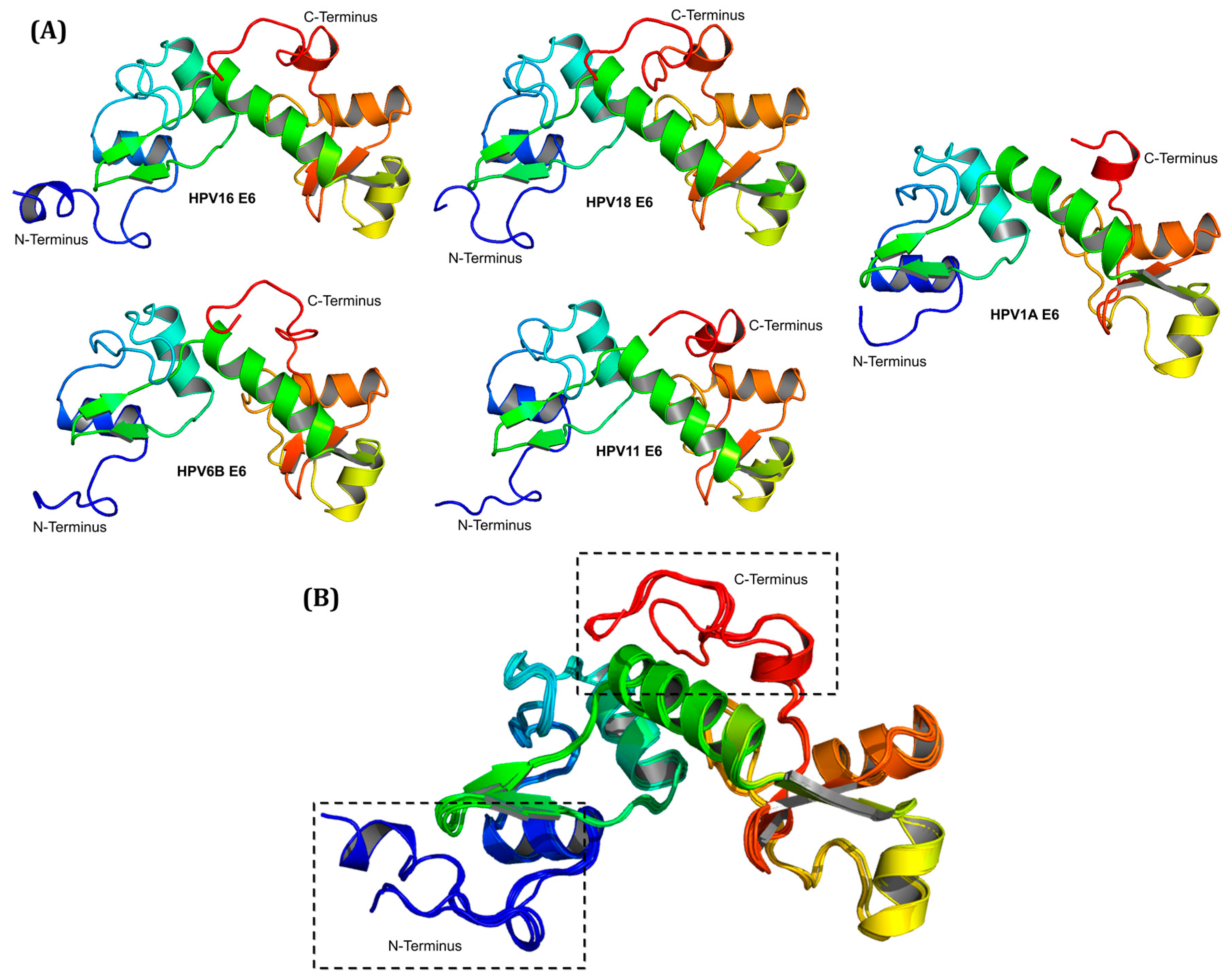
2.2. Molecular Modeling of E6
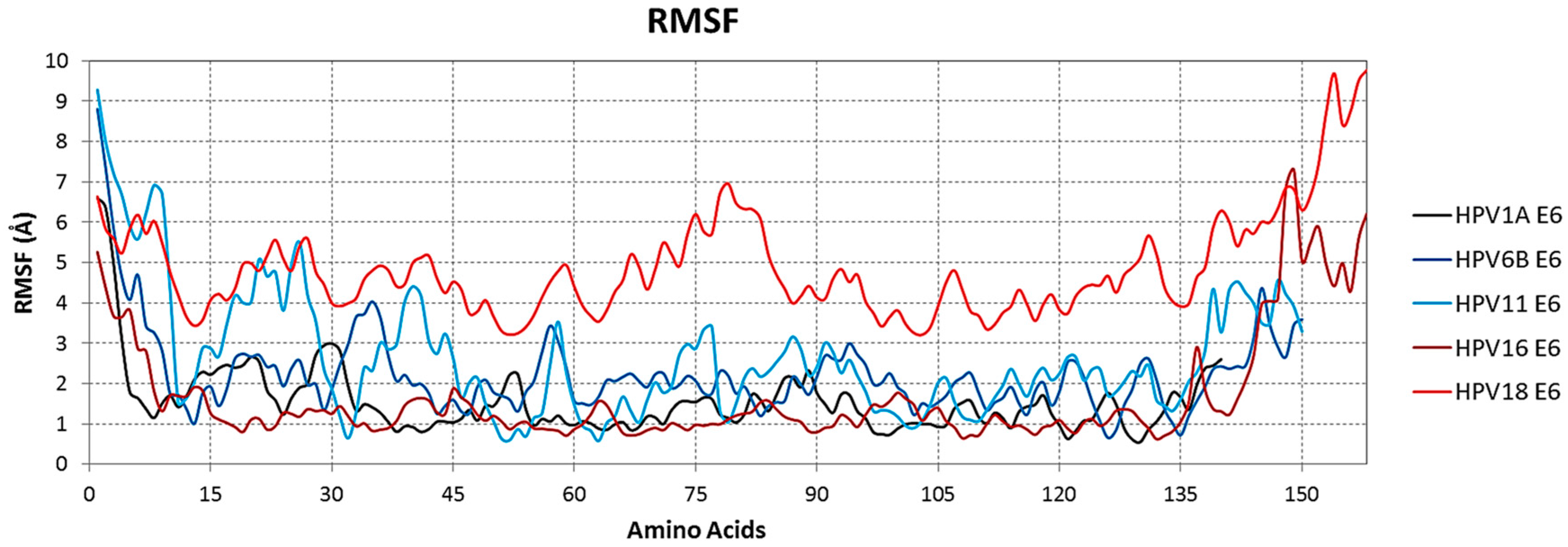
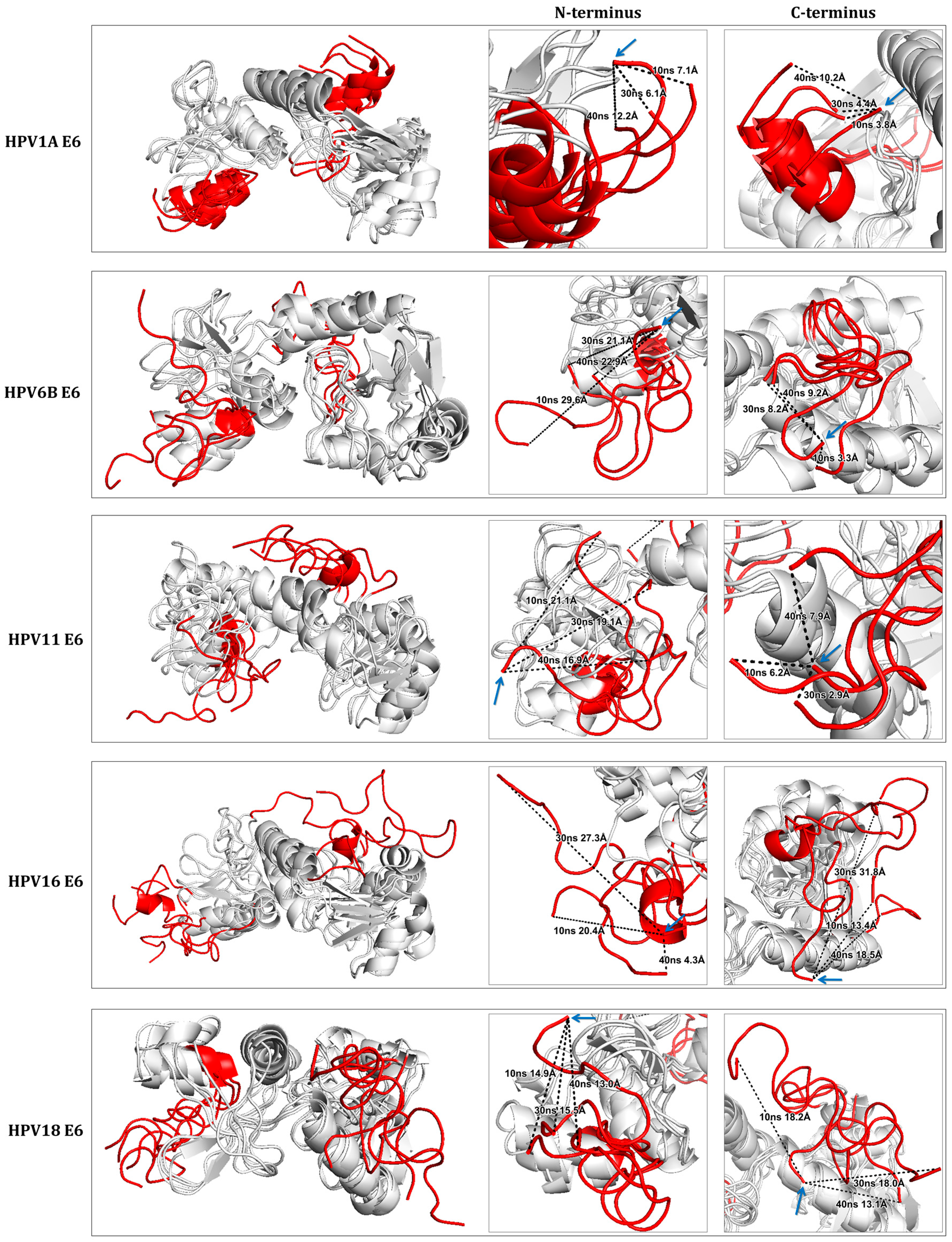
2.3. Simulation of Molecular Dynamics for RMSF Analysis
2.4. Analysis of Electrostatic Potential and Hydrophobicity Profile
3. Discussion
4. Materials and Methods
4.1. Obtaining the Primary Sequences
4.2. Prediction and Analysis of Intrinsic Disorder
4.3. Macromolecular Modeling and Ab Initio Modeling
4.4. Analysis of Electrostatic Potential and Hydrophobicity Profile
4.5. Simulation of Molecular Dynamics
4.6. Visualization and Visual Inspection of Models
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HPV | Human Papillomavirus |
| ID | Intrinsic Disorder |
| RMSF | Root Mean Square Fluctuations |
| Cα | Alpha Carbon |
References
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 2013, 22, 693–724. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Goh, G.K.-M.; Dunker, A.K.; Uversky, V.N. Viral disorder or disordered viruses: Do viral proteins possess unique features? Protein Pept. Lett. 2010, 17, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Berezovsky, I.N. The diversity of physical forces and mechanisms in intermolecular interactions. Phys. Biol. 2011, 8, 035002. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Archaic chaos: Intrinsically disordered proteins in Archaea. BMC Syst. Biol. 2010, 4 (Suppl. 1), S1. [Google Scholar] [CrossRef] [PubMed]
