Transcriptome Profiles Reveal the Crucial Roles of Auxin and Cytokinin in the “Shoot Branching” of Cremastra appendiculata


Abstract
:1. Introduction
2. Results
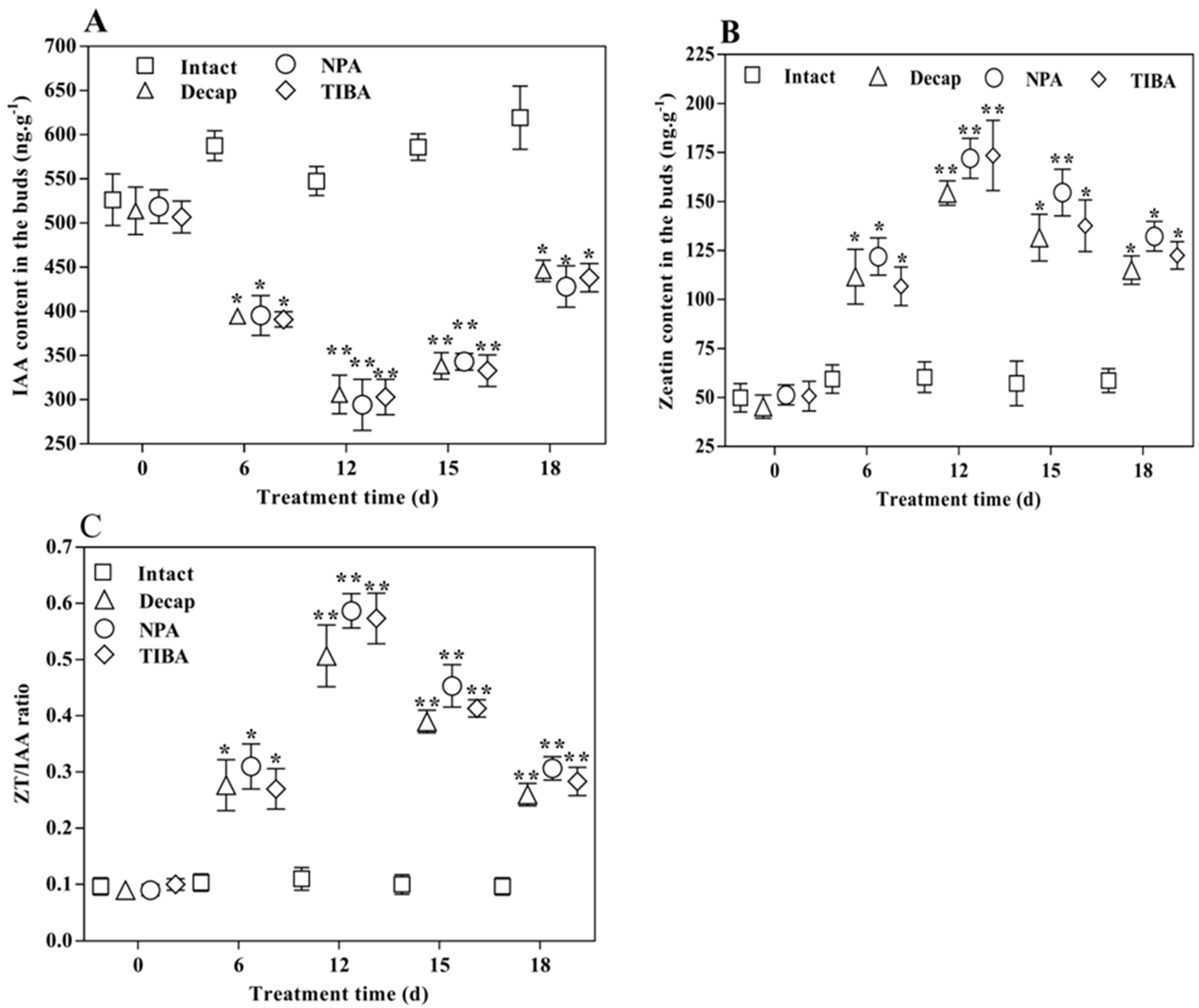
2.1. Decapitation and Auxin Transport Inhibitors Affect Lateral Buds Break
2.2. Content Changes of Hormones in the Lateral