Clinical and Neurobehavioral Features of Three Novel Kabuki Syndrome Patients with Mosaic KMT2D Mutations and a Review of Literature

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Clinical Description
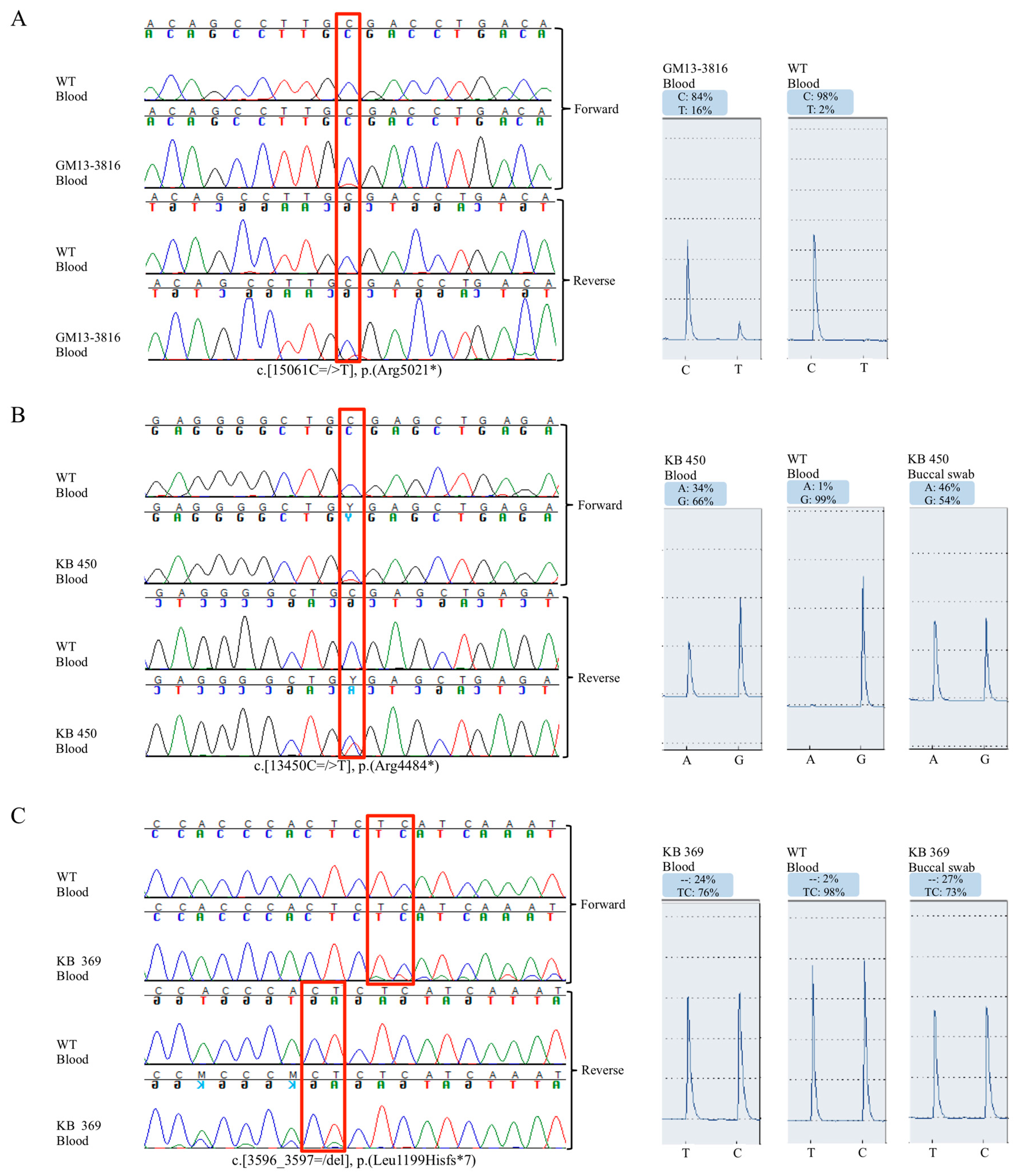
2.2. Mutational Analysis
3. Discussion
4. Materials and Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kuroki, Y.; Suzuki, Y.; Chyo, H.; Hata, A.; Matsui, I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr. 1981, 99, 570–573. [Google Scholar] [CrossRef]
- Niikawa, N.; Matsuura, N.; Fukushima, Y.; Ohsawa, T.; Kajii, T. Kabuki make-up syndrome: A syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 1981, 99, 565–569. [Google Scholar] [CrossRef]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a Histone Demethylase Interacting with MLL2, in Three Patients with Kabuki Syndrome. Am. J. Hum. Genet. 2012, 90, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Bogershausen, N.; Gatinois, V.; Riehmer, V.; Kayserili, H.; Becker, J.; Thoenes, M.; Simsek-Kiper, P.O.; Barat-Houari, M.; Elcioglu, N.H.; Wieczorek, D.; et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum. Mutat. 2016, 37, 847–864. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; Howard, E.; Bunstone, S.; Chandler, K.E.; Kerr, B.; Lachlan, K.; McKee, S.; Mehta, S.G.; Tavares, A.L.; Tolmie, J.; et al. MLL2 mosaic mutations and intragenic deletion-duplications in patients with Kabuki syndrome. Clin. Genet. 2013, 83, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; Orsini, A., Pezzuti, L., Picone, L., Eds.; Italian Edition; Giunti OS: Firenze, Italy, 2012; The Psychological Corporation: San Antonio, TX, USA, 2003. [Google Scholar]
- Harrison, P.L.; Oakland, T. ABAS-II Adaptive Behavior Assessment System, 2nd ed.; Ferrri, R., Orsini, A., Rea, M., Eds.; Italian Edition; Giunti OS: Firenze, Italy, 2014. [Google Scholar]
- Cornoldi, C.; Colpo, G. Prove di Lettura MT-2 per la Scuola Primaria; Giunti OS: Firenze, Italy, 2011. [Google Scholar]
- Tressoldi, P.E.; Cornoldi, C. BVSCO-2 Batteria per la Valutazione della Scrittura e della Competenza Ortografica–2; Giunti OS: Firenze, Italy, 2013. [Google Scholar]
- Kaufman, J.; Birmaher, B.; Rao, U.; Ryan, N. Test K-SADS-PL-Intervista Diagnostica per la Valutazione dei Disturbi Psicopatologici in Bambini e Adolescenti; Edizioni Centro Studi Erickson S.p.A.: Trento, Italy, 2004. [Google Scholar]
- Micale, L.; Augello, B.; Fusco, C.; Selicorni, A.; Loviglio, M.N.; Silengo, M.C.; Reymond, A.; Gumiero, B.; Zucchetti, F.; D’Addetta, E.V.; et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J. Rare Dis. 2011, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Cornoldi, C.; Lucangeli, D.; Bellina, M. Test AC-MT 6–11—Test di Valutazione Delle Abilità di Calcolo e Soluzione di Problemi; Edizioni Centro Studi Erickson S.p.A.: Trento, Italy, 2002. [Google Scholar]