- Betiol, J.; Villa, L.L.; Sichero, L. Impact of HPV infection on the development of head and neck cancer. Braz. J. Med. Biol. Res. 2013, 46, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Bernard, H.-U.; Chen, Z.; de Villiers, E.-M.; zur Hausen, H.; Burk, R.D. Papillomaviruses: Evolution, Linnaean taxonomy and current nomenclature. Trends Microbiol. 2011, 19, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Rietz, A.; Petrov, D.P.; Bartolowits, M.; DeSmet, M.; Davisson, V.J.; Androphy, E.J. Molecular Probing of the HPV-16 E6 Protein Alpha Helix Binding Groove with Small Molecule Inhibitors. PLoS ONE 2016, 11, e0149845. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; Stutz, C.; Kintscher, S.; Reinz, E.; Sehr, P.; Bulkescher, J.; Hoppe-Seyler, K.; Travé, G.; Hoppe-Seyler, F. The E6AP binding pocket of the HPV16 E6 oncoprotein provides a docking site for a small inhibitory peptide unrelated to E6AP, indicating druggability of E6. PLoS ONE 2014, 9, e112514. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Gillison, M.L. Human papillomavirus in head and neck cancer: Its role in pathogenesis and clinical implications. Clin. Cancer Res. 2009, 15, 6758–6762. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.-J.; Liu, H.; Wu, D.; Tang, Z.