Buds during the Bud Elongation Process
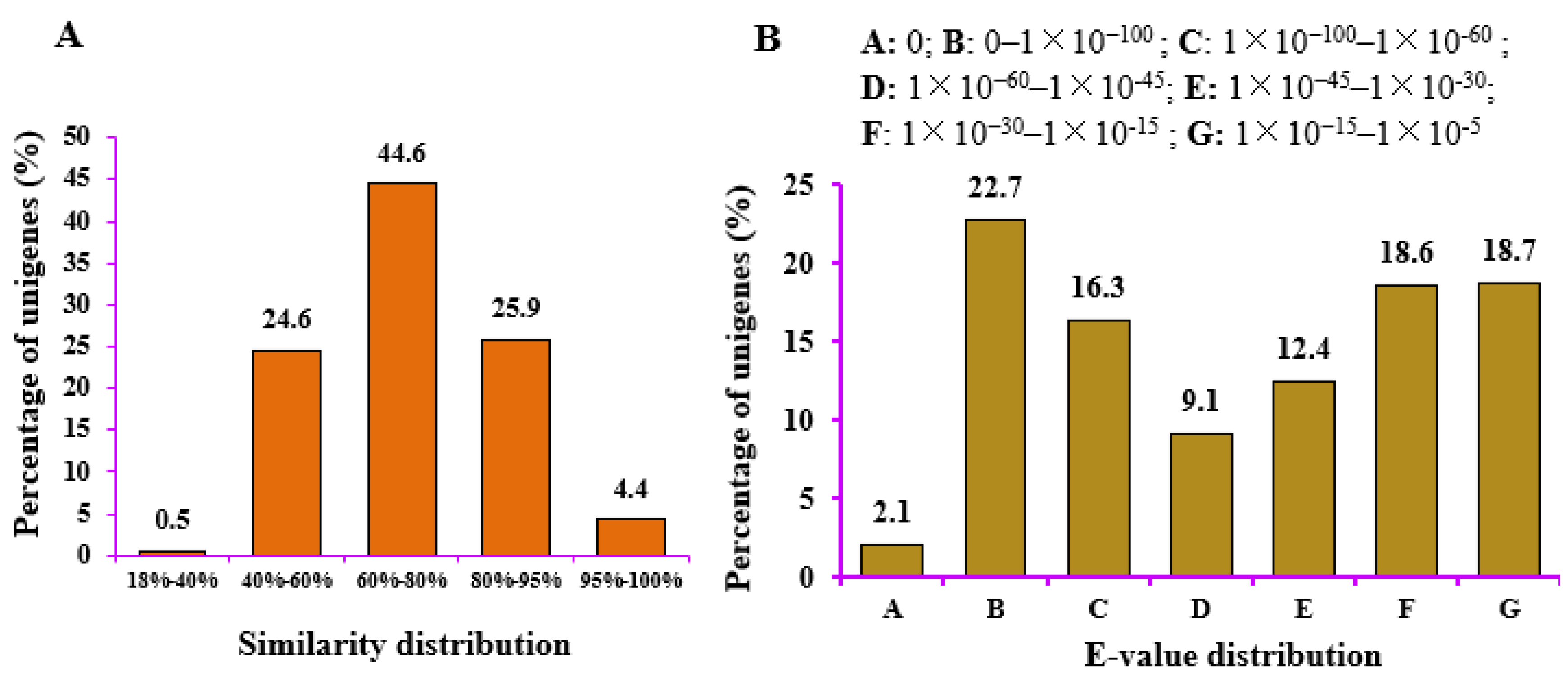
2.3. Sequence Analysis, Read Assembly, and Annotation
2.4. Successive Pairwise