Roles of Mitogen-Activated Protein Kinases in Osteoclast Biology


Abstract
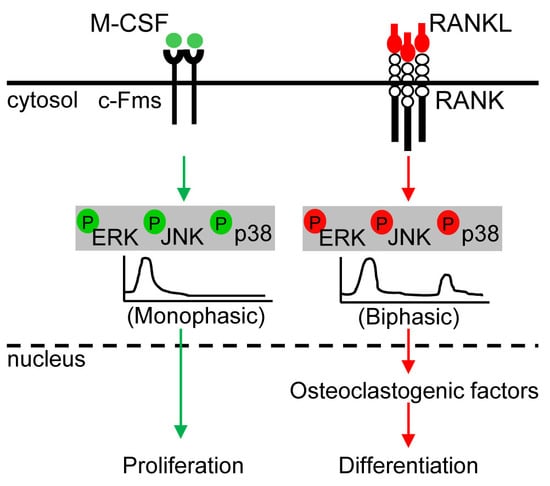
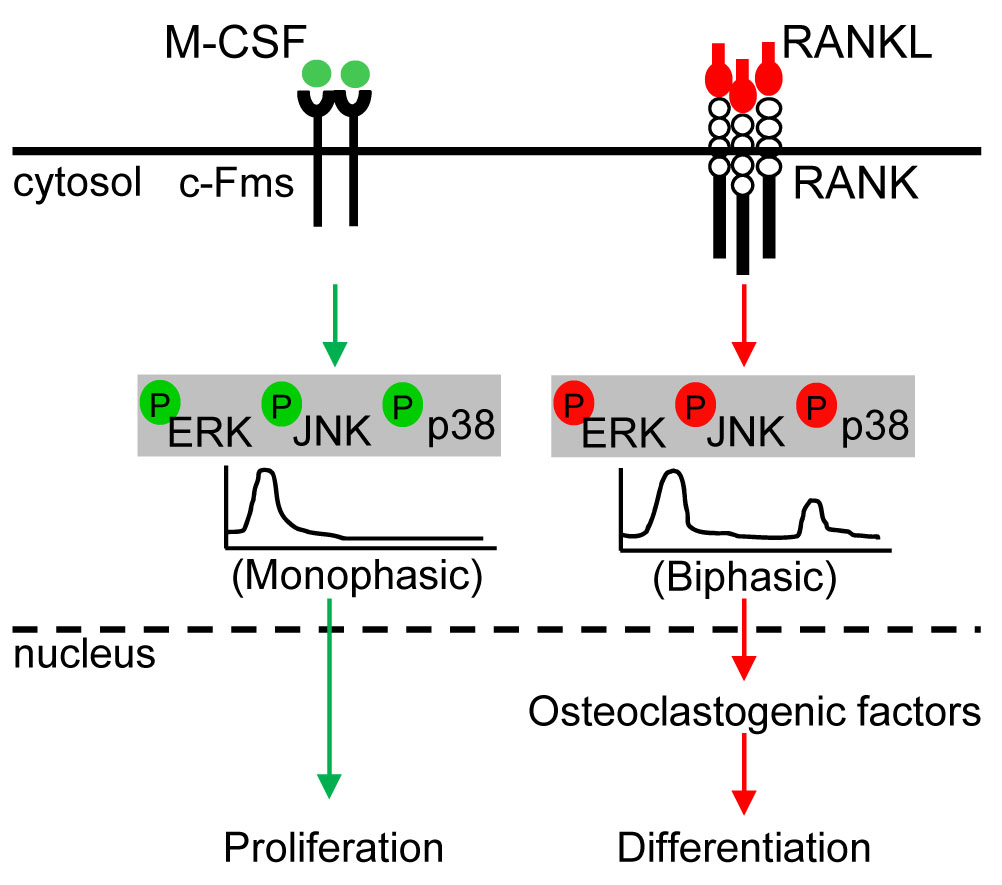
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
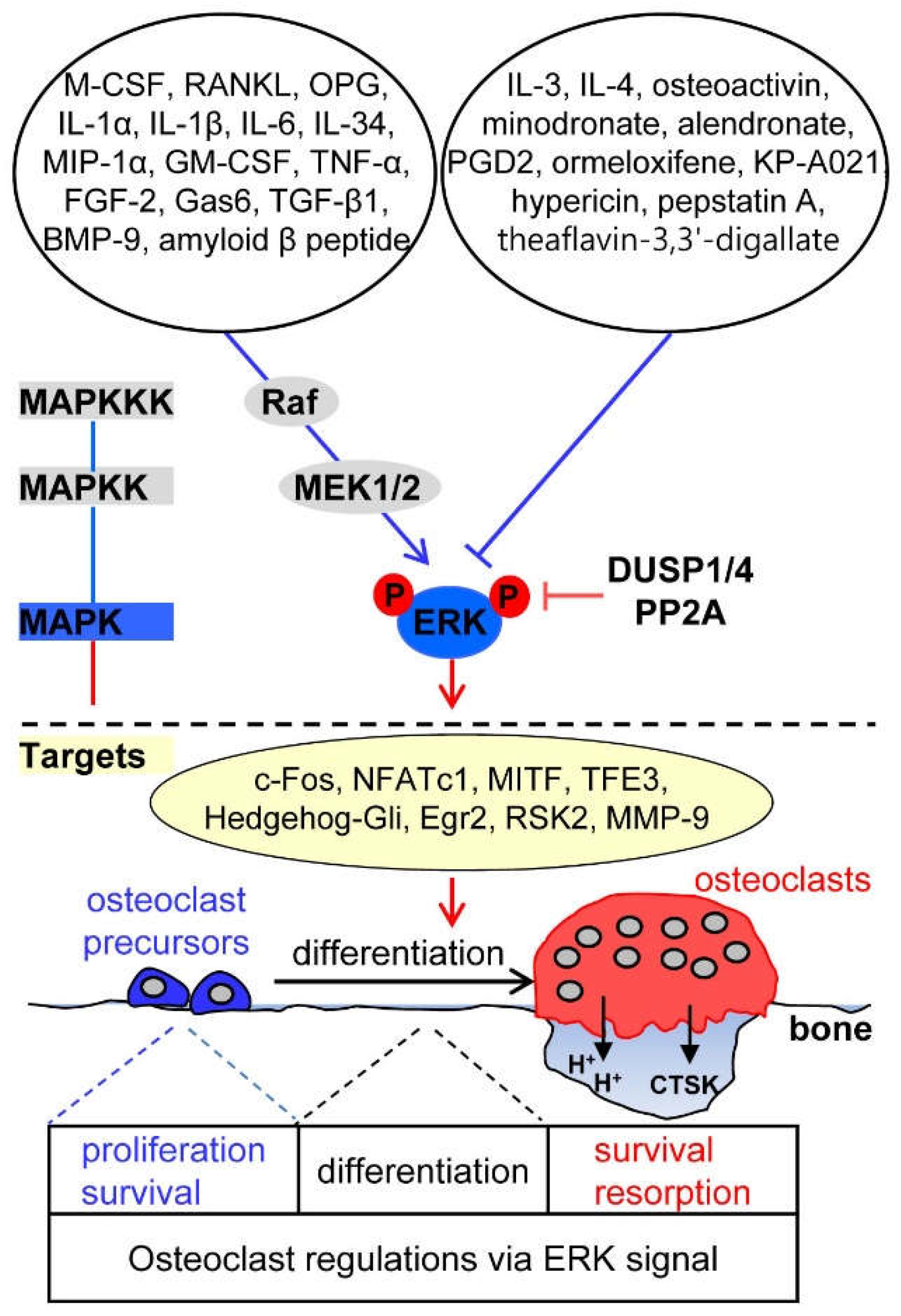
2. ERK Signaling in Osteoclasts
2.1. Upstream Activators of ERK Signaling in Osteoclasts
2.2. Upstream Inhibitors of ERK Signaling in Osteoclasts
2.3. Downstream Targets of ERK Signaling in Osteoclasts
2.4. Phosphatase Regulation of ERK Signaling in Osteoclasts
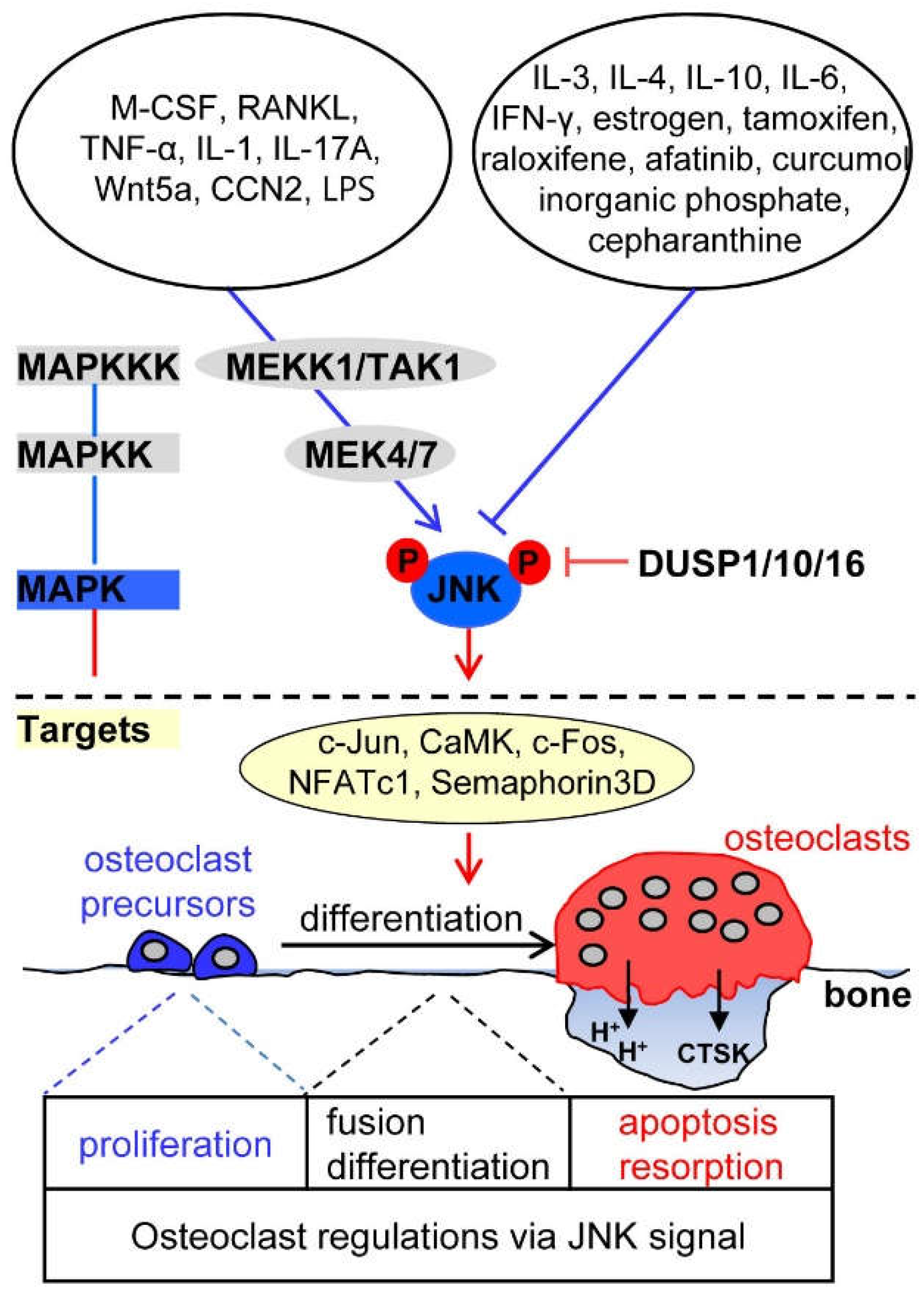
3. JNK Signaling in Osteoclasts
3.1. Upstream Activators of JNK Signaling in Osteoclasts
3.2. Upstream Inhibitors of JNK Signaling in Osteoclasts
3.3. Downstream Targets of JNK Signaling in Osteoclasts
3.4. Phosphatase Regulation of JNK Signaling in Osteoclasts
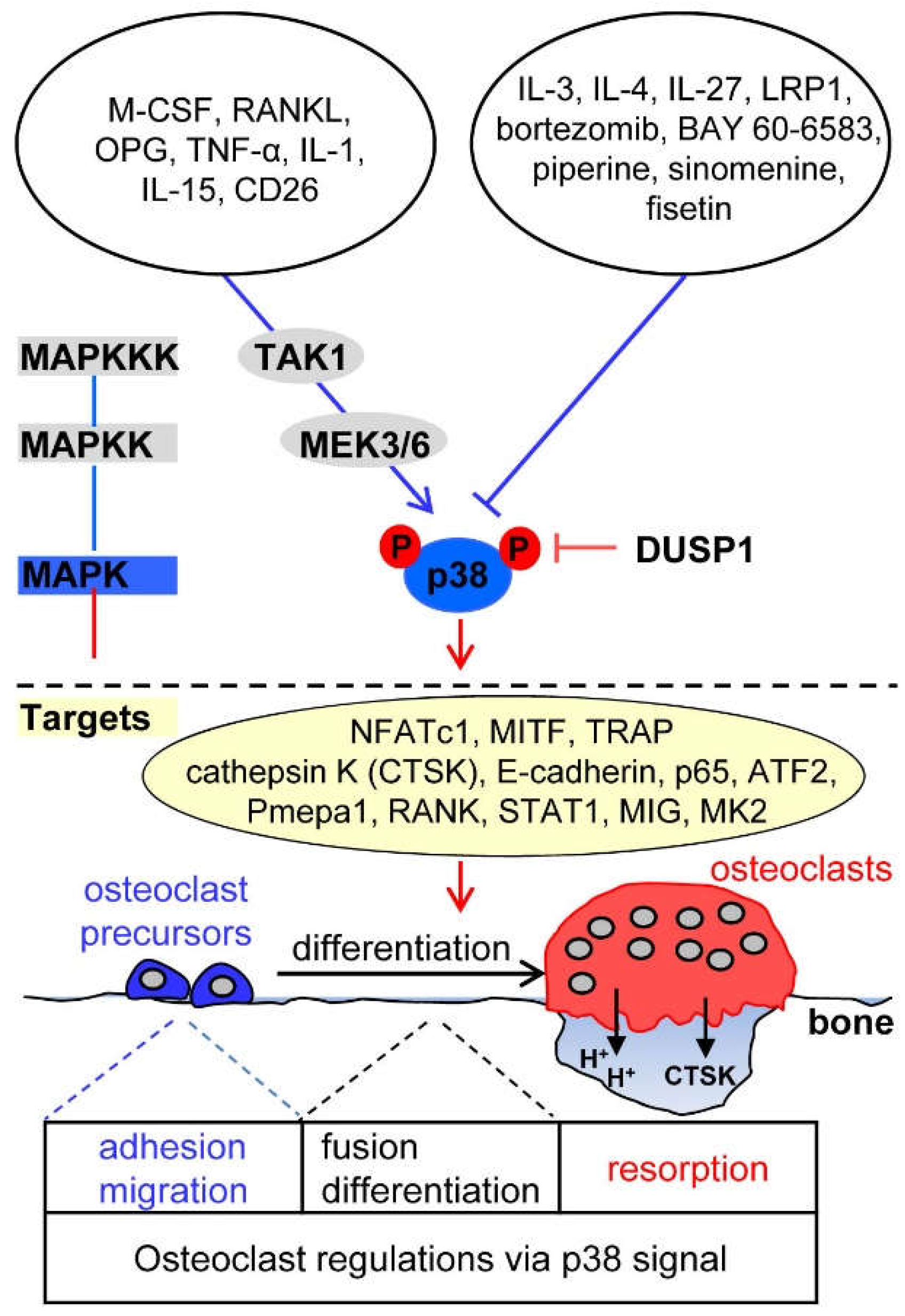
4. p38 Signaling in Osteoclasts
4.1. Upstream Activators of p38 Signaling in Osteoclasts
4.2. Upstream Inhibitors of p38 Signaling in Osteoclasts
4.3. Downstream Targets of p38 in Osteoclasts
4.4. Phosphatase Regulation of p38 Signaling in Osteoclasts
5. Distinct Kinetics of MAPKs and Crosstalk between MAPKs
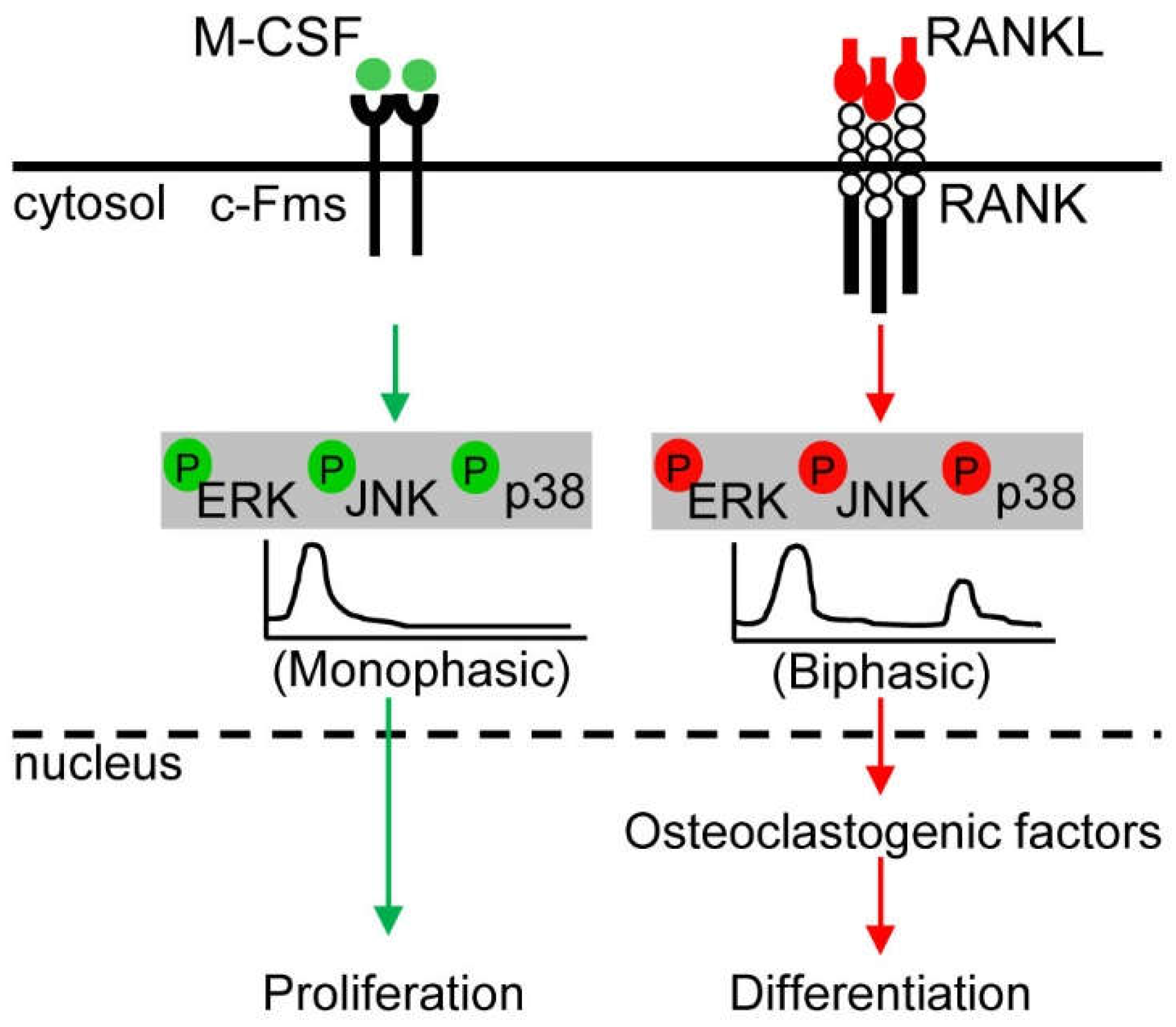
5.1. ERK Kinetics in Osteoclast Metabolism
5.2. JNK Kinetics in Osteoclast Metabolism
5.3. p38 Kinetics in Osteoclast Metabolism
5.4. Crosstalk between ERK and p38 in Osteoclast Metabolism
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Datta, H.K.; Ng, W.F.; Walker, J.A.; Tuck, S.P.; Varanasi, S.S. The cell biology of bone metabolism. J. Clin. Pathol. 2008, 61, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 2007, 13, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J. Regulation of the differentiation and function of osteoclasts. J. Pathol. 2000, 192, 4–13. [Google Scholar] [CrossRef]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, F.P. M-csf, c-fms, and signaling in osteoclasts and their precursors. Ann. N. Y. Acad. Sci. 2006, 1068, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. Rankl-rank signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006, 12, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Chung, Y.H.; Ahn, H.; Kim, H.; Rho, J.; Jeong, D. Selective regulation of mapk signaling mediates rankl-dependent osteoclast differentiation. Int. J. Biol. Sci. 2016, 12, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Matsubara, T.; Tsurukai, T.; Hata, K.; Nishimura, R.; Yoneda, T. Jnk/c-jun signaling mediates an anti-apoptotic effect of rankl in osteoclasts. J. Bone Miner. Res. 2008, 23, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Bohm, C.; Hayer, S.; Kilian, A.; Zaiss, M.M.; Finger, S.; Hess, A.; Engelke, K.; Kollias, G.; Kronke, G.; Zwerina, J.; et al. The alpha-isoform of p38 mapk specifically regulates arthritic bone loss. J. Immunol. 2009, 183, 5938–5947. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian map kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.; Cui, W.; Zhang, Q.; Xu, X.; Mercan, F.; Bennett, A.M.; Vignery, A. Role of mkp-1 in osteoclasts and bone homeostasis. Am. J. Pathol. 2009, 175, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Staser, K.; Rhodes, S.D.; Liu, Y.; Wu, X.; Park, S.J.; Yuan, J.; Yang, X.; Li, X.; Jiang, L.; et al. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS ONE 2011, 6, e24780. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Shirouzu, M.; Kamiya, A.; Hashimoto, K.; Yokoyama, S.; Miyajima, A. Raf/mapk and rapamycin-sensitive pathways mediate the anti-apoptotic function of p21ras in il-3-dependent hematopoietic cells. Oncogene 1997, 15, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of mapks. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Loveridge, C.J.; van‘t Hof, R.J.; Charlesworth, G.; King, A.; Tan, E.H.; Rose, L.; Daroszewska, A.; Prior, A.; Ahmad, I.; Welsh, M.; et al. Analysis of nkx3.1:Cre-driven erk5 deletion reveals a profound spinal deformity which is linked to increased osteoclast activity. Sci. Rep. 2017, 7, 13241. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Gluck, L.; Ebert, R.; Goebeler, M.; Jakob, F.; Schmidt, M. The mek5/erk5 mitogen-activated protein kinase cascade is an effector pathway of bone-sustaining bisphosphonates that regulates osteogenic differentiation and mineralization. Bone 2018, 111, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Amano, S.; Chang, Y.T.; Fukui, Y. Erk5 activation is essential for osteoclast differentiation. PLoS ONE 2015, 10, e0125054. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Colony-stimulating factor-1 receptor. Blood 1990, 75, 1–12. [Google Scholar] [PubMed]