-H.; Shen, Y.-C.; Guo, L. E6/E7 proteins are potential markers for the screening and diagnosis of cervical pre-cancerous lesions and cervical cancer in a Chinese population. Oncol. Lett. 2017, 14, 6251–6258. [Google Scholar] [CrossRef] [PubMed]
- Ruttkay-Nedecky, B.; Jimenez Jimenez, A.M.; Nejdl, L.; Chudobova, D.; Gumulec, J.; Masarik, M.; Adam, V.; Kizek, R. Relevance of infection with human papillomavirus: The role of the p53 tumor suppressor protein and E6/E7 zinc finger proteins (Review). Int. J. Oncol. 2013, 43, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Clifford, G.; Buonaguro, F.M. Classification of weakly carcinogenic human papillomavirus types: Addressing the limits of epidemiology at the borderline. Infect. Agents Cancer 2009, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Cornet, I.; Gheit, T.; Franceschi, S.; Vignat, J.; Burk, R.D.; Sylla, B.S.; Tommasino, M.; Clifford, G.M.; IARC HPV Variant Study Group. Human papillomavirus type 16 genetic variants: Phylogeny and classification based on E6 and LCR. J. Virol. 2012, 86, 6855–6861. [Google Scholar] [CrossRef] [PubMed]
- Nicolau, N.; Giuliatti, S. Modeling and molecular dynamics of the intrinsically disordered e7 proteins from high- and low-risk types of human papillomavirus. J. Mol. Model. 2013, 19, 4025–4037. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Roman, A.; Oldfield, C.J.; Dunker, A.K. Protein intrinsic disorder and human papillomaviruses: Increased amount of disorder in E6 and E7 oncoproteins from high-risk HPVs. J. Proteome Res. 2006, 5, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradović, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Nominé, Y.; Masson, M.; Charbonnier, S.; Zanier, K.; Ristriani, T.; Deryckère, F.; Sibler, A.-P.; Desplancq, D.; Atkinson, R.A.; Weiss, E.; et al. Structural and functional analysis of E6 oncoprotein: Insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol. Cell 2006, 21, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; Charbonnier, S.; Sidi, A.O.; McEwen, A.G.; Ferrario, M.G.; Poussin-Courmontagne, P.; Cura, V.; Brimer, N.; Babah, K.O.; Ansari, T.; et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 2013, 339, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Janert, P.K. Gnuplot in Action: Understanding Data with Graphs; Manning Publications Co.: Greenwich, CT, USA, 2009; ISBN 978-1-933988-39-9. [Google Scholar]
- Lowy, D.R.; Schiller, J.T. Reducing HPV-associated Cancer Globally. Cancer Prev. Res. (Phila.) 2012, 5, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Delano, W. The PyMOL Molecular Graphics System. Available online: http://www.pymol.org (accessed on 29 November 2017).