Comparisons of DEG Profiles
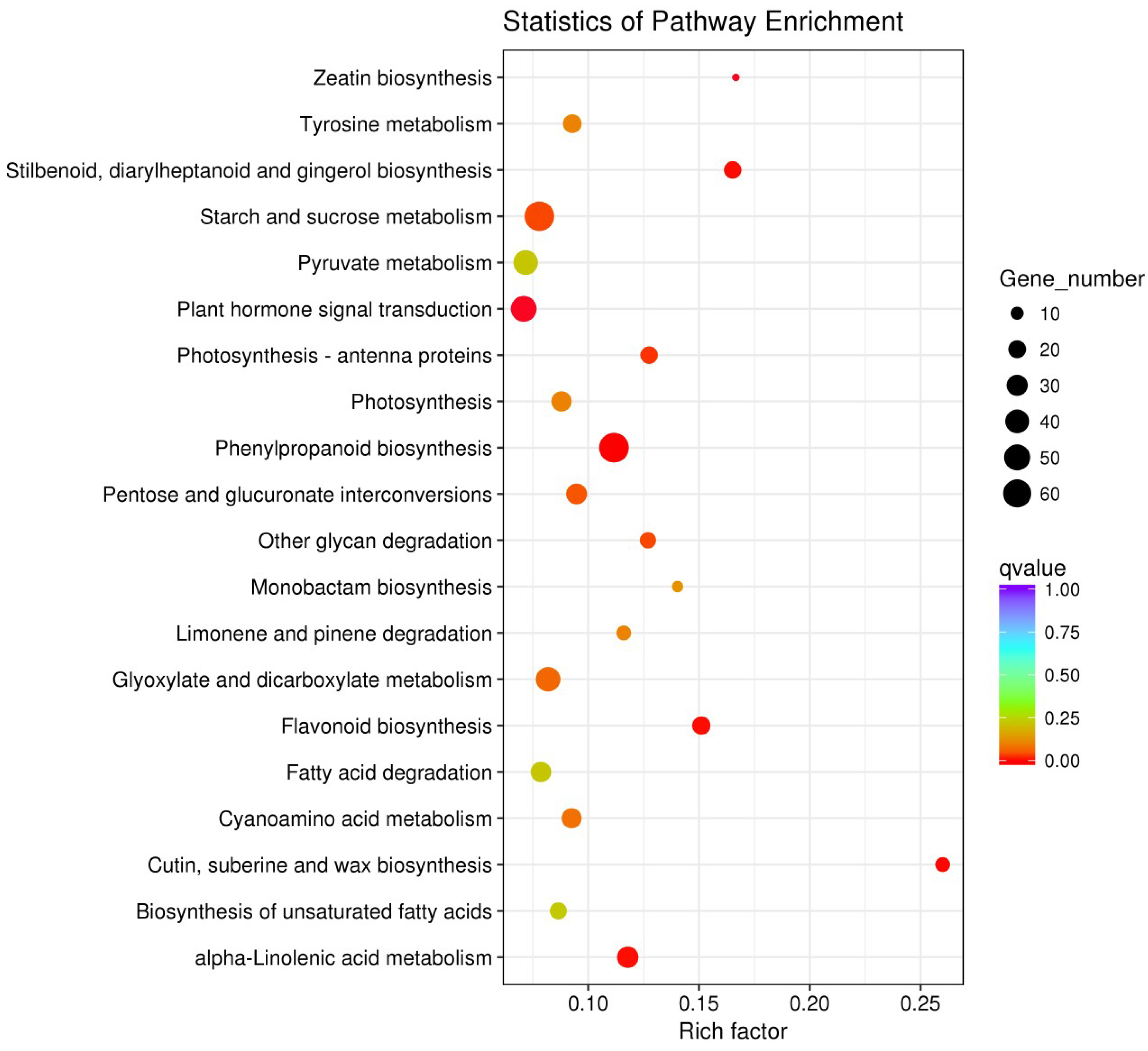
2.5. GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) Enrichment Analyses of All DEGs
