Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer


{kind=link}
{kind=link}
Abstract
:1. Introduction
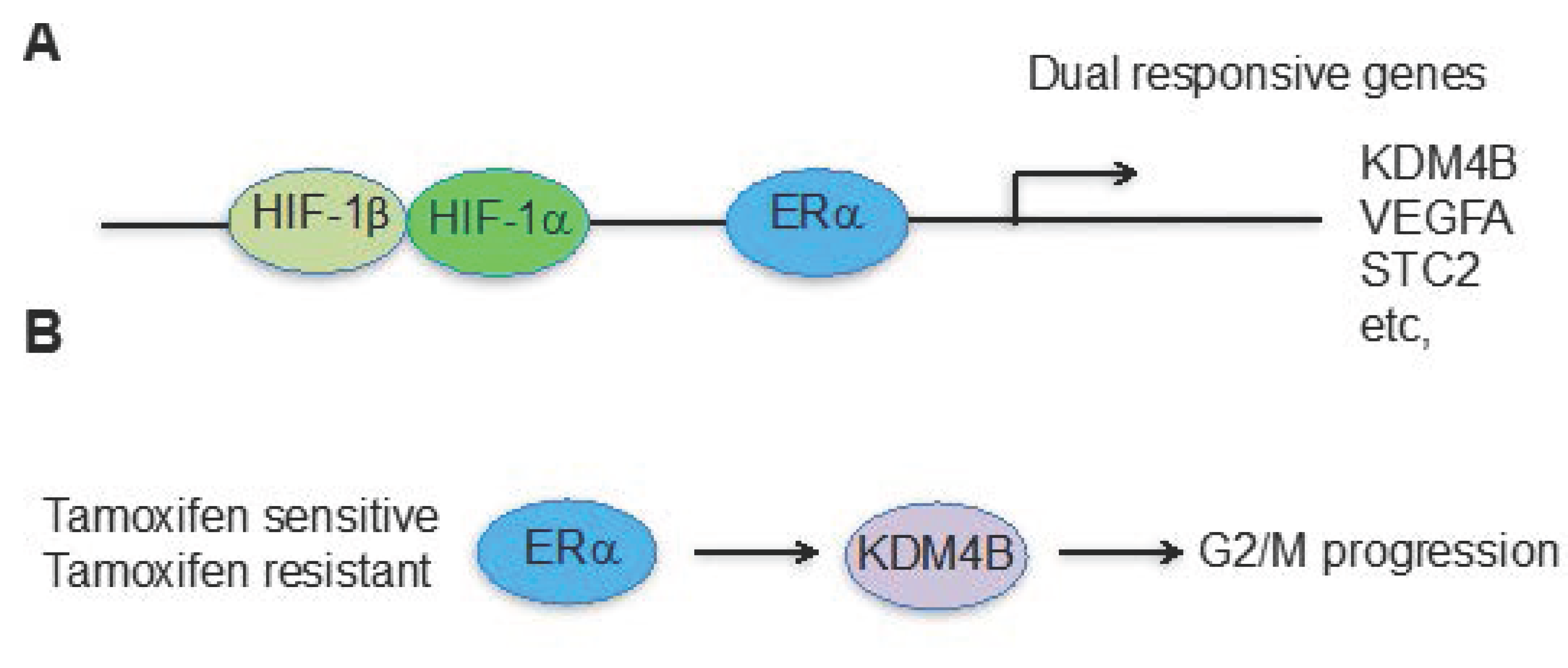
2. The Cross-Talk between Hypoxia and Estrogen
3. The Hypoxia and Estrogen Signaling Pathways Converge on Histone Demethylases
4. Future Prospects
Acknowledgments
Conflicts of Interest
References
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; El-Deiry, W.S. HIF-1 signaling in drug resistance to chemotherapy. Curr. Med. Chem. 2014, 21, 3021–3028. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Tumor hypoxia, drug resistance, and metastases. J. Natl. Cancer Inst. 1990, 82, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Giaccia, A.J. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006, 66, 10238–10241. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Koti, M.; Siemens, D.R.; Graham, C.H. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014, 74, 7185–7190. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1α) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1α) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Pino, M.S.; Zeng, M.; Shirasawa, S.; Chung, D.C. Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1α and -2α in colon cancer. Cancer Res. 2009, 69, 8499–8506. [Google Scholar] [CrossRef] [PubMed]
- Gerald, D.; Berra, E.; Frapart, Y.M.; Chan, D.A.; Giaccia, A.J.; Mansuy, D.; Pouyssegur, J.; Yaniv, M.; Mechta-Grigoriou, F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004, 118, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Vasquez, D.S.; Corbeil, J.; Wu, S.; Leblanc, M.; Wu, C.L.; Vera, D.R.; Shaw, R.J. mTOR and HIF-1α-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 11137–11142. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Simon, M.C.; Rafii, S.; Pandolfi, P.P. PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [PubMed]
- Yang, J.; Ahmed, A.; Poon, E.; Perusinghe, N.; de Haven Brandon, A.; Box, G.; Valenti, M.; Eccles, S.; Rouschop, K.; Wouters, B.; et al. Small-molecule activation of p53 blocks hypoxia-inducible factor 1α and vascular endothelial growth factor expression in vivo and leads to tumor cell apoptosis in normoxia and hypoxia. Mol. Cell. Biol. 2009, 29, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Mirand, E.A.; Gordon, A.S. Mechanism of estrogen action in erythropoiesis. Endocrinology 1966, 78, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Peschle, C.; Rappaport, I.A.; Sasso, G.F.; Condorelli, M.; Gordon, A.S. The role of estrogen in the regulation of erythropoietin production. Endocrinology 1973, 92, 358–362. [Google Scholar] [PubMed]
- Anagnostou, A.; Zander, A.; Barone, J.; Fried, W. Mechanism of the increased splenic erythropoiesis in mice treated with estradiol benzoate. J. Lab. Clin. Med. 1976, 88, 700–706. [Google Scholar] [PubMed]
- Yasuda, Y.; Masuda, S.; Chikuma, M.; Inoue, K.; Nagao, M.; Sasaki, R. Estrogen-dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. J. Biol. Chem. 1998, 273, 25381–25387. [Google Scholar] [CrossRef] [PubMed]
- Ruohola, J.K.; Valve, E.M.; Karkkainen, M.J.; Joukov, V.; Alitalo, K.; Harkonen, P.L. Vascular endothelial growth factors are differentially regulated by steroid hormones and antiestrogens in breast cancer cells. Mol. Cell. Endocrinol. 1999, 149, 29–40. [Google Scholar] [CrossRef]
- Bausero, P.; Ben-Mahdi, M.; Mazucatelli, J.; Bloy, C.; Perrot-Applanat, M. Vascular endothelial growth factor is modulated in vascular muscle cells by estradiol, tamoxifen, and hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2033–H2042. [Google Scholar] [CrossRef] [PubMed]
