Cytotoxicity of Selenium Immunoconjugates against Triple Negative Breast Cancer Cells


Abstract
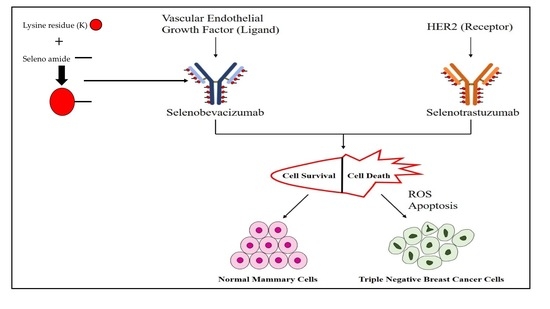
:
1. Introduction
2. Results
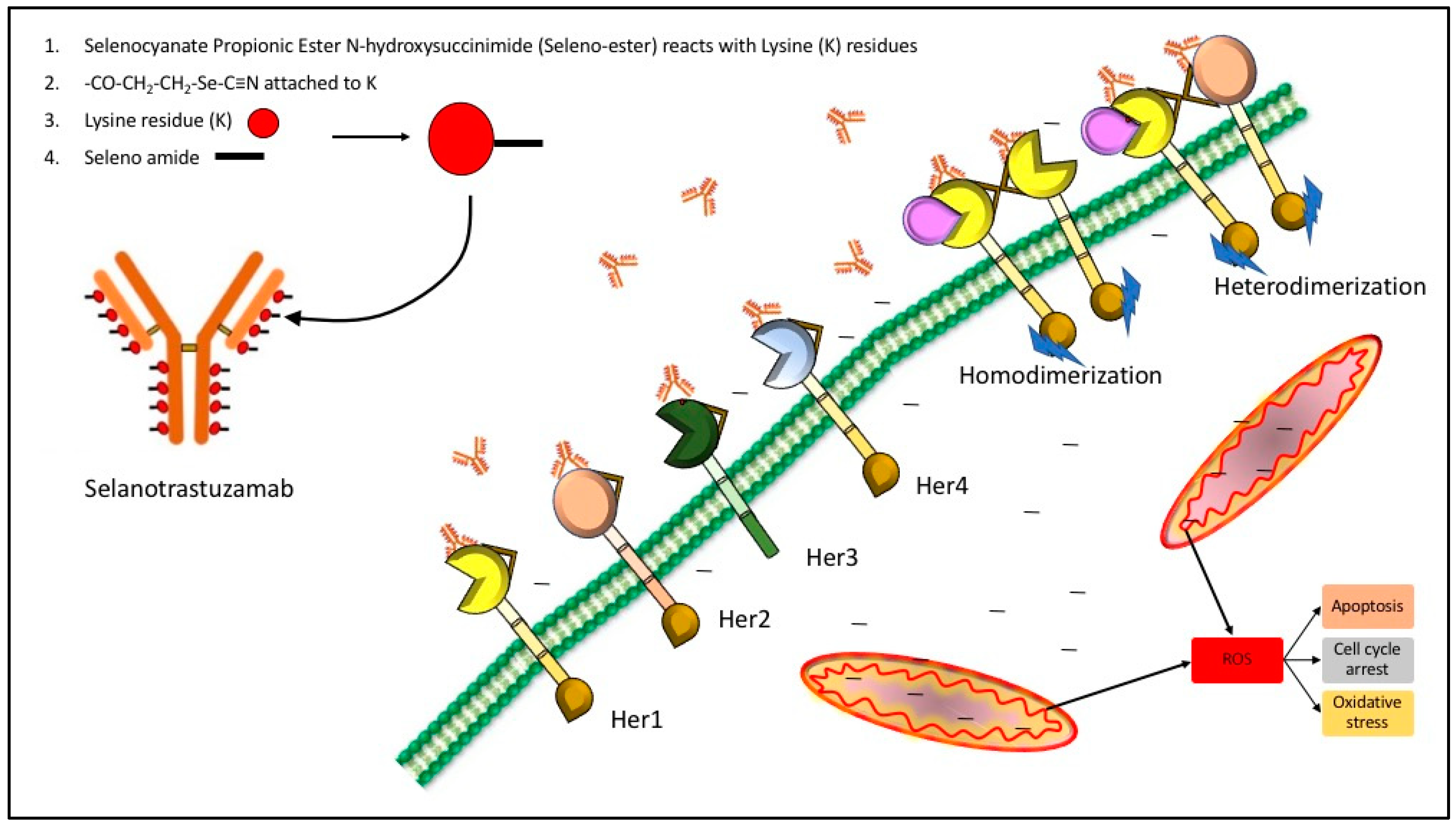
2.1. Analysis of Selenium in Control and Conjugated Antibodies
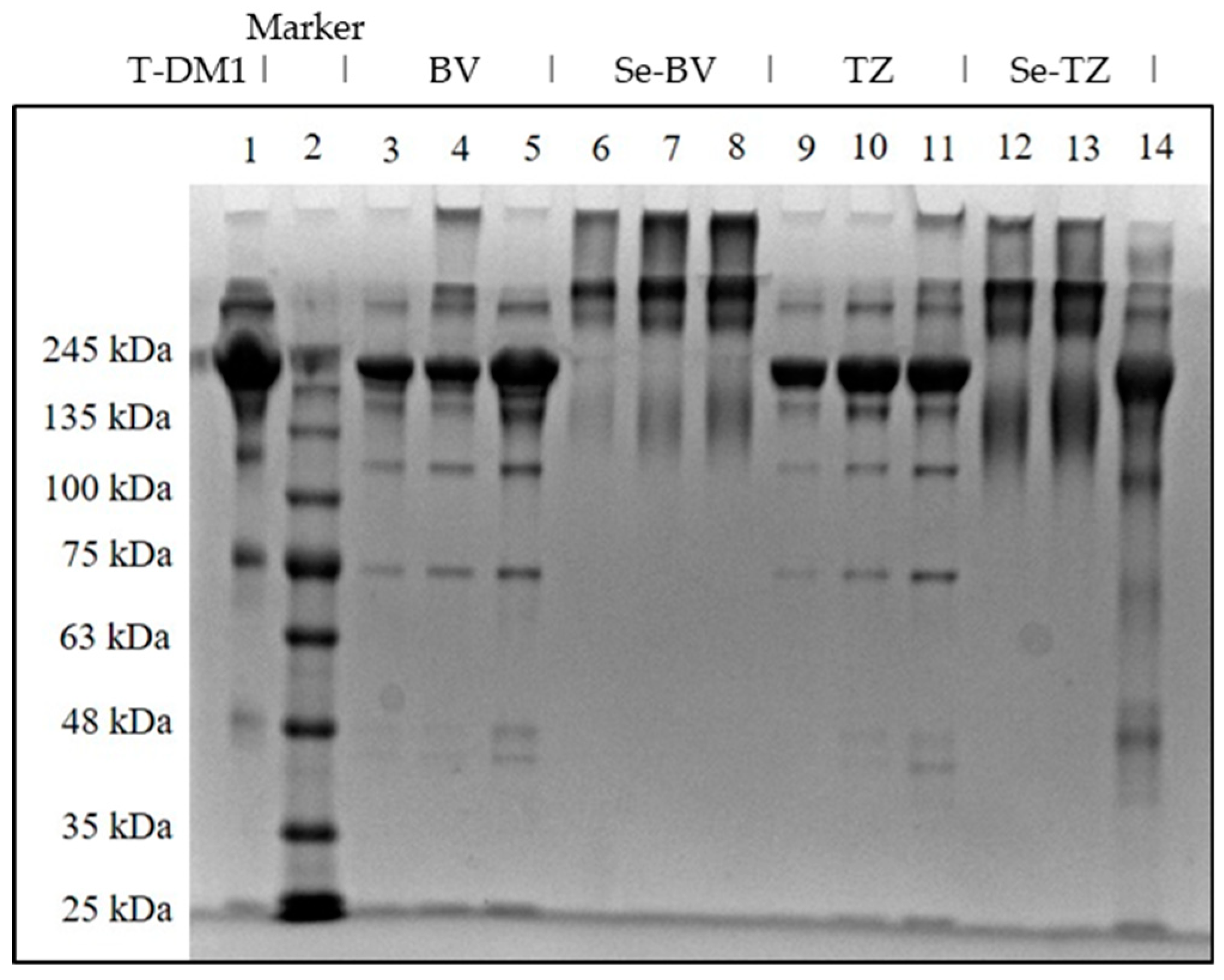
2.2. Homogenity and Stability of Selenium Immunoconjugates
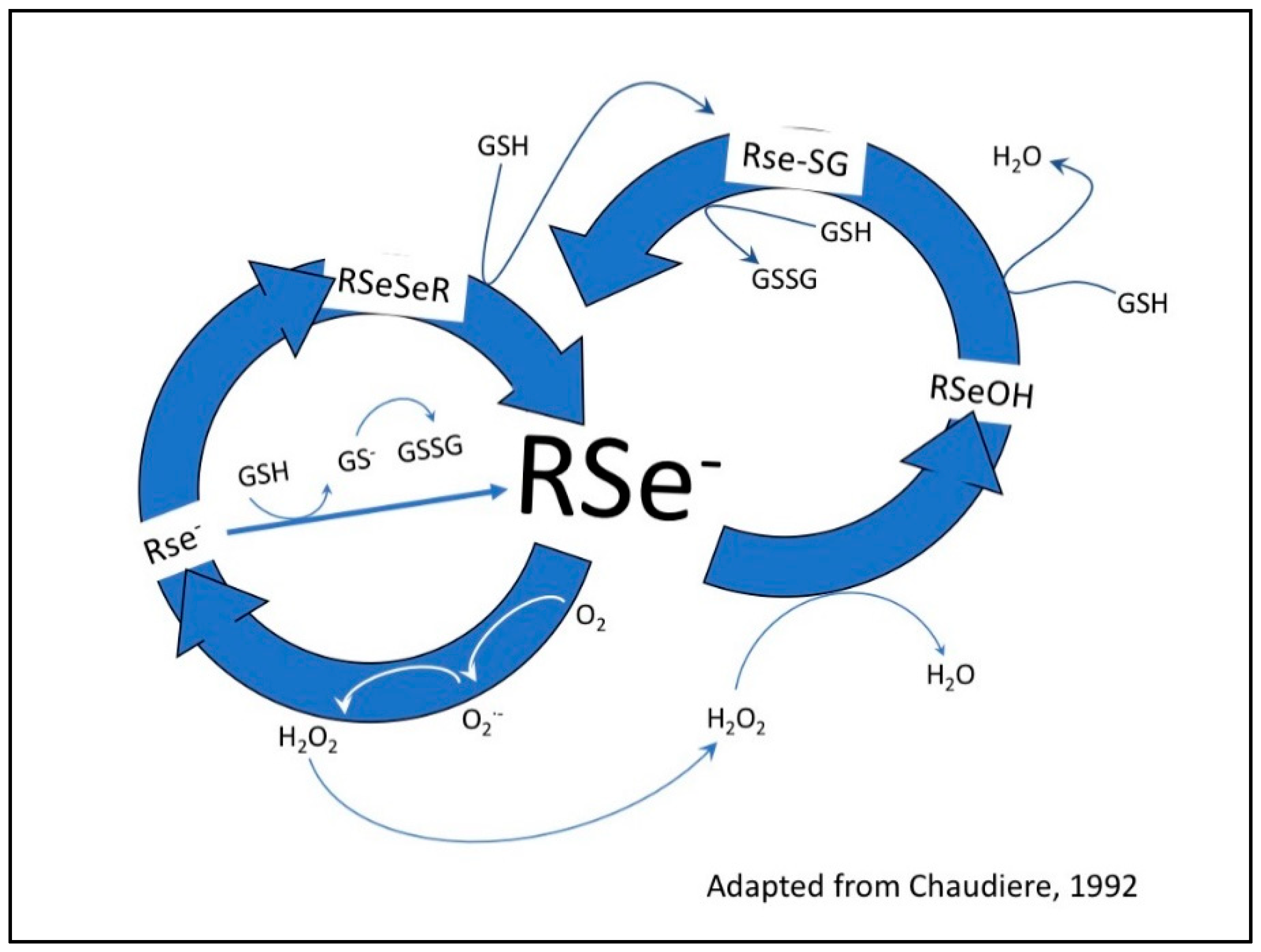
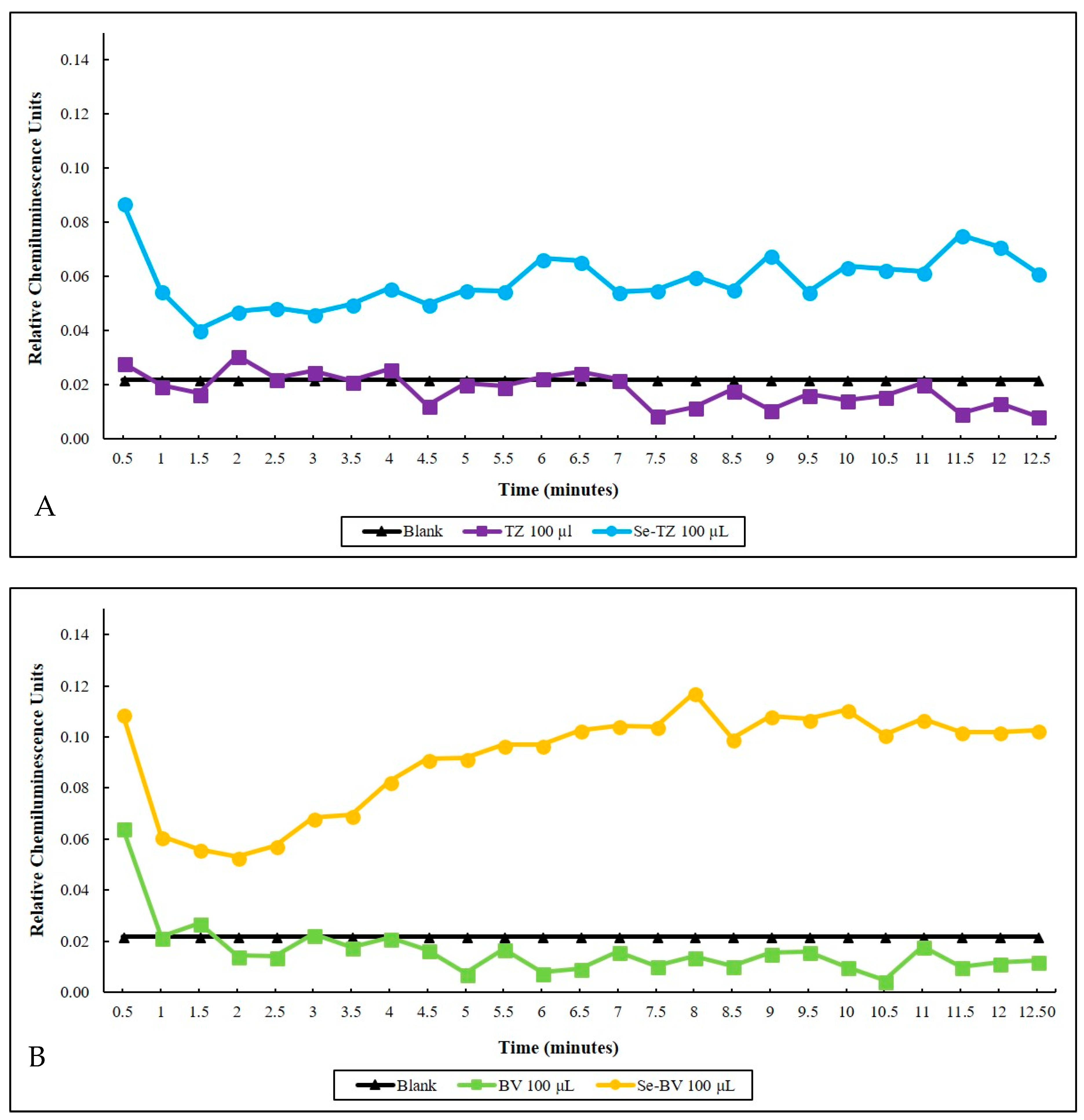
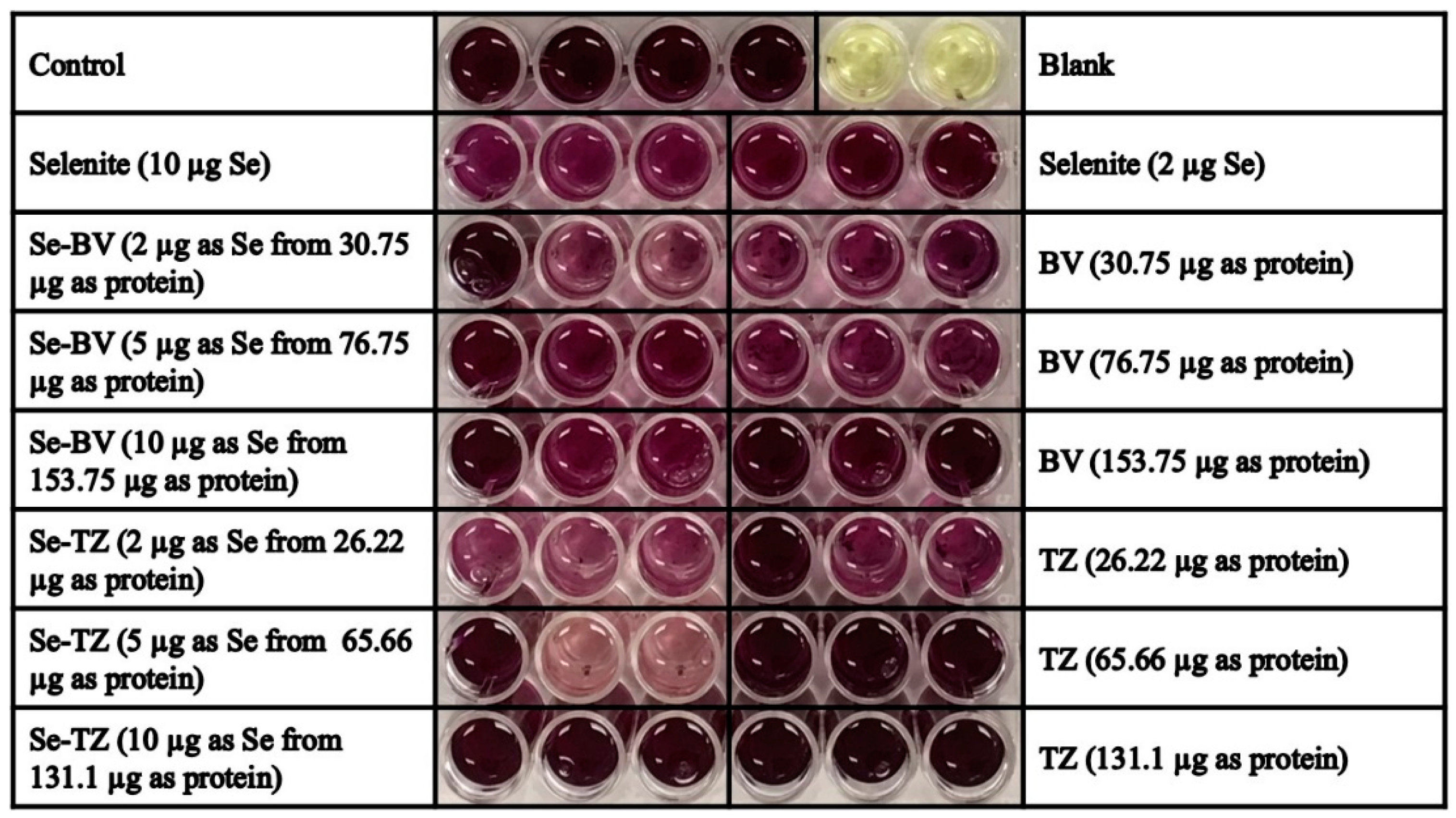
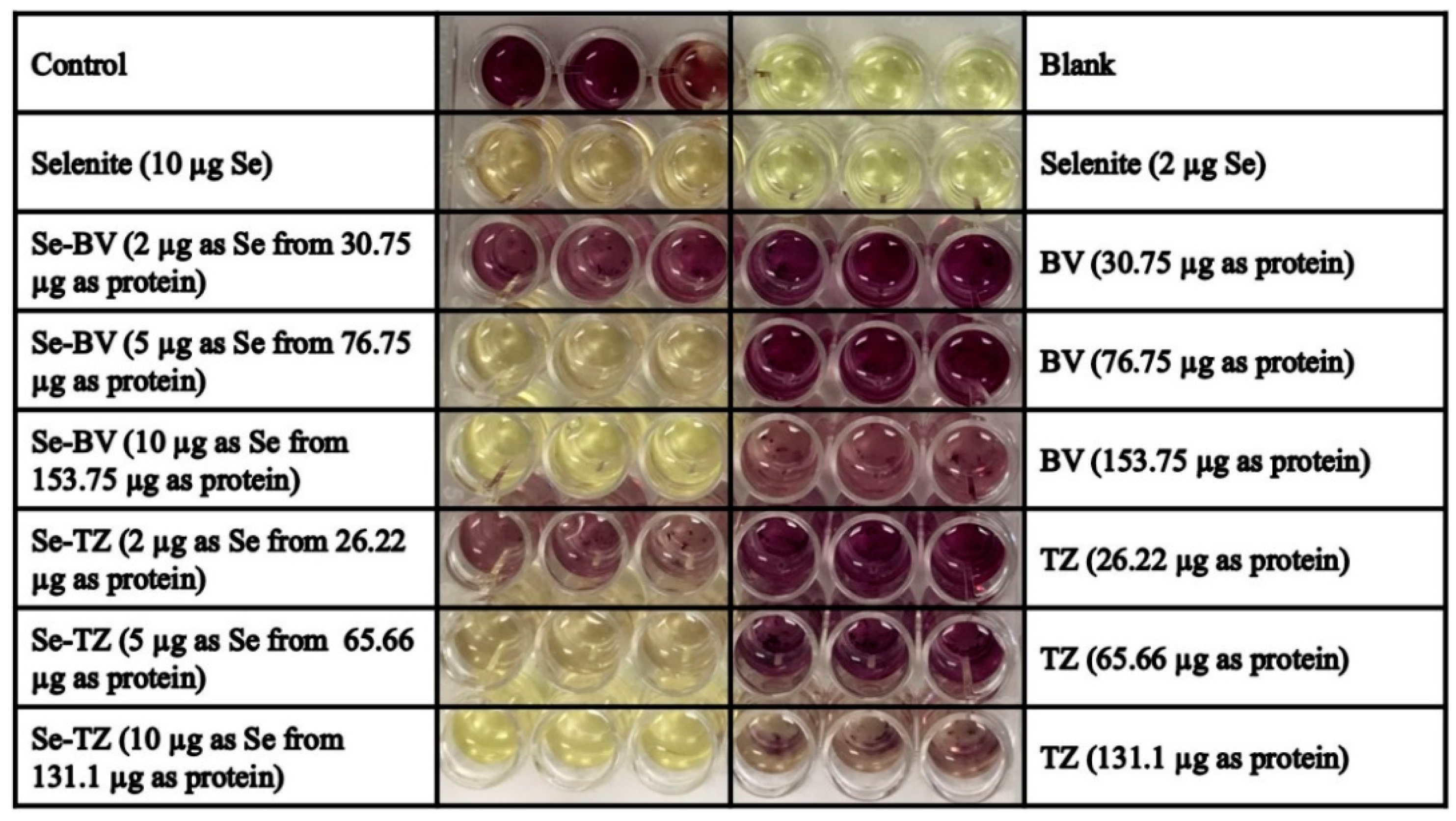
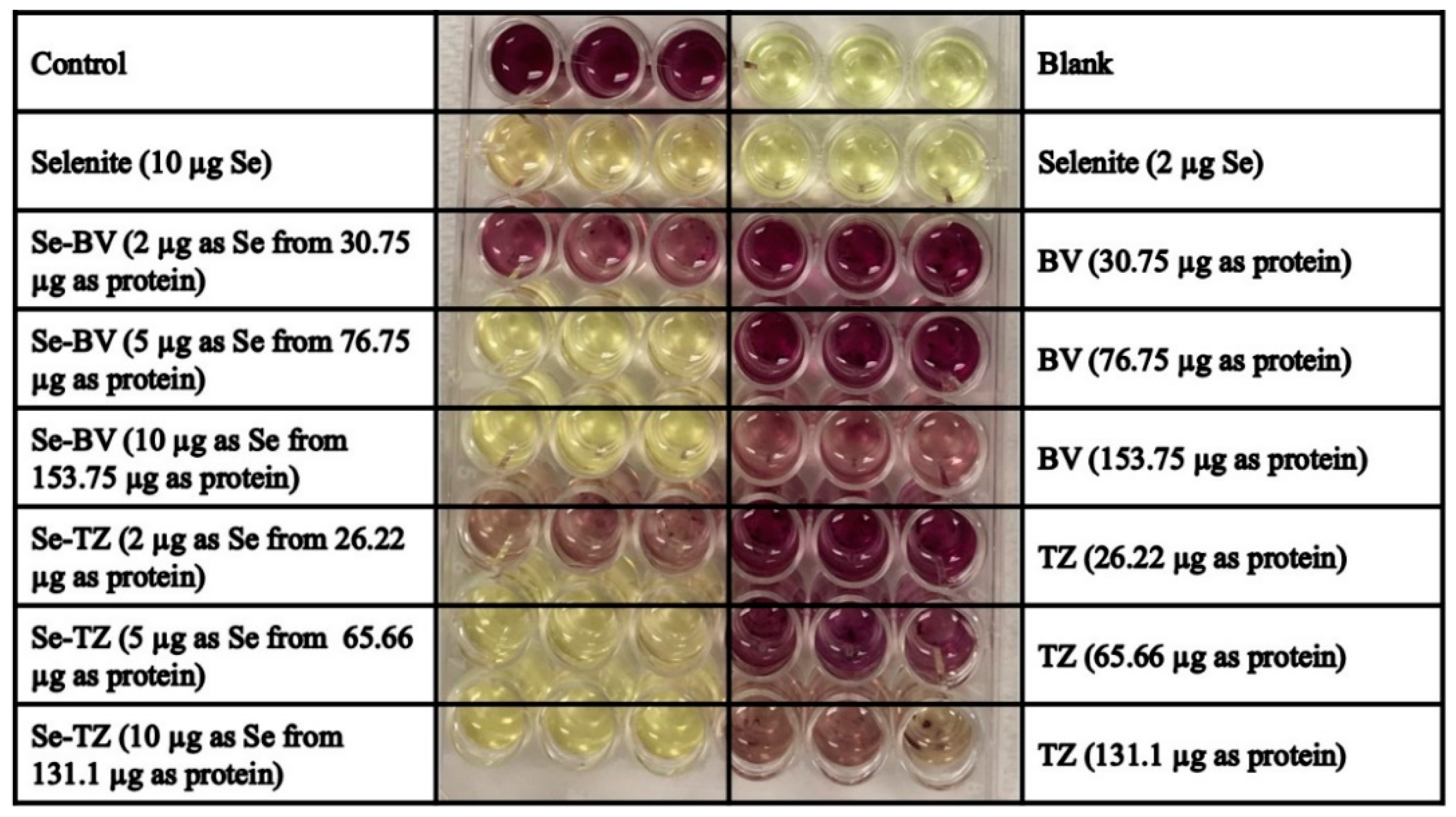
2.3. Detection of Superoxide In Vitro and In Situ
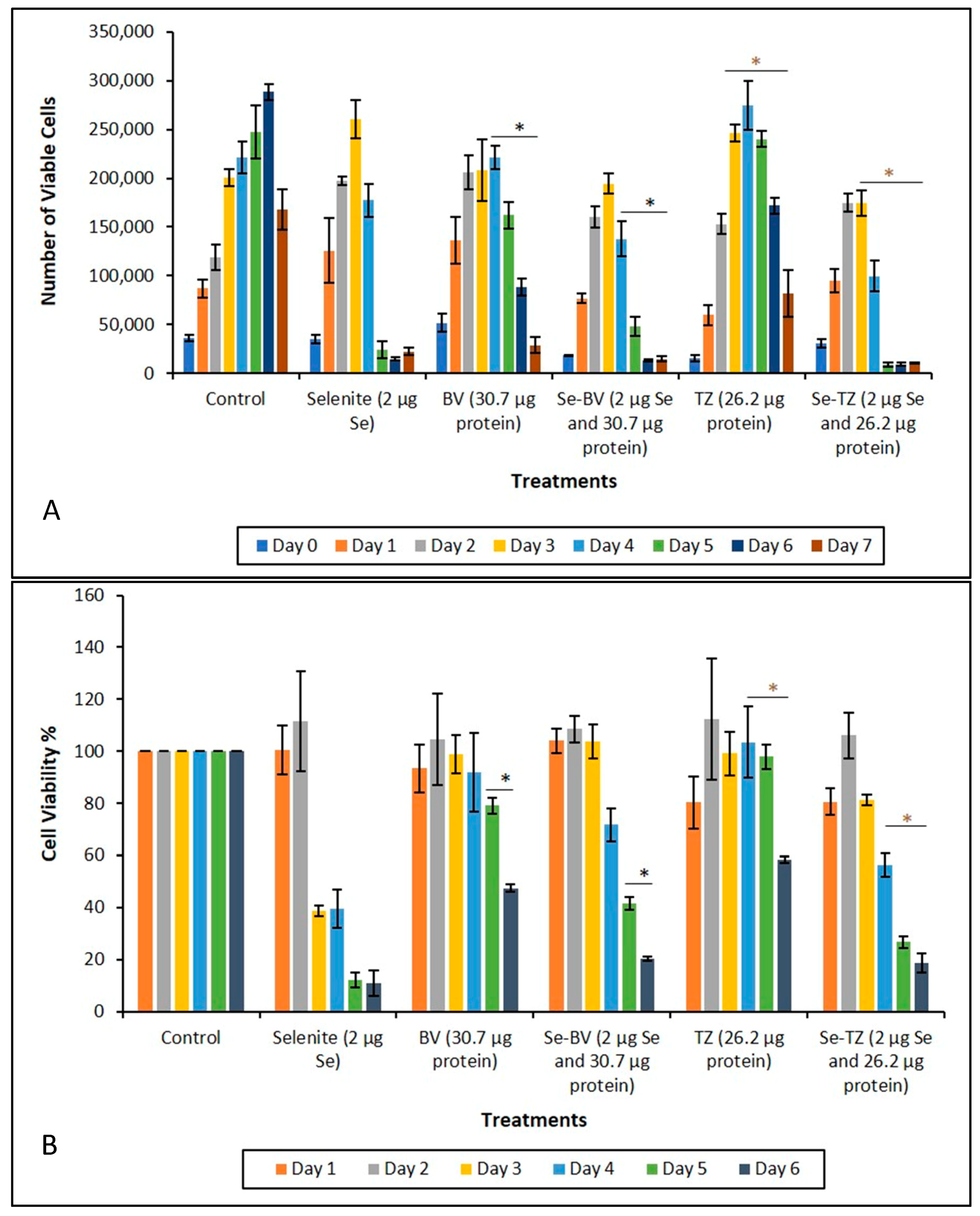
2.4. Evaluating the Cytotoxicity of the Se-Immunoconjugates
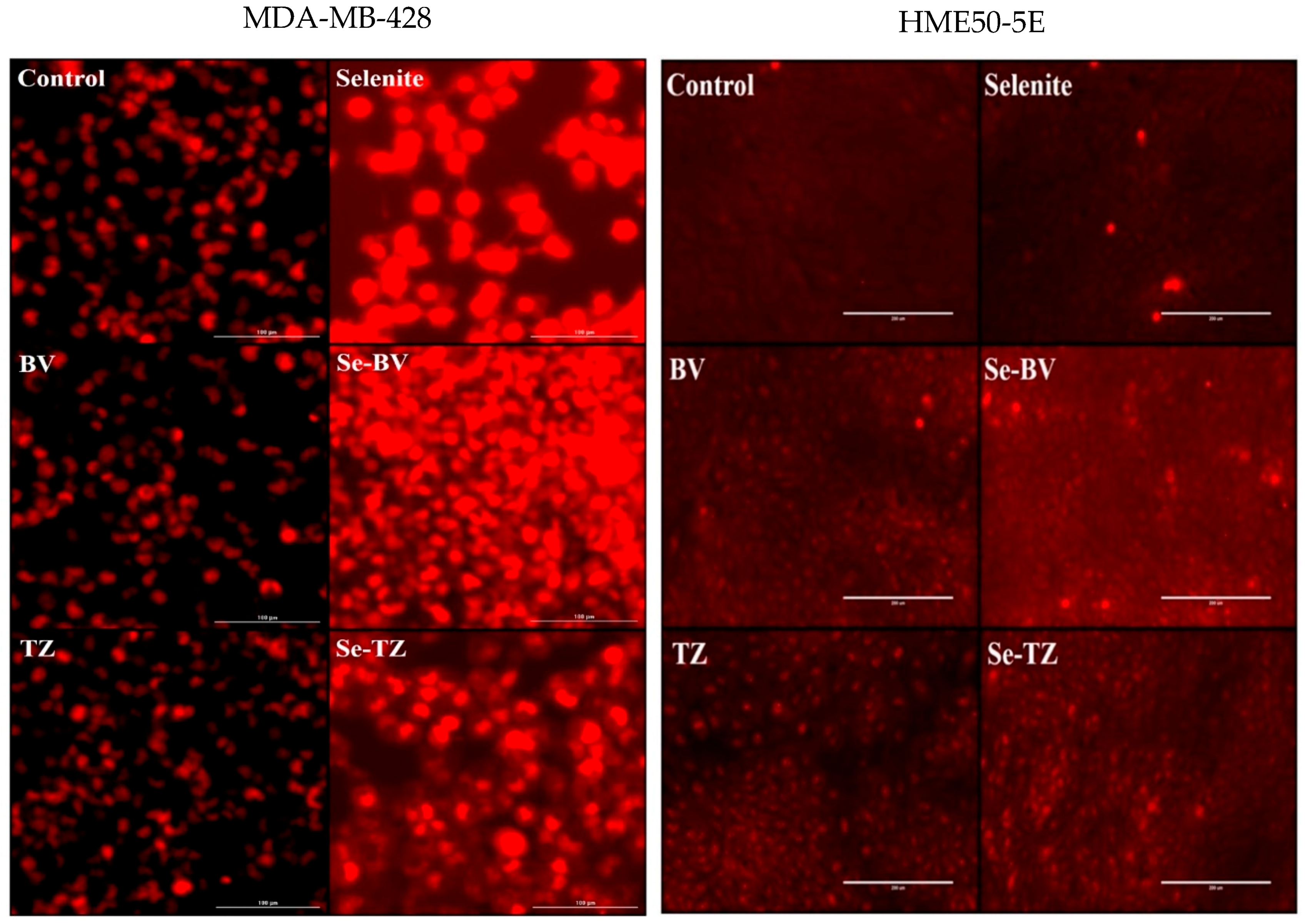
2.5. Morphological Changes in Triple Negative Breast Cancer (TNBC) Cells and HME50-5E Cells after Se-Trastuzumab (TZ) or Se-Bevacizumab (BV) Treatment
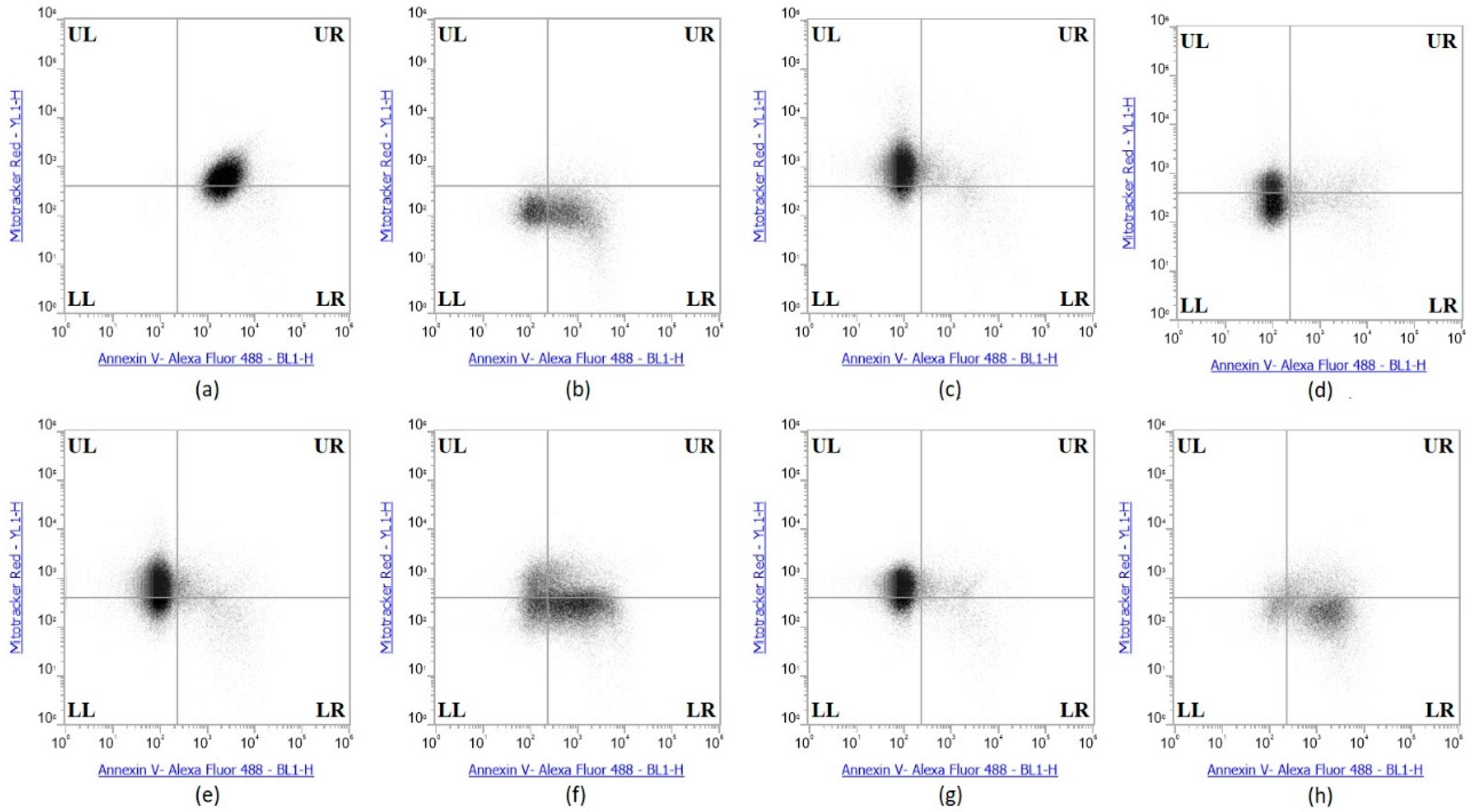
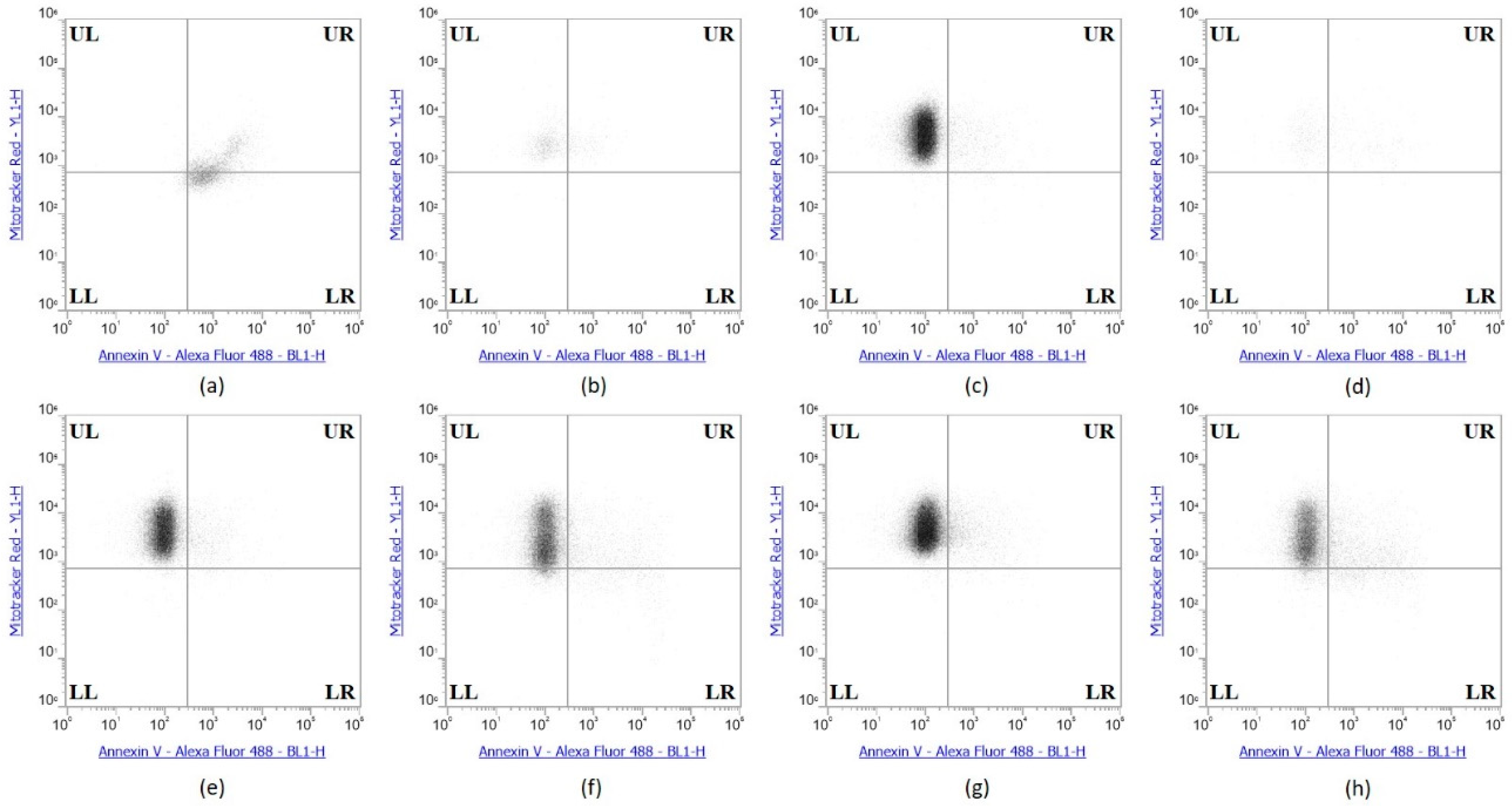
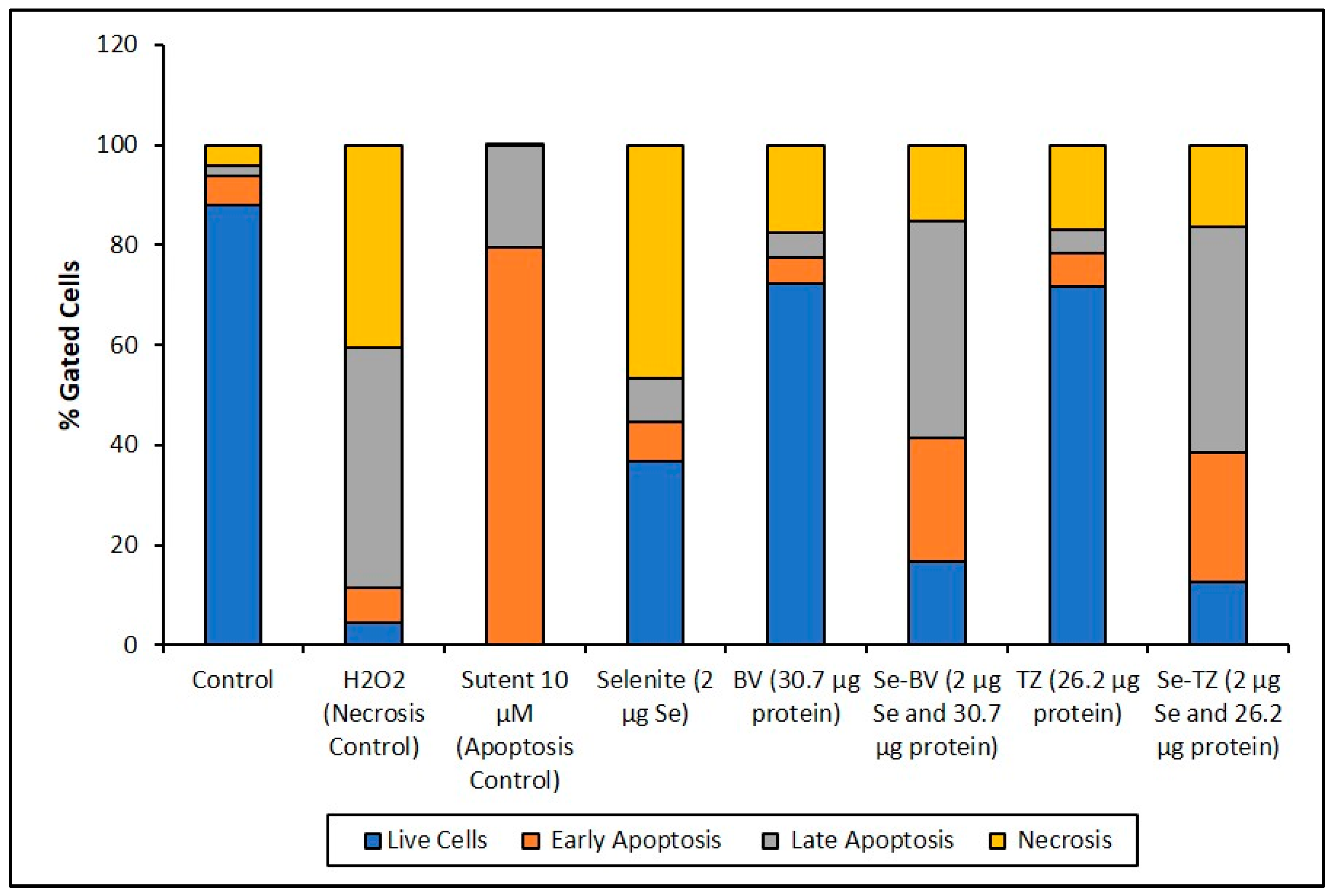
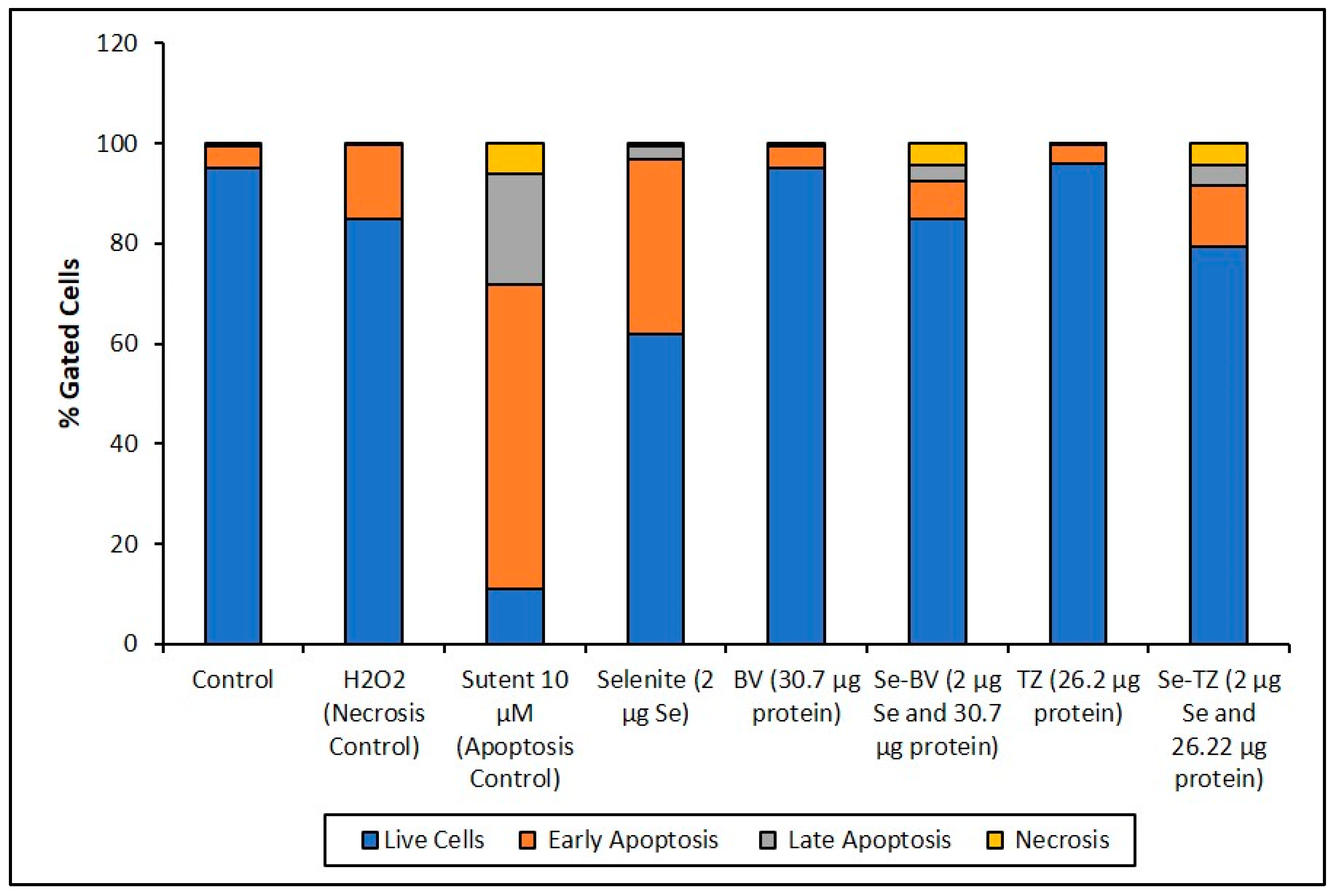
2.6. Selenium Treatment Induced Apoptosis
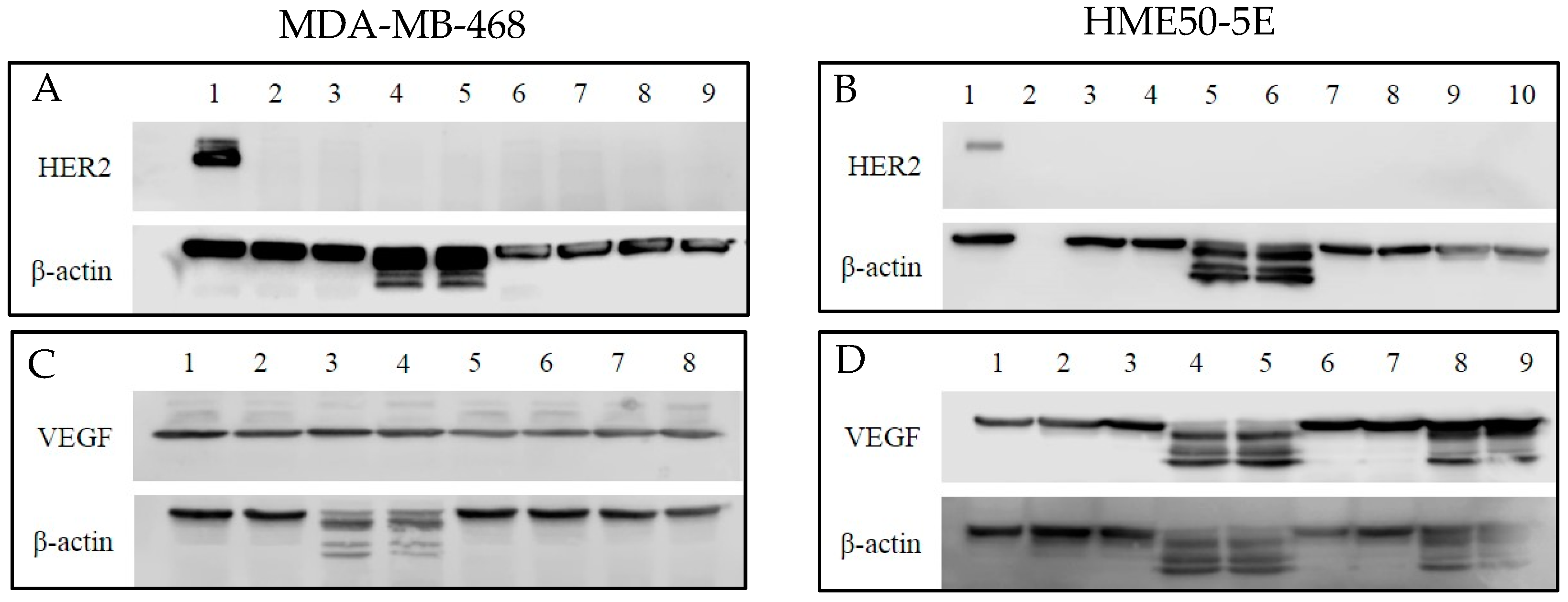
2.7. Human Epidermal Growth Factor 2 (HER2) and Vascular Endothelial Growth Factor (VEGF) Protein Expression
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
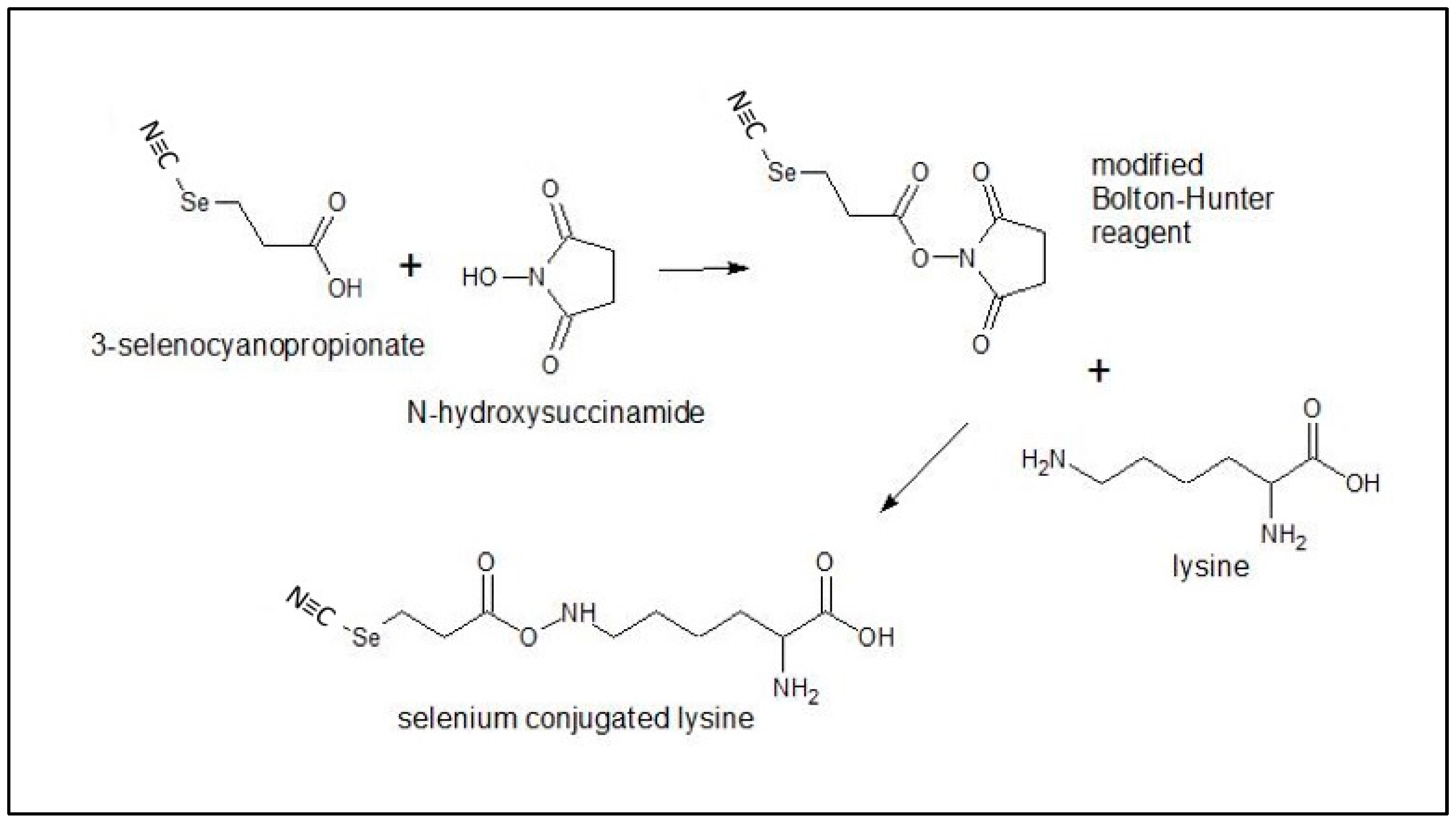
4.2.1. Conjugation of Redox Se to Monoclonal Antibodies
4.2.2. Analysis of Se-TZ and Se-BV
4.2.3. Detection of Superoxide in Vitro by BV, Se-BV, TZ and Se-TZ
4.2.4. Cell Culture
4.2.5. Cell Treatments
4.2.6. Photographic Assessment of Cellular Morphology
4.2.7. Cell Viability Measured by Trypan Blue Exclusion
- Cell type: BrCa cells
- Minimum diameter (microns): 12
- Maximum diameter (microns): 50
- Cell brightness (%): 85
- Cell sharpness: 100
- Viable cell spot brightness (%): 65
- Viable cell spot area (%): 5