- Mancini, A.; Niedenthal, R.; Joos, H.; Koch, A.; Trouliaris, S.; Niemann, H.; Tamura, T. Identification of a second grb2 binding site in the v-fms tyrosine kinase. Oncogene 1997, 15, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. Opgl is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Theoleyre, S.; Wittrant, Y.; Couillaud, S.; Vusio, P.; Berreur, M.; Dunstan, C.; Blanchard, F.; Redini, F.; Heymann, D. Cellular activity and signaling induced by osteoprotegerin in osteoclasts: Involvement of receptor activator of nuclear factor kappab ligand and mapk. Biochim. Biophys. Acta 2004, 1644, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, X.; Zou, H.; Dai, N.; Yao, L.; Gao, Q.; Liu, W.; Gu, J.; Yuan, Y.; Bian, J.; et al. Osteoprotegerin induces podosome disassembly in osteoclasts through calcium, erk, and p38 mapk signaling pathways. Cytokine 2015, 71, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Choi, B.; Song, D.H.; Song, Y.; Kang, S.W.; Yoon, S.Y.; Kim, S.W.; Lee, H.K.; Chang, E.J. Interleukin-1beta promotes the lc3-mediated secretory function of osteoclast precursors by stimulating the ca(2)(+)-dependent activation of erk. Int. J. Biochem. Cell Biol. 2014, 54, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.H.; Lee, S.E.; Kim, C.W.; Lee, S.H.; Kim, S.W.; Kwack, K.; Walsh, K.; Kim, H.H. Il-1alpha stimulation of osteoclast survival through the pi 3-kinase/akt and erk pathways. J. Biochem. 2002, 131, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Liu, H.; Luo, T.; Liu, D.; Du, J.; Sun, J.; Wang, W.; Han, X.; Yang, K.; Guo, J.; et al. Combination of il-6 and sil-6r differentially regulate varying levels of rankl-induced osteoclastogenesis through nf-kappab, erk and jnk signaling pathways. Sci. Rep. 2017, 7, 41411. [Google Scholar] [CrossRef] [PubMed]
- Baud’huin, M.; Renault, R.; Charrier, C.; Riet, A.; Moreau, A.; Brion, R.; Gouin, F.; Duplomb, L.; Heymann, D. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in rankl-induced osteoclastogenesis. J. Pathol. 2010, 221, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Kato, C.; Isono, A.; Kaneko, J.; Isozaki, M.; Satou, T.; Itoh, T.; Kidera, Y.; Tanimori, Y.; Yanae, M.; et al. Macrophage inflammatory protein-1alpha induces osteoclast formation by activation of the mek/erk/c-fos pathway and inhibition of the p38mapk/irf-3/ifn-beta pathway. J. Cell. Biochem. 2010, 111, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kim, H.S.; Yeon, J.T.; Choi, S.W.; Chun, C.H.; Kwak, H.B.; Oh, J. Gm-csf regulates fusion of mononuclear osteoclasts into bone-resorbing osteoclasts by activating the ras/erk pathway. J. Immunol. 2009, 183, 3390–3399. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Katagiri, M.; Chikazu, D. Osteoclastic bone resorption through receptor tyrosine kinase and extracellular signal-regulated kinase signaling in mature osteoclasts. Mod. Rheumatol. 2004, 14, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Chung, W.J.; Kwak, H.B.; Chung, C.H.; Kwack, K.B.; Lee, Z.H.; Kim, H.H. Tumor necrosis factor-alpha supports the survival of osteoclasts through the activation of akt and erk. J. Biol. Chem. 2001, 276, 49343–49349. [Google Scholar] [CrossRef] [PubMed]