- Maity, A.; Sall, W.; Koch, C.J.; Oprysko, P.R.; Evans, S.M. Low pO2 and β-estradiol induce VEGF in MCF-7 and MCF-7-5C cells: Relationship to in vivo hypoxia. Breast Cancer Res. Treat. 2001, 67, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.A.; Koos, R.D. Estrogen-induced activation of hypoxia-inducible factor-1α, vascular endothelial growth factor expression, and edema in the uterus are mediated by the phosphatidylinositol 3-kinase/Akt pathway. Endocrinology 2007, 148, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Mukundan, H.; Kanagy, N.L.; Resta, T.C. 17-beta estradiol attenuates hypoxic induction of HIF-1α and erythropoietin in Hep3B cells. J. Cardiovasc. Pharmacol. 2004, 44, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Seifeddine, R.; Dreiem, A.; Tomkiewicz, C.; Fulchignoni-Lataud, M.C.; Brito, I.; Danan, J.L.; Favaudon, V.; Barouki, R.; Massaad-Massade, L. Hypoxia and estrogen co-operate to regulate gene expression in T-47D human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2007, 104, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Sato, Y.; Iwasaki, R.; Kobayashi, T.; Watanabe, R.; Oike, T.; Miyamoto, K.; Toyama, Y.; Matsumoto, M.; Nakamura, M.; et al. Selective estrogen receptor modulators suppress Hif1α protein accumulation in mouse osteoclasts. PLoS ONE 2016, 11, e0165922. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Brown, M. Estrogen receptor target gene: An evolving concept. Mol. Endocrinol. 2006, 20, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, D.P.; Norris, J.D. Connections and regulation of the human estrogen receptor. Science 2002, 296, 1642–1644. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Third annual William L. McGuire Memorial Lecture. "Studies on the estrogen receptor in breast cancer"–20 years as a target for the treatment and prevention of cancer. Breast Cancer Res. Treat. 1995, 36, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Jaiyesimi, I.A.; Buzdar, A.U.; Decker, D.A.; Hortobagyi, G.N. Use of tamoxifen for breast cancer: Twenty-eight years later. J. Clin. Oncol. 1995, 13, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Osborne, C.K.; Morris, C.; Wakeling, A.E. ICI 182,780 (Faslodex): Development of a novel, “pure” antiestrogen. Cancer 2000, 89, 817–825. [Google Scholar]
- Osborne, C.K. Aromatase inhibitors in relation to other forms of endocrine therapy for breast cancer. Endocr. Relat. Cancer 1999, 6, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.; Buzdar, A.; Cuzick, J.; Forbes, J.; Houghton, J.; Howell, A.; Sahmoud, T.; Group, A.T. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: Results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer 2003, 98, 1802–1810. [Google Scholar] [PubMed]
- Massarweh, S.; Osborne, C.K.; Jiang, S.; Wakeling, A.E.; Rimawi, M.; Mohsin, S.K.; Hilsenbeck, S.; Schiff, R. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006, 66, 8266–8273. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; DeFriend, D.; Robertson, J.; Blamey, R.; Walton, P. Response to a specific antioestrogen (ICI 182780) in tamoxifen-resistant breast cancer. Lancet 1995, 345, 29–30. [Google Scholar] [CrossRef]
- Clarke, R.; Leonessa, F.; Welch, J.N.; Skaar, T.C. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol. Rev. 2001, 53, 25–71. [Google Scholar] [PubMed]
- Groner, A.C.; Brown, M. Role of steroid receptor and coregulator mutations in hormone-dependent cancers. J. Clin. Investig. 2017, 127, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.Y.; Hsu, H.K.; Lan, X.; Juan, L.; Yan, P.S.; Labanowska, J.; Heerema, N.; Hsiao, T.H.; Chiu, Y.C.; Chen, Y.; et al. Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer. Cancer Cell 2013, 24, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; Zhong, H.; Hanrahan, C.F.; Mommers, E.C.; Semenza, G.L.; Pinedo, H.M.; Abeloff, M.D.; Simons, J.W.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1 α during breast carcinogenesis. J. Natl. Cancer Inst. 2001, 93, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Brunnberg, S.; Pettersson, K.; Rydin, E.; Matthews, J.; Hanberg, A.; Pongratz, I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 6517–6522. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Buffa, F.M.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Phosphorylated ERα, HIF-1α, and MAPK signaling as predictors of primary endocrine treatment response and resistance in patients with breast cancer. J. Clin. Oncol. 2009, 27, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Wigfield, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Hypoxia-inducible factor-1alpha expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin. Cancer Res. 2006, 12, 4562–4568. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.L.; Baban, D.; et al. Estrogen receptor-alpha directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef] [PubMed]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ledaki, I.; Turley, H.; Gatter, K.C.; Montero, J.C.; Li, J.L.; Harris, A.L. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann. N. Y. Acad. Sci. 2009, 1177, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Southey, M.C.; Chang, A.C.; Reddel, R.R.; Willhite, D.; Glynne, R.; Henderson, M.A.; Armes, J.E.; Venter, D.J. Stanniocalcin 2 is an estrogen-responsive gene coexpressed with the estrogen receptor in human breast cancer. Cancer Res. 2002, 62, 1289–1295. [Google Scholar] [PubMed]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.D.; Vigne, J.L.; Minchenko, A.; Lebovic, D.I.; Leitman, D.C.; Taylor, R.N. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors α and β. Proc. Natl. Acad. Sci. USA 2000, 97, 10972–10977. [Google Scholar] [CrossRef] [PubMed]
- Law, A.Y.; Wong, C.K. Stanniocalcin-2 is a HIF-1 target gene that promotes cell proliferation in hypoxia. Exp. Cell Res. 2010, 316, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Jeselsohn, R.; Pereira, R.; Hollingsworth, E.F.; Creighton, C.J.; Li, F.; Shea, M.; Nardone, A.; De Angelis, C.; Heiser, L.M.; et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E6600–E6609. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Hojfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Whetstine, J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 2007, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef] [PubMed]
- Wellmann, S.; Bettkober, M.; Zelmer, A.; Seeger, K.; Faigle, M.; Eltzschig, H.K.; Buhrer, C. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem. Biophys. Res. Commun. 2008, 372, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Das, P.P.; Shao, Z.; Beyaz, S.; Apostolou, E.; Pinello, L.; De Los Angeles, A.; O’Brien, K.; Atsma, J.M.; Fujiwara, Y.; Nguyen, M.; et al. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol. Cell 2014, 53, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Wu, Y.; Guo, L.; et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2012, 45, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Pryor, J.G.; Brown-Kipphut, B.A.; Iqbal, A.; Scott, G.A. Microarray comparative genomic hybridization detection of copy number changes in desmoplastic melanoma and malignant peripheral nerve sheath tumor. Am. J. Dermatopathol. 2011, 33, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Walters, Z.S.; Villarejo-Balcells, B.; Olmos, D.; Buist, T.W.; Missiaglia, E.; Allen, R.; Al-Lazikani, B.; Garrett, M.D.; Blagg, J.; Shipley, J. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2014, 33, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Kawazu, M.; Saso, K.; Tong, K.I.; McQuire, T.; Goto, K.; Son, D.O.; Wakeham, A.; Miyagishi, M.; Mak, T.W.; Okada, H. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS ONE 2011, 6, e17830. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Berry, W.L.; Kim, T.D.; Janknecht, R. Stimulation of beta-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int. J. Oncol. 2014, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.T.; Fan, Q.; Zhu, Z.; Kwan, S.Y.; Chen, L.; Chen, J.H.; Ying, Z.L.; Zhou, Y.; Gu, W.; Wang, L.H.; et al. KDM4B and KDM4A promote endometrial cancer progression by regulating androgen receptor, c-myc, and p27kip1. Oncotarget 2015, 6, 31702–31720. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Cho, H.S.; Iwai, Y.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Shimizu, N.; Tanaka, H.; Tsunoda, T.; et al. The histone demethylase JMJD2B plays an essential role in human carcinogenesis through positive regulation of cyclin-dependent kinase 6. Cancer Prev. Res. 2011, 4, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Bur, H.; Haapasaari, K.M.; Turpeenniemi-Hujanen, T.; Kuittinen, O.; Auvinen, P.; Marin, K.; Soini, Y.; Karihtala, P. Strong KDM4B and KDM4D Expression associates with radioresistance and aggressive phenotype in classical hodgkin lymphoma. Anticancer Res. 2016, 36, 4677–4683. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Qiu, L.; Hong, Y.; Karnik, T.; Tadros, G.; Mau, B.; Ma, T.; Mu, Y.; New, J.; Louie, R.J.; et al. The histone demethylase KDM4B regulates peritoneal seeding of ovarian cancer. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, W.; Zang, W.; Liu, Z.; Xu, X.; Yu, H.; Yang, Q.; Jia, J. JMJD2B promotes epithelial-mesenchymal transition by cooperating with β-catenin and enhances gastric cancer metastasis. Clin. Cancer Res. 2013, 19, 6419–6429. [Google Scholar] [CrossRef] [PubMed]
- Castellini, L.; Moon, E.J.; Razorenova, O.V.; Krieg, A.J.; von Eyben, R.; Giaccia, A.J. KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res. 2017, 45, 3674–3692. [Google Scholar] [CrossRef] [PubMed]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chen, L.; Pledger, W.J.; Fang, J.; Chen, J. p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene 2014, 33, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; AlTahan, A.M.; Hu, D.; Wang, Y.; Cheng, P.H.; Morton, C.L.; Qu, C.; Nathwani, A.C.; Shohet, J.M.; Fotsis, T.; et al. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. J. Natl. Cancer Inst. 2015, 107, djv080. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.H.; Wang, L.Y.; Hsu, K.C.; Chen, C.C.; Cheng, H.H.; Wang, S.M.; Wu, C.M.; Chen, T.J.; Li, L.T.; Liu, R.; et al. KDM4B as a target for prostate cancer: Structural analysis and selective inhibition by a novel inhibitor. J. Med. Chem. 2014, 57, 5975–5985. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Milasta, S.; Hu, D.; AlTahan, A.M.; Interiano, R.B.; Zhou, J.; Davidson, J.; Low, J.; Lin, W.; Bao, J.; et al. Targeting histone demethylases in MYC-driven neuroblastomas with ciclopirox. Cancer Res. 2017, 77, 4626–4638. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Stepputtis, S.S.; Strietz, J.; Preca, B.T.; Urban, S.; Willmann, D.; Allen, A.; Zenk, F.; Iovino, N.; Bronsert, P.; et al. KDM4 inhibition targets breast cancer stem-like cells. Cancer Res. 2017, 77, 5900–5912. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.K.; Bonaldi, T.; Cuomo, A.; del Rosario, J.R.; Hosfield, D.J.; Kanouni, T.; Kao, S.C.; Lai, C.; Lobo, N.A.; Matuszkiewicz, J.; et al. Design of KDM4 inhibitors with antiproliferative effects in cancer models. ACS Med. Chem. Lett. 2017, 8, 869–874. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Harris, A.L.; Davidoff, A.M. Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. Int. J. Mol. Sci. 2018, 19, 240. https://doi.org/10.3390/ijms19010240
Yang J, Harris AL, Davidoff AM. Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. International Journal of Molecular Sciences. 2018; 19(1):240. https://doi.org/10.3390/ijms19010240
Chicago/Turabian StyleYang, Jun, Adrian L. Harris, and Andrew M. Davidoff. 2018. "Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer" International Journal of Molecular Sciences 19, no. 1: 240. https://doi.org/10.3390/ijms19010240
APA StyleYang, J., Harris, A. L., & Davidoff, A. M. (2018). Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. International Journal of Molecular Sciences, 19(1), 240. https://doi.org/10.3390/ijms19010240