4.2.8. Measuring Cell Proliferation Using MTT Assay
4.2.9. MitoTracker® Red and Annexin V Staining
4.2.10. Detection of Intracellular Superoxide Generation
4.2.11. BCA Assay for Protein Determination of mAbs
4.2.12. PAGE Under Denaturing Conditions
4.2.13. PAGE Under Non-Denaturing Conditions
4.2.14. Western Blotting
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BV | Bevacizumab |
| BCA | Bicinchoninic Acid |
| CL | Chemiluminescence |
| DCC | N,N′-dicyclohexylcarbodimide |
| DHE | Dihydroethidium |
| ECL | Enhanced chemiluminescence |
| EGFR | Epidermal growth factor receptor |
| ESR1 | Estrogen receptor α |
| FDA | Food and drug administration |
| HER2 | Human epidermal receptor 2 |
| mAb(s) | Monoclonal antibody(ies) |
| MTT | Tetrazolium dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PAGE | Polyacrylamide gel electrophoresis |
| PR | Progesterone receptor |
| ROS | Reactive oxygen specieis |
| Se | Selenium |
| Se-BV | Selenobevacizumab |
| Se-TZ | Selenotrastuzumab |
| THF | Tetrahydrofuran |
| TNBC | Triple negative breast cancer |
| TZ | Trastuzumab |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
Appendix A





References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Podo, F.; Buydens, L.; Degani, H.; Hilhorst, R.; Klipp, E.; Gribbestad, I.S.; Van Huffel, S.; Van Laarhoven, H.; Luts, J.; Monleon, D.; et al. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 2010, 4, 209–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, D.P.; Guadalupe Cabral, M.; Jacinto, A.; Brago, S. How many diseases is triple negative breast cancer: The protagonism of the immune microenvironment. ESMO Open 2017, 2, e000208. [Google Scholar] [CrossRef] [PubMed]
- Jitariu, A.A.; Cîmpean, A.M.; Ribatti, D.; Raica, M. Triple negative breast cancer: The kiss of death. Oncotarget 2017, 8, 46652. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dustin, D.; Fuqua, S.A. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr. Opin. Pharmacol. 2016, 31, 97–103. [Google Scholar] [CrossRef] [PubMed]