- Banka, S.; Veeramachaneni, R.; Reardon, W.; Howard, E.; Bunstone, S.; Ragge, N.; Parker, M.J.; Crow, Y.J.; Kerr, B.; Kingston, H.; et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet. 2012, 20, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, A.D.; Stegmann, A.P.; Blok, M.J.; Tserpelis, D.; Posma-Velter, C.; Detisch, Y.; Smeets, E.E.; Wagemans, A.; Schrander, J.J.; van den Boogaard, M.J.; et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum. Mutat. 2011, 32, E2018–E2025. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N.; Mazery, A.C.; Visier, A.; Baumann, C.; Lachesnais, D.; Capri, Y.; Toutain, A.; Odent, S.; Mikaty, M.; Goizet, C.; et al. Molecular, clinical and neuropsychological study in 31 patients with Kabuki syndrome and KMT2D mutations. Clin. Genet. 2017, 92, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Caciolo, C.; Alfieri, P.; Piccini, G.; Digilio, M.C.; Lepri, F.R.; Tartaglia, M.; Menghini, D.; Vicari, S. Neurobehavioral features in individuals with Kabuki Syndrome. Mol. Genet. Genom. Med. 2017, in press. [Google Scholar]
- Fusco, C.; Micale, L.; Pellico, M.T.; D’Addetta, E.V.; Augello, B.; Mandriani, B.; De Nittis, P.; Cocciadiferro, D.; Malerba, N.; Sacco, M.; et al. Genomic and Genetic Disorders Biobank. Genetic Biobank from patients with Williams-Beuren syndrome and other genomic and genetic disorders. Open J. Bioresour. 2015, 2. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Features | GM13-3816 | KB450 | KB369 | Prevalence of KS Features |
|---|---|---|---|---|
| Gender | F | F | F | |
| Age (years) | 6.1 | 8 | 17 | |
| Facial anomalies | + | + | + | 87 5 |
| Elongated palpebral fissures | + | + | + | 95 18 |
| Sparse eyebrows | + | + | + | 82 18 |
| Palpebral ptosis | + | − | + | 52 18 |
| Broad nasal tip | − | + | + | 69 18 |
| Thin upper and full lower lip | + | + | + | 71 18 |
| Large dysmorhic ears | + | + | − | 90 18 |
| Short stature | − | − | + | 70 5 |
| Feeding difficulties | − | − | − | 82 5 |
| Cleft palate | − | − | − | 37 5 |
| Cardiac defects | − | + | − | 46 5 |
| Urogenital anomalies | − | − | − | 42 5 |
| IQ Impairment | Borderline | Borderline | Moderate | 99 5 |
| Compromised adaptive functioning | Mild | On average | Mild | 99 5 |
| Genotype | c.15061C=/>T, p.R5021 * | c.13450C=/>T, p.R4484 * | c.3596_3597=/delTC, p.L1199Hfs *7 | |
| Leukocytes mosaicism (%) | 32 | 68 | 40 |