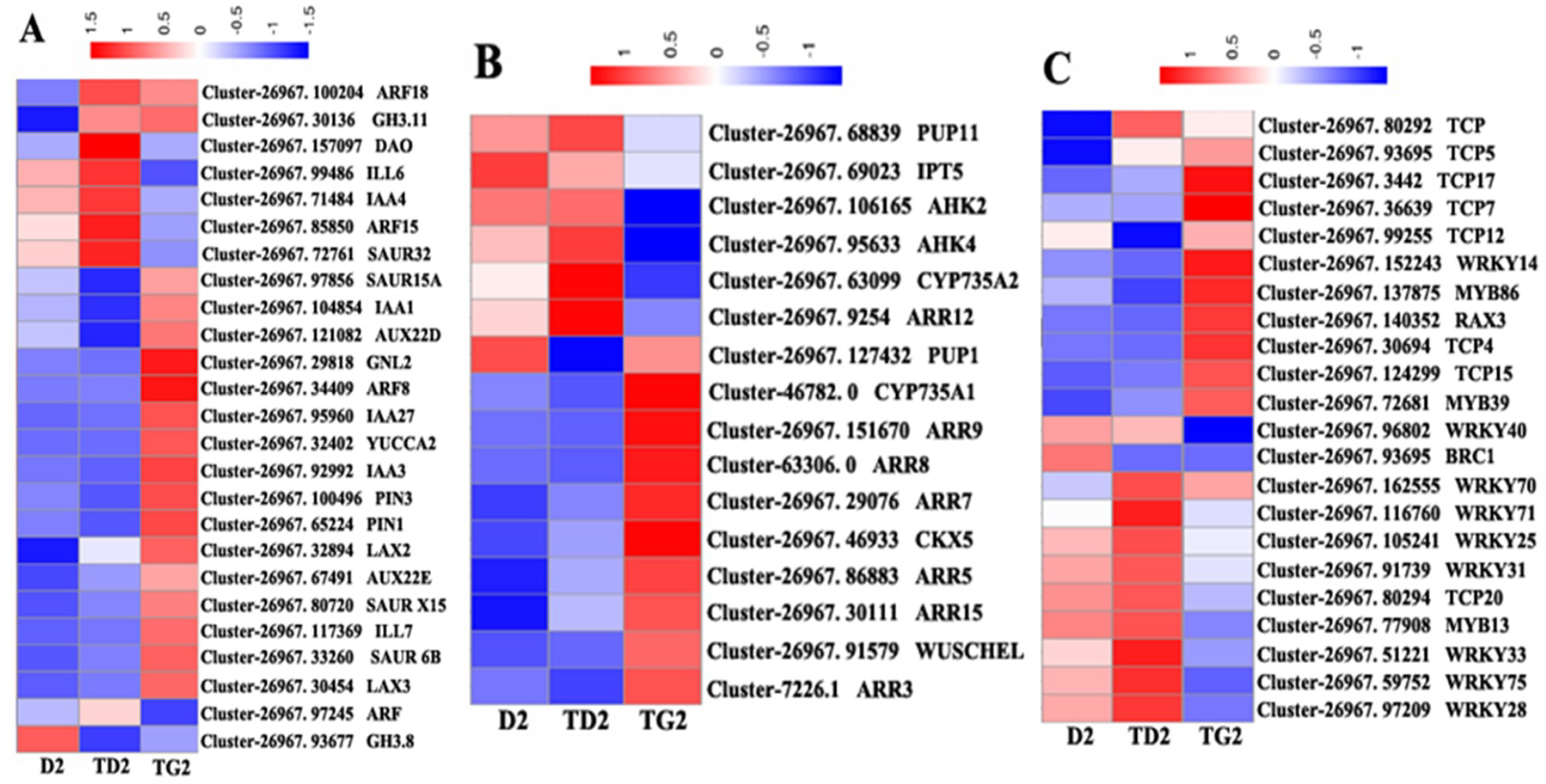
2.6. Cluster Analysis of Hormone- and Transcription Factor-Regulated DEGs during the Bud Elongation Process