- De Laurentiis, M.; Cianniello, D.; Caputo, R.; Stanzione, B.; Arpino, G.; Cinieri, S.; Lorusso, V.; de Placido, S. Treatment of triple negative breast cancer (tnbc): Current options and future perspectives. Cancer Treat. Rev. 2010, 36, S80–S86. [Google Scholar] [CrossRef]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer 2016, 8, 93. [Google Scholar] [PubMed]
- Dieci, M.V.; Orvieto, E.; Dominici, M.; Conte, P.; Guarneri, V. Rare breast cancer subtypes: Histological, molecular, and clinical peculiarities. Oncologist 2014, 19, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch. Pathol. Lab. Med. 2010, 134, e48–e72. [Google Scholar] [PubMed]
- Hanna, W.M.; Rüschoff, J.; Bilous, M.; Coudry, R.A.; Dowsett, M.; Osamura, R.Y.; Penault-Llorca, F.; Van de Vijver, M.; Viale, G. HER2 in situ hybridization in breast cancer: Clinical implications of polysomy 17 and genetic heterogeneity. Mod. Pathol. 2014, 27, 4. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Wicha, M.S. HER2 and breast cancer stem cells: More than meets the eye. Cancer Res. 2013, 73, 3489–3493. [Google Scholar] [CrossRef] [PubMed]
- Maughan, K.L.; Lutterbie, M.A.; Ham, P.S. Treatment of breast cancer. Chemotherapy 2010, 51, 53. [Google Scholar]
- Carmeliet, P. Vegf as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Duda, D.G.; Clark, J.W.; Loeffler, J.S. Lessons from phase iii clinical trials on anti-vegf therapy for cancer. Nat. Rev. Clin. Oncol. 2006, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wu, S.; Dahut, W.L.; Parikh, C.R. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: Systematic review and meta-analysis. Am. J. Kidney Dis. 2007, 49, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Diana, A.; Franzese, E.; Centonze, S.; Carlino, F.; Della Corte, C.M.; Ventriglia, J.; Petrillo, A.; de Vita, F.; Alfano, R.; Ciardiello, F.; et al. Triple-negative breast cancers: Systematic review of the literature on molecular and clinical features with a focus on treatment with innovative drugs. Curr. Oncol. Rep. 2018, 20, 76. [Google Scholar] [CrossRef] [PubMed]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: Synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Ip, C. Lessons from basic research in selenium and cancer prevention. J. Nutr. 1998, 128, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar] [PubMed]
- Kieliszek, M.; Lipinski, B.; Błażejak, S. Application of sodium selenite in the prevention and treatment of cancers. Cells 2017, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Brozmanová, J.; Mániková, D.; Vlčková, V.; Chovanec, M. Selenium: A double-edged sword for defense and offence in cancer. Arch. Toxicol. 2010, 84, 919–938. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.A.; Dubowchik, G.M.; Lowinger, T.B. Antibody drug conjugates for treatment of breast cancer: Novel targets and diverse approaches in adc design. Pharmacol. Ther. 2017, 181, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H.; Kerr, J.R.; Currie, A. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [PubMed]
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Saito, Y.; Kitahara, J.; Imura, N. Active oxygen generation by the reaction of selenite with reduced glutathione in vitro. In Selenium in Biology and Medicine; Springer: Berlin, Germany, 1989; pp. 70–73. [Google Scholar]
- Chaudiere, J.; Courtin, O.; Leclaire, J. Glutathione oxidase activity of selenocystamine: A mechanistic study. Arch. Biochem. Biophys. 1992, 296, 328–336. [Google Scholar] [CrossRef]
- Bapat, P. Cytotoxic Effects of Selenium Conjugated Trastuzumab on HER2+ Breast Cancer Cell Lines. Ph.D. Thesis, Texas Tech University, Lubbock, TX, USA, May 2015. [Google Scholar]