- Houde, N.; Chamoux, E.; Bisson, M.; Roux, S. Transforming growth factor-beta1 (tgf-beta1) induces human osteoclast apoptosis by up-regulating bim. J. Biol. Chem. 2009, 284, 23397–23404. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.; Bisson, M.; Laberge, G.; McManus, S.; Grenier, G.; Faucheux, N.; Roux, S. Bone morphogenetic protein-9 activates smad and erk pathways and supports human osteoclast function and survival in vitro. Cell Signal 2013, 25, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, B.; Zhang, L.; Rong, L. Amyloid beta peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone 2014, 61, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, B.; Teguh, D.; Zhou, L.; Xu, J.; Rong, L. Amyloid beta peptide enhances rankl-induced osteoclast activation through nf-kappab, erk, and calcium oscillation signaling. Int. J. Mol. Sci. 2016, 17, 1683. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seong, S.; Kim, J.H.; Kim, K.; Kim, I.; Jeong, B.C.; Nam, K.I.; Kim, K.K.; Hennighausen, L.; Kim, N. Stat5 is a key transcription factor for il-3-mediated inhibition of rankl-induced osteoclastogenesis. Sci. Rep. 2016, 6, 30977. [Google Scholar] [CrossRef] [PubMed]
- Hirose, J.; Masuda, H.; Tokuyama, N.; Omata, Y.; Matsumoto, T.; Yasui, T.; Kadono, Y.; Hennighausen, L.; Tanaka, S. Bone resorption is regulated by cell-autonomous negative feedback loop of stat5-dusp axis in the osteoclast. J. Exp. Med. 2014, 211, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wang, M.W.; Teitelbaum, S.L.; Ross, F.P. Interleukin-4 reversibly inhibits osteoclastogenesis via inhibition of nf-kappa b and mitogen-activated protein kinase signaling. J. Biol. Chem. 2002, 277, 6622–6630. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Haroun, S.; Parent, J.L.; de Brum-Fernandes, A.J. Prostaglandin d(2) induces apoptosis of human osteoclasts through erk1/2 and akt signaling pathways. Bone 2014, 60, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.A.; Annis, M.G.; Dong, Z.; Pepin, F.; Hallett, M.; Park, M.; Siegel, P.M. Adam10 releases a soluble form of the gpnmb/osteoactivin extracellular domain with angiogenic properties. PLoS ONE 2010, 5, e12093. [Google Scholar] [CrossRef] [PubMed]
- Sondag, G.R.; Mbimba, T.S.; Moussa, F.M.; Novak, K.; Yu, B.; Jaber, F.A.; Abdelmagid, S.M.; Geldenhuys, W.J.; Safadi, F.F. Osteoactivin inhibition of osteoclastogenesis is mediated through cd44-erk signaling. Exp. Mol. Med. 2016, 48, e257. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.J.; Gordon, S.; Benford, H.L.; Coxon, F.P.; Luckman, S.P.; Monkkonen, J.; Frith, J.C. Cellular and molecular mechanisms of action of bisphosphonates. Cancer 2000, 88, 2961–2978. [Google Scholar] [CrossRef]
- Tsubaki, M.; Komai, M.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Takeda, T.; Ogawa, N.; Mashimo, K.; Fujiwara, D.; et al. Nitrogen-containing bisphosphonates inhibit rankl- and m-csf-induced osteoclast formation through the inhibition of erk1/2 and akt activation. J. Biomed. Sci. 2014, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Misra, N.C.; Nigam, P.K.; Gupta, R.; Agarwal, A.K.; Kamboj, V.P. Centchroman--a non-steroidal anti-cancer agent for advanced breast cancer: Phase-ii study. Int. J. Cancer 1989, 43, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Kharkwal, G.; Chandra, V.; Fatima, I.; Dwivedi, A. Ormeloxifene inhibits osteoclast differentiation in parallel to downregulating rankl-induced ros generation and suppressing the activation of erk and jnk in murine raw264.7 cells. J. Mol. Endocrinol. 2012, 48, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.H.; Wang, Y. Recent researches in triazole compounds as medicinal drugs. Curr. Med. Chem. 2012, 19, 239–280. [Google Scholar] [CrossRef] [PubMed]
- Ihn, H.J.; Lee, D.; Lee, T.; Shin, H.I.; Bae, Y.C.; Kim, S.H.; Park, E.K. The 1,2,3-triazole derivative kp-a021 suppresses osteoclast differentiation and function by inhibiting rankl-mediated mek-erk signaling pathway. Exp. Biol. Med. (Maywood) 2015, 240, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Zhai, Z.; Li, H.; Liu, X.; Qu, X.; Li, X.; Fan, Q.; Tang, T.; Qin, A.; Dai, K. Hypericin suppresses osteoclast formation and wear particle-induced osteolysis via modulating erk signalling pathway. Biochem. Pharmacol. 2014, 90, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ping, Z.; Gan, M.; Tao, Y.; Wang, L.; Shi, J.; Wu, X.; Zhang, W.; Yang, H.; Xu, Y.; et al. Theaflavin-3,3′-digallate represses osteoclastogenesis and prevents wear debris-induced osteolysis via suppression of erk pathway. Acta Biomater. 2017, 48, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okamoto, K.; Iwamoto, T.; Sakai, E.; Kanaoka, K.; Hu, J.P.; Shibata, M.; Hotokezaka, H.; Nishishita, K.; Mizuno, A.; et al. Pepstatin a, an aspartic proteinase inhibitor, suppresses rankl-induced osteoclast differentiation. J. Biochem. 2006, 139, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Amcheslavsky, A.; Bar-Shavit, Z. Toll-like receptor 9 ligand blocks osteoclast differentiation through induction of phosphatase. J. Bone Miner. Res. 2007, 22, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Matsuo, K. Signalling in osteoclasts and the role of fos/ap1 proteins. Ann. Rheum. Dis. 2003, 62 Suppl. 2, ii83–85. [Google Scholar] [CrossRef]