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Oldfield, C.J.; Xue, B.; Hsu, W.-L.; Meng, J.; Liu, X.; Shen, L.; Romero, P.; Uversky, V.N.; Dunker, A. Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinform. 2014, 15 (Suppl. 17), S4. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Terakawa, T.; Takada, S. p53 dynamics upon response element recognition explored by molecular simulations. Sci. Rep. 2015, 5, 17107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinen, A.; Kremer, D.; Göttle, P.; Kruse, F.; Hasse, B.; Lehmann, H.; Hartung, H.P.; Küry, P. The cyclin-dependent kinase inhibitor p57kip2 is a negative regulator of Schwann cell differentiation and in vitro myelination. Proc. Natl. Acad. Sci. USA 2008, 105, 8748–8753. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, L.; Qing, Y.; Li, C.; Ren, T.; Li, Q.; Li, M.; Zhang, S.; Shan, J.; Wang, G.; et al. Polymorphisms of BCL2 and BAX Genes Associate with Outcomes in Advanced Non-small cell lung cancer Patients treated with platinum-based Chemotherapy. Sci. Rep. 2015, 5, 17766. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P. Ewing sarcoma protein: A key player in human cancer. Int. J. Cell Biol. 2013, 2013, 642853. [Google Scholar] [CrossRef] [PubMed]
- Cino, E.A.; Karttunen, M.; Choy, W.-Y. Effects of molecular crowding on the dynamics of intrinsically disordered proteins. PLoS ONE 2012, 7, e49876. [Google Scholar] [CrossRef] [PubMed]
- Marasco, D.; Scognamiglio, P.L. Identification of inhibitors of biological interactions involving intrinsically disordered proteins. Int. J. Mol. Sci. 2015, 16, 7394–7412. [Google Scholar] [CrossRef] [PubMed]
- Pushker, R.; Mooney, C.; Davey, N.E.; Jacqué, J.-M.; Shields, D.C. Marked variability in the extent of protein disorder within and between viral families. PLoS ONE 2013, 8, e60724. [Google Scholar] [CrossRef] [PubMed]
- Pelka, P.; Ablack, J.N.G.; Fonseca, G.J.; Yousef, A.F.; Mymryk, J.S. Intrinsic structural disorder in adenovirus E1A: A viral molecular hub linking multiple diverse processes. J. Virol. 2008, 82, 7252–7263. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Nakahara, T.; Tanaka, K.; Inagawa, Y.; Narisawa-Saito, M.; Yugawa, T.; Ohno, S.-I.; Fujita, M.; Nakagama, H.; Kiyono, T. Roles of the PDZ-binding motif of HPV 16 E6 protein in oncogenic transformation of human cervical keratinocytes. Cancer Sci. 2017, 108, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Spanos, W.C.; Hoover, A.; Harris, G.F.; Wu, S.; Strand, G.L.; Anderson, M.E.; Klingelhutz, A.J.; Hendriks, W.; Bossler, A.D.; Lee, J.H. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J. Virol. 2008, 82, 2493–2500. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Yoshimatsu, Y.; Ohno, S.; Yugawa, T.; Egawa, N.; Fujita, M.; Hirohashi, S.; Kiyono, T. An in vitro multistep carcinogenesis model for human cervical cancer. Cancer Res. 2008, 68, 5699–5705. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 2005, 61 (Suppl. 7), 176–182. [Google Scholar] [CrossRef] [PubMed]
- Pons, J.-L.; Labesse, G. @TOME-2: A new pipeline for comparative modeling of protein-ligand complexes. Nucleic Acids Res. 2009, 37, W485–W491. [Google Scholar] [CrossRef] [PubMed]
- Sali, A. Comparative protein modeling by satisfaction of spatial restraints. Mol. Med. Today 1995, 1, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Canutescu, A.A.; Dunbrack, R.L. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003, 12, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein structure prediction using Rosetta. Meth. Enzymol. 2004, 383, 66–93. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics 2008, 24, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Tosatto, S.C.E.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins 2008, 71, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–28. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPV Type | Classification | #Residues | % ID | Disordered Amino Acids N-Terminal | Disordered Amino Acids C-Terminal |