- Yan, L.; Yee, J.A.; Boylan, M.; Spallholz, J.E. Effect of selenium compounds and thiols on human mammary tumor cells. Biol. Trace Elem. Res. 1991, 30, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Spallholz, J.E. Generation of reactive oxygen species from the reaction of selenium compounds with thiols and mammary tumor cells. Biochem. Pharmacol. 1993, 45, 429–437. [Google Scholar] [CrossRef]
- Li, J.; Zuo, L.; Shen, T.; Xu, C.M.; Zhang, Z.N. Induction of apoptosis by sodium selenite in human acute promyelocytic leukemia nb4 cells: Involvement of oxidative stress and mitochondria. J. Trace Elem. Med. Biol. 2003, 17, 19–26. [Google Scholar] [CrossRef]
- Zou, Y.; Niu, P.; Yang, J.; Yuan, J.; Wu, T.; Chen, X. The jnk signaling pathway is involved in sodium-selenite-induced apoptosis mediated by reactive oxygen in hepg2 cells. Cancer Biol. Ther. 2008, 7, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S. Mitochondrial regulation of apoptotic cell death. Toxicol. Lett. 2004, 149, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.D.; Grubb, D.; Lawen, A. The mitochondrial membrane potential (δψm) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Sanmartín, C.; Plano, D.; Sharma, A.K.; Palop, J.A. Selenium compounds, apoptosis and other types of cell death: An overview for cancer therapy. Int. J. Mol. Sci. 2012, 13, 9649–9672. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Rho gtpases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Brodin, O.; Eksborg, S.; Wallenberg, M.; Asker-Hagelberg, C.; Larsen, E.H.; Mohlkert, D.; Lenneby-Helleday, C.; Jacobsson, H.; Linder, S.; Misra, S.; et al. Pharmacokinetics and toxicity of sodium selenite in the treatment of patients with carcinoma in a phase i clinical trial: The secar study. Nutrients 2015, 7, 4978–4994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinchar, E.; Makey, K.L.; Gibson, J.; Chen, F.; Cole, S.A.; Megason, G.C.; Vijayakumar, S.; Miele, L.; Gu, J.W. Sunitinib