| Cognitive Profile | GM13-3816, c.15061C=/>T, p.R5021X | KB450, c.13450C=/>T (p.R4484X) | KB369, c.3596_3597=/Del (p.L1199HfsX7) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cognitive Profile | ||||||||||||||
| WISC III/IV | TIQ 87 | VCI 108 | PRI 87 | WMI 82 | PSI 76 | TIQ 84 | VCI 76 | PRI 87 | WMI 91 | PSI 74 | TIQ * 46 | VIQ * 58 | PIQ * 46 | |
| Adaptive Behavior | ||||||||||||||
| ABAS II | GAC 65 | CON 65 | SO 81 | PR 68 | GAC 115 | CON 112 | SO 108 | PR 120 | GAC 68 | CON 72 | SO 88 | PR 58 | ||
| Learning Abilities | ||||||||||||||
| Reading skills: MT-2 task | Speed: <2 s.d., Accuracy: <2 s.d. | Speed: <1 s.d., Accuracy: <1 s.d. | Not measurable | |||||||||||
| Writing skills: BVSCO-2 task | Accuracy: <2 s.d. | Accuracy: <2 s.d. | Not measurable | |||||||||||
| Reading comprehension skills: MT-2 task | Accuracy: <2 s.d. | Accuracy: <1 s.d. | Not measurable | |||||||||||
| Psychopathological profile | ||||||||||||||
| K-SADS and psychiatric evaluation |
|
|
| |||||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lepri, F.R.; Cocciadiferro, D.; Augello, B.; Alfieri, P.; Pes, V.; Vancini, A.; Caciolo, C.; Squeo, G.M.; Malerba, N.; Adipietro, I.; et al. Clinical and Neurobehavioral Features of Three Novel Kabuki Syndrome Patients with Mosaic KMT2D Mutations and a Review of Literature. Int. J. Mol. Sci. 2018, 19, 82. https://doi.org/10.3390/ijms19010082
Lepri FR, Cocciadiferro D, Augello B, Alfieri P, Pes V, Vancini A, Caciolo C, Squeo GM, Malerba N, Adipietro I, et al. Clinical and Neurobehavioral Features of Three Novel Kabuki Syndrome Patients with Mosaic KMT2D Mutations and a Review of Literature. International Journal of Molecular Sciences. 2018; 19(1):82. https://doi.org/10.3390/ijms19010082
Chicago/Turabian StyleLepri, Francesca Romana, Dario Cocciadiferro, Bartolomeo Augello, Paolo Alfieri, Valentina Pes, Alessandra Vancini, Cristina Caciolo, Gabriella Maria Squeo, Natascia Malerba, Iolanda Adipietro, and et al. 2018. "Clinical and Neurobehavioral Features of Three Novel Kabuki Syndrome Patients with Mosaic KMT2D Mutations and a Review of Literature" International Journal of Molecular Sciences 19, no. 1: 82. https://doi.org/10.3390/ijms19010082
APA StyleLepri, F. R., Cocciadiferro, D., Augello, B., Alfieri, P., Pes, V., Vancini, A., Caciolo, C., Squeo, G. M., Malerba, N., Adipietro, I., Novelli, A., Sotgiu, S., Gherardi, R., Digilio, M. C., Dallapiccola, B., & Merla, G. (2018). Clinical and Neurobehavioral Features of Three Novel Kabuki Syndrome Patients with Mosaic KMT2D Mutations and a Review of Literature. International Journal of Molecular Sciences, 19(1), 82. https://doi.org/10.3390/ijms19010082