- Weilbaecher, K.N.; Motyckova, G.; Huber, W.E.; Takemoto, C.M.; Hemesath, T.J.; Xu, Y.; Hershey, C.L.; Dowland, N.R.; Wells, A.G.; Fisher, D.E. Linkage of m-csf signaling to mitf, tfe3, and the osteoclast defect in mitf(mi/mi) mice. Mol. Cell 2001, 8, 749–758. [Google Scholar] [CrossRef]
- Li, X.; Jie, Q.; Zhang, H.; Zhao, Y.; Lin, Y.; Du, J.; Shi, J.; Wang, L.; Guo, K.; Li, Y.; et al. Disturbed mek/erk signaling increases osteoclast activity via the hedgehog-gli pathway in postmenopausal osteoporosis. Prog. Biophys. Mol. Biol. 2016, 122, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.W.; Ruan, M.M.; Oursler, M.J. Novel pro-survival functions of the kruppel-like transcription factor egr2 in promotion of macrophage colony-stimulating factor-mediated osteoclast survival downstream of the mek/erk pathway. J. Biol. Chem. 2008, 283, 8055–8064. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Iorio, C.; Yan, K.; Yang, H.; Takeshita, S.; Kang, S.; Neel, B.G.; Yang, W. A erk/rsk-mediated negative feedback loop regulates m-csf-evoked pi3k/akt activation in macrophages. FASEB J. 2018, 32, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Engsig, M.T.; Chen, Q.J.; Vu, T.H.; Pedersen, A.C.; Therkidsen, B.; Lund, L.R.; Henriksen, K.; Lenhard, T.; Foged, N.T.; Werb, Z.; et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J. Cell Biol. 2000, 151, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, K.; Nishimura, R.; Senn, J.; Youssef, R.F.; London, S.D.; Reddy, S.V. Rank ligand signaling modulates the matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp. Cell Res. 2007, 313, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Caunt, C.J.; Keyse, S.M. Dual-specificity map kinase phosphatases (mkps): Shaping the outcome of map kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Akira, S. Functional roles of stat family proteins: Lessons from knockout mice. Stem. Cells 1999, 17, 138–146. [Google Scholar] [CrossRef] [PubMed]
- David, J.P.; Sabapathy, K.; Hoffmann, O.; Idarraga, M.H.; Wagner, E.F. Jnk1 modulates osteoclastogenesis through both c-jun phosphorylation-dependent and -independent mechanisms. J. Cell Sci. 2002, 115, 4317–4325. [Google Scholar] [CrossRef] [PubMed]
- Otero, J.E.; Dai, S.; Foglia, D.; Alhawagri, M.; Vacher, J.; Pasparakis, M.; Abu-Amer, Y. Defective osteoclastogenesis by ikkbeta-null precursors is a result of receptor activator of nf-kappab ligand (rankl)-induced jnk-dependent apoptosis and impaired differentiation. J. Biol. Chem. 2008, 283, 24546–24553. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.J.; Ha, J.; Huang, H.; Kim, H.J.; Woo, J.H.; Lee, Y.; Lee, Z.H.; Kim, J.H.; Kim, H.H. The jnk-dependent camk pathway restrains the reversion of committed cells during osteoclast differentiation. J. Cell Sci. 2008, 121, 2555–2564. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Cong, Q.; Li, P.; Ma, G.; Guo, X.; Yeh, J.; Xie, M.; Schneider, M.D.; Liu, H.; Li, B. Ablation of tak1 in osteoclast progenitor leads to defects in skeletal growth and bone remodeling in mice. Sci. Rep. 2014, 4, 7158. [Google Scholar] [CrossRef] [PubMed]
- Stanley, E.R.; Chitu, V. Csf-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef] [PubMed]
- Gangoiti, P.; Granado, M.H.; Wang, S.W.; Kong, J.Y.; Steinbrecher, U.P.; Gomez-Munoz, A. Ceramide 1-phosphate stimulates macrophage proliferation through activation of the pi3-kinase/pkb, jnk and erk1/2 pathways. Cell Signal. 2008, 20, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Sang, C.; Zhang, J.; Zhang, Y.; Chen, F.; Cao, X.; Guo, L. Tnf-alpha promotes osteoclastogenesis through jnk signaling-dependent induction of semaphorin3d expression in estrogen-deficiency induced osteoporosis. J. Cell. Physiol. 2017, 232, 3396–3408. [Google Scholar] [CrossRef] [PubMed]