significantly suppresses the proliferation, migration, apoptosis resistance, tumor angiogenesis and growth of triple-negative breast cancers but increases breast cancer stem cells. Vasc. Cell 2014, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pledgie-Tracy, A.; Sobolewski, M.D.; Davidson, N. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol. Cancer Ther. 2007, 6, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.P.; Hastings, J.F.; Han, J.Z.R.; Croucher, D.R. The under-appreciated promiscuity of the epidermal growth factor receptor family. Front. Cell Dev. Biol. 2016, 4, 88. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.; FitzGerald, D. Immunotoxintherapies for the treatment of epidermal growth factor receptor-dependent cancers. Toxins 2016, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Darius, J.R.; Richardson, D.R. William hunter and radioiodination: Revolutions in the labelling of proteins with radionuclides of iodine. Biochem. J. 2011, 1–4. [Google Scholar]
- Winer, E.; Gralow, J.; Diller, L.; Karlan, B.; Loehrer, P.; Pierce, L.; Demetri, G.; Ganz, P.; Kramer, B.; Kris, M.; et al. Clinical cancer advances 2008: Major research advances in cancer treatment, prevention, and screening—A report from the american society of clinical oncology. J. Clin. Oncol. 2008, 27, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Crampsie, M.A.; Pandey, M.K.; Desai, D.; Spallholz, J.; Amin, S.; Sharma, A.K. Phenylalkyl isoselenocyanates vs phenylalkyl isothiocyanates: Thiol reactivity and its implications. Chem.-Biol. Interact. 2012, 200, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TZ | Se-TZ | BV | Se-BV | |
|---|---|---|---|---|
| Selenium Concentration (mg/L) | <0.0200 | 88.00 | <0.0200 | 81.3 |
| Protein Concentration (mg/mL) | 2.82 | 2.32 | 2.73 | 2.53 |