|---|---|---|---|---|---|
| HPV1A | Control | 140 | 8.6 | 6 | 6 |
| HPV16 | High-Risk | 158 | 39.9 | 23 | 40 |
| HPV18 | High-Risk | 158 | 27.2 | 14 | 29 |
| HPV31 | High-Risk | 149 | 38.9 | 20 | 38 |
| HPV33 | High-Risk | 149 | 35.6 | 16 | 37 |
| HPV35 | High-Risk | 149 | 33.6 | 13 | 37 |
| HPV39 | High-Risk | 158 | 31.6 | 8 | 42 |
| HPV45 | High-Risk | 158 | 31.6 | 13 | 37 |
| HPV51 | High-Risk | 151 | 33.1 | 18 | 32 |
| HPV52 | High-Risk | 148 | 45.9 | 35 | 33 |
| HPV56 | High-Risk | 155 | 27.1 | 20 | 22 |
| HPV58 | High-Risk | 149 | 36.9 | 18 | 37 |
| HPV59 | High-Risk | 160 | 25.6 | 15 | 26 |
| HPV68 | High-Risk | 158 | 25.3 | 11 | 29 |
| HPV73 | High-Risk | 148 | 14.2 | 14 | 7 |
| HPV82 | High-Risk | 151 | 36.4 | 13 | 42 |
| HPV6B | Low-Risk | 150 | 6.7 | 9 | 1 |
| HPV11 | Low-Risk | 150 | 7.3 | 10 | 1 |
| HPV40 | Low-Risk | 154 | 3.9 | 6 | 0 |
| HPV42 | Low-Risk | 150 | 11.3 | 10 | 7 |
| HPV43 | Low-Risk | 155 | 11.0 | 7 | 10 |
| HPV44 | Low-Risk | 150 | 7.3 | 11 | 0 |
| HPV53 | Low-Risk | 154 | 14.3 | 20 | 2 |
| HPV54 | Low-Risk | 144 | 13.9 | 12 | 8 |
| HPV61 | Low-Risk | 146 | 4.8 | 3 | 4 |
| HPV66 | Low-Risk | 155 | 10.3 | 11 | 5 |
| HPV72 | Low-Risk | 148 | 4.7 | 6 | 1 |
| HPV81 | Low-Risk | 154 | 7.8 | 10 | 2 |
| HPV Type | Classification | ID UniProtKB | #Residues |
|---|---|---|---|
| HPV1A | Control | P06929 | 140 |
| HPV16 | High-Risk | P03126 | 158 |
| HPV18 | High-Risk | P06463 | 158 |
| HPV31 | High-Risk | P17386 | 149 |
| HPV33 | High-Risk | P06427 | 149 |
| HPV35 | High-Risk | P27228 | 149 |
| HPV39 | High-Risk | P24835 | 158 |
| HPV45 | High-Risk | P21735 | 158 |
| HPV51 | High-Risk | P26554 | 151 |
| HPV52 | High-Risk | P36814 | 148 |
| HPV56 | High-Risk | P24836 | 155 |
| HPV58 | High-Risk | P26555 | 149 |
| HPV59 | High-Risk | B9UPD9 | 160 |
| HPV68 | High-Risk | Q7KYK8 | 158 |
| HPV73 | High-Risk | Q82005 | 148 |
| HPV82 | High-Risk | Q9IR59 | 151 |
| HPV6B | Low-Risk | P06462 | 150 |
| HPV11 | Low-Risk | P04019 | 150 |
| HPV40 | Low-Risk | P36812 | 154 |
| HPV42 | Low-Risk | P27229 | 150 |
| HPV43 | Low-Risk | P19709 | 155 |
| HPV44 | Low-Risk | P19710 | 150 |
| HPV53 | Low-Risk | Q17UC4 | 154 |
| HPV54 | Low-Risk | Q81018 | 144 |
| HPV61 | Low-Risk | Q80948 | 146 |
| HPV66 | Low-Risk | Q80955 | 155 |
| HPV72 | Low-Risk | Q81997 | 148 |
| HPV81 | Low-Risk | Q705E9 | 154 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamarozzi, E.R.; Giuliatti, S. Understanding the Role of Intrinsic Disorder of Viral Proteins in the Oncogenicity of Different Types of HPV. Int. J. Mol. Sci. 2018, 19, 198. https://doi.org/10.3390/ijms19010198
Tamarozzi ER, Giuliatti S. Understanding the Role of Intrinsic Disorder of Viral Proteins in the Oncogenicity of Different Types of HPV. International Journal of Molecular Sciences. 2018; 19(1):198. https://doi.org/10.3390/ijms19010198
Chicago/Turabian StyleTamarozzi, Elvira Regina, and Silvana Giuliatti. 2018. "Understanding the Role of Intrinsic Disorder of Viral Proteins in the Oncogenicity of Different Types of HPV" International Journal of Molecular Sciences 19, no. 1: 198. https://doi.org/10.3390/ijms19010198
APA StyleTamarozzi, E. R., & Giuliatti, S. (2018). Understanding the Role of Intrinsic Disorder of Viral Proteins in the Oncogenicity of Different Types of HPV. International Journal of Molecular Sciences, 19(1), 198. https://doi.org/10.3390/ijms19010198