- Hotokezaka, H.; Sakai, E.; Ohara, N.; Hotokezaka, Y.; Gonzales, C.; Matsuo, K.; Fujimura, Y.; Yoshida, N.; Nakayama, K. Molecular analysis of rankl-independent cell fusion of osteoclast-like cells induced by tnf-alpha, lipopolysaccharide, or peptidoglycan. J. Cell Biochem. 2007, 101, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Akiyama, S.; Tsurukai, T.; Okahashi, N.; Kobayashi, K.; Udagawa, N.; Nishihara, T.; Takahashi, N.; Suda, T. Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J. Immunol. 1999, 163, 434–442. [Google Scholar] [PubMed]
- Ke, D.; Fu, X.; Xue, Y.; Wu, H.; Zhang, Y.; Chen, X.; Hou, J. Il-17a regulates the autophagic activity of osteoclast precursors through rankl-jnk1 signaling during osteoclastogenesis in vitro. Biochem. Biophys. Res. Commun. 2018, 497, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Kobayashi, Y.; Udagawa, N.; Uehara, S.; Ishihara, A.; Mizoguchi, T.; Kikuchi, Y.; Takada, I.; Kato, S.; Kani, S.; et al. Wnt5a-ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 2012, 18, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, E.; Kubota, S.; Khattab, H.M.; Nishida, T.; Takigawa, M. Ccn2 enhances rankl-induced osteoclast differentiation via direct binding to rank and opg. Bone 2015, 73, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Noh, A.L.; Kang, J.H.; Sim, J.S.; Lee, D.S.; Yim, M. Peroxiredoxin ii negatively regulates lipopolysaccharide-induced osteoclast formation and bone loss via jnk and stat3. Antioxid. Redox Signal. 2015, 22, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Khapli, S.M.; Mangashetti, L.S.; Yogesha, S.D.; Wani, M.R. Il-3 acts directly on osteoclast precursors and irreversibly inhibits receptor activator of nf-kappa b ligand-induced osteoclast differentiation by diverting the cells to macrophage lineage. J. Immunol. 2003, 171, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Khapli, S.M.; Tomar, G.B.; Barhanpurkar, A.P.; Gupta, N.; Yogesha, S.D.; Pote, S.T.; Wani, M.R. Irreversible inhibition of rank expression as a possible mechanism for il-3 inhibition of rankl-induced osteoclastogenesis. Biochem. Biophys. Res. Commun. 2010, 399, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.G.; Sugiyama, E.; Shinoda, K.; Taki, H.; Hounoki, H.; Abdel-Aziz, H.O.; Maruyama, M.; Kobayashi, M.; Ogawa, H.; Miyahara, T. Interleukin-10 inhibits rankl-mediated expression of nfatc1 in part via suppression of c-fos and c-jun in raw264.7 cells and mouse bone marrow cells. Bone 2007, 41, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Yoshitake, F.; Itoh, S.; Narita, H.; Ishihara, K.; Ebisu, S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of nf-kappab signaling pathways. J. Biol. Chem. 2008, 283, 11535–11540. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between rankl and ifn-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Maeda, H.; Roccisana, J.L.; Kawanabe, N.; Reddy, S.V. Cytokineregulation and the signaling mechanism of osteoclast inhibitory peptide-1 (oip-1/hsca) to inhibit osteoclast formation. J. Bone Miner. Res. 2003, 18, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Shevde, N.K.; Bendixen, A.C.; Dienger, K.M.; Pike, J.W. Estrogens suppress rank ligand-induced osteoclast differentiation via a stromal cell independent mechanism involving c-jun repression. Proc. Natl. Acad. Sci. USA 2000, 97, 7829–7834. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Toraldo, G.; Weitzmann, M.N.; Cenci, S.; Ross, F.P.; Pacifici, R. Estrogen decreases osteoclast formation by down-regulating receptor activator of nf-kappa b ligand (rankl)-induced jnk activation. J. Biol. Chem. 2001, 276, 8836–8840. [Google Scholar] [CrossRef] [PubMed]
- Wind, S.; Schnell, D.; Ebner, T.; Freiwald, M.; Stopfer, P. Clinical pharmacokinetics and pharmacodynamics of afatinib. Clin. Pharmacokinet. 2017, 56, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Ihn, H.J.; Kim, J.A.; Bae, Y.C.; Shin, H.I.; Baek, M.C.; Park, E.K. Afatinib ameliorates osteoclast differentiation and function through downregulation of rank signaling pathways. BMB Rep. 2017, 50, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Rogosnitzky, M.; Danks, R. Therapeutic potential of the biscoclaurine alkaloid, cepharanthine, for a range of clinical conditions. Pharmacol. Rep. 2011, 63, 337–347. [Google Scholar] [CrossRef]