| Selenium Concentration/ mg of protein (µg/mg) | <0.007 | 38.00 | <0.007 | 32.12 |
| Experiment Name | Treatments | Treatment Day | F Value | p Value |
|---|---|---|---|---|
| Trypan Blue | Control, Selenite, TZ and Se-TZ | Day 1 | F (3,8) = 4 | 0.052 |
| Trypan Blue | Control, Selenite, TZ and Se-TZ | Day 1 | F (3,8) = 12.52 | 0.0022 |
| Trypan Blue | Control, Selenite, TZ and Se-TZ | Day 2 | F (3,8) = 105.45 | 0.0000009 |
| Trypan Blue | Control, Selenite, TZ and Se-TZ | Day 3 | F (3,8) = 164.18 | 0.00000015 |
| Trypan Blue | Control, Selenite, TZ and Se-TZ | Day 4 | F (3,8) = 58.78 | 0.0000085 |
| Trypan Blue | Control, Selenite, TZ and Se-TZ | Day 5 | F (3,8) = 427.72 | 0.00000000361 |
| Trypan Blue | Control, Selenite, TZ and Se-TZ | Day 6 | F (3,8) = 242.71 | 0.0000000341 |
| Trypan Blue | Control, Selenite, TZ and Se-TZ | Day 7 | F (3,8) = 5.5 | 0.0241 |
| Experiment Name | Treatments | Treatment Day | F Value | p Value |
|---|---|---|---|---|
| MTT | Control, Selenite, TZ and Se-TZ | Day 1 | F (3,8) = 2.41 | 0.142 |
| MTT | Control, Selenite, TZ and Se-TZ | Day 2 | F (3,8) = 0.19 | 0.9018 |
| MTT | Control, Selenite, TZ and Se-TZ | Day 3 | F (3,8) = 2.13 | 0.175 |
| MTT | Control, Selenite, TZ and Se-TZ | Day 4 | F (3,8) = 6.19 | 0.013 |
| MTT | Control, Selenite, TZ and Se-TZ | Day 5 | F (3,8) = 45.69 | 0.000022 |
| MTT | Control, Selenite, TZ and Se-TZ | Day 6 | F (3,8) = 58.78 | 0.00000857 |
| Experiment Name | Treatments | Treatment Day | F Value | p Value |
|---|---|---|---|---|
| Trypan Blue | Control, Selenite, BV and Se-BV | Day 0 | F (3,8) = 6.24 | 0.0173 |
| Trypan Blue | Control, Selenite, BV and Se-BV | Day 1 | F (3,8) = 15.43 | 0.0010909 |
| Trypan Blue | Control, Selenite, BV and Se-BV | Day 2 | F (3,8) = 71.05 | 0.00000415 |
| Trypan Blue | Control, Selenite, BV and Se-BV | Day 3 | F (3,8) = 33.96 | 0.000067 |
| Trypan Blue | Control, Selenite, BV and Se-BV | Day 4 | F (3,8) = 69.1 | 0.000004 |
| Trypan Blue | Control, Selenite, BV and Se-BV | Day 5 | F (3,8) = 50.47 | 0.000015 |
| Trypan Blue | Control, Selenite, BV and Se-BV | Day 6 | F (3,8) = 40.38 | 0.000035 |
| Trypan Blue | Control, Selenite, BV and Se-BV | Day 7 | F (3,8) = 2.79 | 0.1096 |
| Experiment Name | Treatments | Treatment Day | F Value | p Value |
|---|---|---|---|---|
| MTT | Control, Selenite, BV and Se-BV | Day 1 | F (3,8) = 0.41 | 0.7479 |
| MTT | Control, Selenite, BV and Se-BV | Day 2 | F (3,8) = 0.15 | 0.9278 |
| MTT | Control, Selenite, BV and Se-BV | Day 3 | F (3,8) = 0.1 | 0.9594 |
| MTT | Control, Selenite, BV and Se-BV | Day 4 | F (3,8) = 2.02 | 0.1894 |
| MTT | Control, Selenite, BV and Se-BV | Day 5 | F (3,8) = 29.58 | 0.000111 |
| MTT | Control, Selenite, BV and Se-BV | Day 6 | F (3,8) = 69.38 | 0.0000045 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khandelwal, S.; Boylan, M.; Spallholz, J.E.; Gollahon, L. Cytotoxicity of Selenium Immunoconjugates against Triple Negative Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3352. https://doi.org/10.3390/ijms19113352
Khandelwal S, Boylan M, Spallholz JE, Gollahon L. Cytotoxicity of Selenium Immunoconjugates against Triple Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2018; 19(11):3352. https://doi.org/10.3390/ijms19113352
Chicago/Turabian StyleKhandelwal, Soni, Mallory Boylan, Julian E. Spallholz, and Lauren Gollahon. 2018. "Cytotoxicity of Selenium Immunoconjugates against Triple Negative Breast Cancer Cells" International Journal of Molecular Sciences 19, no. 11: 3352. https://doi.org/10.3390/ijms19113352
APA StyleKhandelwal, S., Boylan, M., Spallholz, J. E., & Gollahon, L. (2018). Cytotoxicity of Selenium Immunoconjugates against Triple Negative Breast Cancer Cells. International Journal of Molecular Sciences, 19(11), 3352. https://doi.org/10.3390/ijms19113352