- Zhou, C.H.; Meng, J.H.; Yang, Y.T.; Hu, B.; Hong, J.Q.; Lv, Z.T.; Chen, K.; Heng, B.C.; Jiang, G.Y.; Zhu, J.; et al. Cepharanthine prevents estrogen deficiency-induced bone loss by inhibiting bone resorption. Front. Pharmacol. 2018, 9, 210. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, X.; Lv, C.; Yi, X.; Zhang, Y.; Xue, M.; He, S.; Zhu, G.; Wang, H. Curcumol suppresses rankl-induced osteoclast formation by attenuating the jnk signaling pathway. Biochem. Biophys. Res. Commun. 2014, 447, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Vascular calcification: In vitro evidence for the role of inorganic phosphate. J. Am. Soc. Nephrol. 2003, 14, S300–S304. [Google Scholar] [CrossRef]
- Mozar, A.; Haren, N.; Chasseraud, M.; Louvet, L.; Maziere, C.; Wattel, A.; Mentaverri, R.; Morliere, P.; Kamel, S.; Brazier, M.; et al. High extracellular inorganic phosphate concentration inhibits rank-rankl signaling in osteoclast-like cells. J. Cell. Physiol. 2008, 215, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, A.E.; Wang, Z.Q.; Cecchini, M.G.; Hofstetter, W.; Felix, R.; Fleisch, H.A.; Wagner, E.F. C-fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994, 266, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Vaira, S.; Alhawagri, M.; Anwisye, I.; Kitaura, H.; Faccio, R.; Novack, D.V. Rela/p65 promotes osteoclast differentiation by blocking a rankl-induced apoptotic jnk pathway in mice. J. Clin. Investig. 2008, 118, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Cong, Q.; Jia, H.; Li, P.; Qiu, S.; Yeh, J.; Wang, Y.; Zhang, Z.L.; Ao, J.; Li, B.; Liu, H. P38alpha mapk regulates proliferation and differentiation of osteoclast progenitors and bone remodeling in an aging-dependent manner. Sci. Rep. 2017, 7, 45964. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Sudo, T.; Saito, T.; Osada, H.; Tsujimoto, M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of nf-kappa b ligand (rankl). J. Biol. Chem. 2000, 275, 31155–31161. [Google Scholar] [CrossRef] [PubMed]
- Lamothe, B.; Lai, Y.; Xie, M.; Schneider, M.D.; Darnay, B.G. Tak1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol. Cell. Biol. 2013, 33, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ryu, J.; Ha, J.; Chang, E.J.; Kim, H.J.; Kim, H.M.; Kitamura, T.; Lee, Z.H.; Kim, H.H. Osteoclast differentiation requires tak1 and mkk6 for nfatc1 induction and nf-kappab transactivation by rankl. Cell Death Differ. 2006, 13, 1879–1891. [Google Scholar] [CrossRef] [PubMed]
- Boyle, D.L.; Hammaker, D.; Edgar, M.; Zaiss, M.M.; Teufel, S.; David, J.P.; Schett, G.; Firestein, G.S. Differential roles of mapk kinases mkk3 and mkk6 in osteoclastogenesis and bone loss. PLoS ONE 2014, 9, e84818. [Google Scholar] [CrossRef]
- Zhu, N.; Cui, J.; Qiao, C.; Li, Y.; Ma, Y.; Zhang, J.; Shen, B. Camp modulates macrophage development by suppressing m-csf-induced mapks activation. Cell Mol. Immunol. 2008, 5, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Jang, Y.; Choi, B.; Song, D.H.; Lee, E.J.; Kim, S.M.; Song, Y.; Kang, S.W.; Yoon, S.Y.; Chang, E.J. Beclin-1 is required for rankl-induced osteoclast differentiation. J. Cell. Physiol. 2014, 229, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Sudo, T.; Maruyama, M.; Osada, H.; Tsujimoto, M. Activation of p38 mitogen-activated protein kinase is crucial in osteoclastogenesis induced by tumor necrosis factor. FEBS Lett. 2000, 486, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The mechanism of osteoclast differentiation induced by il-1. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Okabe, I.; Kikuchi, T.; Mogi, M.; Takeda, H.; Aino, M.; Kamiya, Y.; Fujimura, T.; Goto, H.; Okada, K.; Hasegawa, Y.; et al. Il-15 and rankl play a synergistically important role in osteoclastogenesis. J. Cell Biochem. 2017, 118, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Suzuki, H.; Madokoro, H.; Hayashi, M.; Morimoto, C.; Sakamoto, M.; Yamada, T. Blockade of cd26 signaling inhibits human osteoclast development. J. Bone Miner. Res. 2014, 29, 2439–2455. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.D.; Park-Min, K.H.; Shen, Z.; Fajardo, R.J.; Goldring, S.R.; McHugh, K.P.; Ivashkiv, L.B. Inhibition of rank expression and osteoclastogenesis by tlrs and ifn-gamma in human osteoclast precursors. J. Immunol. 2009, 183, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Lee, H.Y.; Kim, H.J.; Park, Y.J.; Seo, J.K.; Park, J.S.; Bae, Y.S. Serum amyloid a inhibits rankl-induced osteoclast formation. Exp. Mol. Med. 2015, 47, e194. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Zhao, B.; Triantafyllopoulou, A.; Park-Min, K.H.; Ivashkiv, L.B. Interleukin-27 inhibits human osteoclastogenesis by abrogating rankl-mediated induction of nuclear factor of activated t cells c1 and suppressing proximal rank signaling. Arthritis Rheum. 2010, 62, 402–413. [Google Scholar] [PubMed]
- Lu, D.; Li, J.; Liu, H.; Foxa, G.E.; Weaver, K.; Li, J.; Williams, B.O.; Yang, T. Lrp1 suppresses bone resorption in mice by inhibiting the rankl-stimulated nf-kappab and p38 pathways during osteoclastogenesis. J Bone Miner. Res. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- von Metzler, I.; Krebbel, H.; Hecht, M.; Manz, R.A.; Fleissner, C.; Mieth, M.; Kaiser, M.; Jakob, C.; Sterz, J.; Kleeberg, L.; et al. Bortezomib inhibits human osteoclastogenesis. Leukemia 2007, 21, 2025–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.H.; Oh, J.H.; Lee, N.K. The inactivation of erk1/2, p38 and nf-kb is involved in the down-regulation of osteoclastogenesis and function by a2b adenosine receptor stimulation. Mol. Cells 2017, 40, 752–760. [Google Scholar] [PubMed]
- Choi, S.W.; Son, Y.J.; Yun, J.M.; Kim, S.H. Fisetin inhibits osteoclast differentiation via downregulation of p38 and c-fos-nfatc1 signaling pathways. Evid. Based Complement. Alternat. Med. 2012, 2012, 810563. [Google Scholar] [CrossRef] [PubMed]
- Deepak, V.; Kruger, M.C.; Joubert, A.; Coetzee, M. Piperine alleviates osteoclast formation through the p38/c-fos/nfatc1 signaling axis. Biofactors 2015, 41, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, L.; Hu, Y.; Duan, H.; Li, X.; Tan, S.; Zou, M.; Gu, C.; Zeng, X.; Yu, L.; et al. Sinomenine suppresses osteoclast formation and mycobacterium tuberculosis h37ra-induced bone loss by modulating rankl signaling pathways. PLoS ONE 2013, 8, e74274. [Google Scholar] [CrossRef] [PubMed]
- Mansky, K.C.; Sankar, U.; Han, J.; Ostrowski, M.C. Microphthalmia transcription factor is a target of the p38 mapk pathway in response to receptor activator of nf-kappa b ligand signaling. J. Biol. Chem. 2002, 277, 11077–11083. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential role of p38 mitogen-activated protein kinase in cathepsin k gene expression during osteoclastogenesis through association of nfatc1 and pu.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Udagawa, N.; Itoh, K.; Suda, K.; Murase, Y.; Nishihara, T.; Suda, T.; Takahashi, N. P38 mapk-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology 2002, 143, 3105–3113. [Google Scholar] [CrossRef] [PubMed]
- Funakubo, N.; Xu, X.; Kukita, T.; Nakamura, S.; Miyamoto, H.; Kukita, A. Pmepa1 induced by rankl-p38 mapk pathway has a novel role in osteoclastogenesis. J. Cell. Physiol. 2018, 233, 3105–3118. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.B.; Lee, S.W.; Jin, H.M.; Ha, H.; Lee, S.H.; Takeshita, S.; Tanaka, S.; Kim, H.M.; Kim, H.H.; Lee, Z.H. Monokine induced by interferon-gamma is induced by receptor activator of nuclear factor kappa b ligand and is involved in osteoclast adhesion and migration. Blood 2005, 105, 2963–2969. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.A.; Valerio, M.S.; Gaestel, M.; Kirkwood, K.L. Sexual dimorphism in mapk-activated protein kinase-2 (mk2) regulation of rankl-induced osteoclastogenesis in osteoclast progenitor subpopulations. PLoS ONE 2015, 10, e0125387. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Li, F.; Kirkwood, K.L. Map kinase phosphatase-1 protects against inflammatory bone loss. J. Dent. Res. 2009, 88, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular interpretation of erk signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; Mano, H.; Yuasa, T.; Mori, Y.; Miyazawa, K.; Shiokawa, M.; Nakamaru, Y.; Hiroi, E.; Hiura, K.; Kameda, A.; et al. Estrogen inhibits bone resorption by directly inducing apoptosis of the bone-resorbing osteoclasts. J. Exp. Med. 1997, 186, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.R.; Plotkin, L.I.; Aguirre, J.I.; Han, L.; Jilka, R.L.; Kousteni, S.; Bellido, T.; Manolagas, S.C. Transient versus sustained phosphorylation and nuclear accumulation of erks underlie anti-versus pro-apoptotic effects of estrogens. J. Biol. Chem. 2005, 280, 4632–4638. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Fujiwara, T.; Zhou, J.; Varughese, K.I.; Zhao, H. Lis1 regulates osteoclastogenesis through modulation of m-scf and rankl signaling pathways and cdc42. Int. J. Biol. Sci. 2016, 12, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Hotokezaka, H.; Sakai, E.; Kanaoka, K.; Saito, K.; Matsuo, K.; Kitaura, H.; Yoshida, N.; Nakayama, K. U0126 and pd98059, specific inhibitors of mek, accelerate differentiation of raw264.7 cells into osteoclast-like cells. J. Biol. Chem. 2002, 277, 47366–47372. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Matsumura, S.; Ibaragi, S.; Isowa, S.; Kishimoto, K.; Mese, H.; Nishiyama, A.; Sasaki, A. Specific inhibitor of mek-mediated cross-talk between erk and p38 mapk during differentiation of human osteosarcoma cells. J. Cell Commun. Signal. 2007, 1, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Leitch, J.; Armstrong, C.; Dixon, J.; Cohen, P. Effects of map kinase cascade inhibitors on the mkk5/erk5 pathway. FEBS Lett. 2001, 502, 21–24. [Google Scholar] [CrossRef]
- Jaworowski, A.; Wilson, N.J.; Christy, E.; Byrne, R.; Hamilton, J.A. Roles of the mitogen-activated protein kinase family in macrophage responses to colony stimulating factor-1 addition and withdrawal. J. Biol. Chem. 1999, 274, 15127–15133. [Google Scholar] [CrossRef] [PubMed]
- Valledor, A.F.; Comalada, M.; Xaus, J.; Celada, A. The differential time-course of extracellular-regulated kinase activity correlates with the macrophage response toward proliferation or activation. J. Biol. Chem. 2000, 275, 7403–7409. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, K.L.; Li, F.; Rogers, J.E.; Otremba, J.; Coatney, D.D.; Kreider, J.M.; D’Silva, N.J.; Chakravarty, S.; Dugar, S.; Higgins, L.S.; et al. A p38alpha selective mitogen-activated protein kinase inhibitor prevents periodontal bone loss. J. Pharmacol. Exp. Ther. 2007, 320, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.E.; Li, F.; Coatney, D.D.; Otremba, J.; Kriegl, J.M.; Protter, T.A.; Higgins, L.S.; Medicherla, S.; Kirkwood, K.L. A p38 mitogen-activated protein kinase inhibitor arrests active alveolar bone loss in a rat periodontitis model. J. Periodontol. 2007, 78, 1992–1998. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, K.L.; Rossa, C., Jr. The potential of p38 mapk inhibitors to modulate periodontal infections. Curr. Drug Metab. 2009, 10, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.R.; Dean, J.L. The p38 mapk pathway in rheumatoid arthritis: A. sideways look. Open Rheumatol. J. 2012, 6, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Siegal, G.P. P38 mapk as a potential therapeutic target for inflammatory osteolysis. Adv. Anat. Pathol. 2007, 14, 42–45. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.; Seo, I.; Choi, M.H.; Jeong, D. Roles of Mitogen-Activated Protein Kinases in Osteoclast Biology. Int. J. Mol. Sci. 2018, 19, 3004. https://doi.org/10.3390/ijms19103004
Lee K, Seo I, Choi MH, Jeong D. Roles of Mitogen-Activated Protein Kinases in Osteoclast Biology. International Journal of Molecular Sciences. 2018; 19(10):3004. https://doi.org/10.3390/ijms19103004
Chicago/Turabian StyleLee, Kyunghee, Incheol Seo, Mun Hwan Choi, and Daewon Jeong. 2018. "Roles of Mitogen-Activated Protein Kinases in Osteoclast Biology" International Journal of Molecular Sciences 19, no. 10: 3004. https://doi.org/10.3390/ijms19103004
APA StyleLee, K., Seo, I., Choi, M. H., & Jeong, D. (2018). Roles of Mitogen-Activated Protein Kinases in Osteoclast Biology. International Journal of Molecular Sciences, 19(10), 3004. https://doi.org/10.3390/ijms19103004