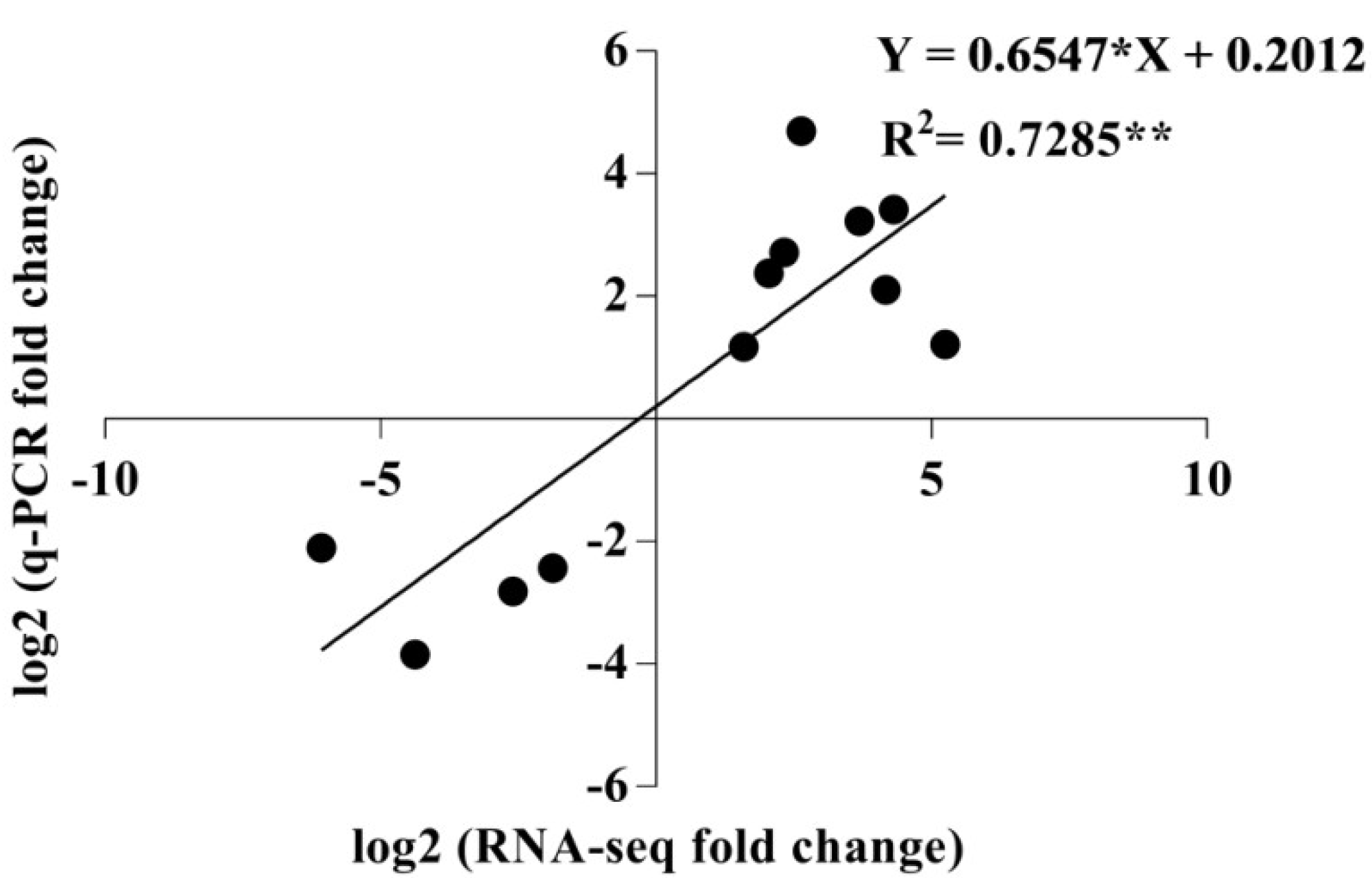
2.7. qRT-PCR Validation of Differentially Expressed Transcripts from Transcriptome Analysis
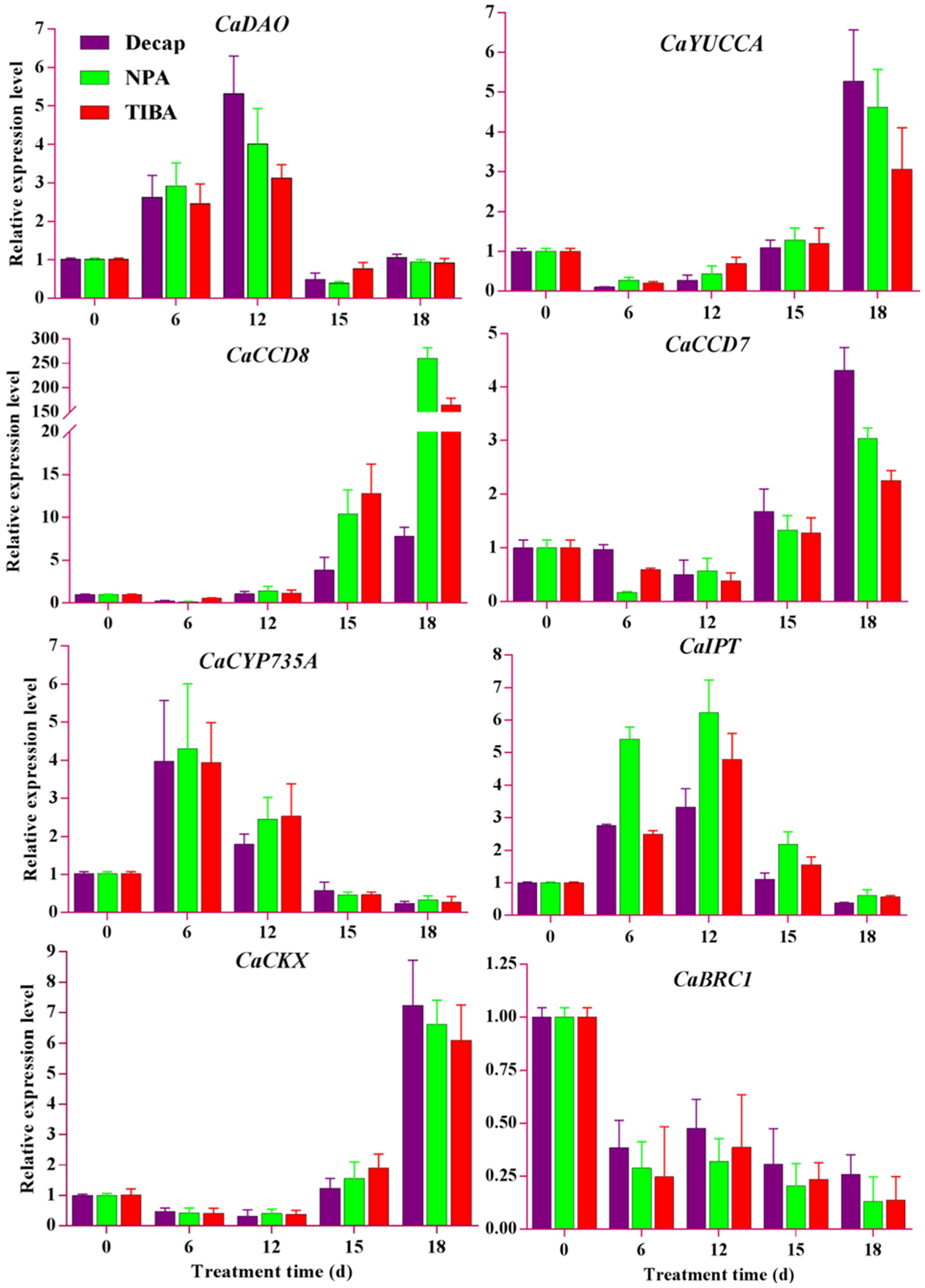
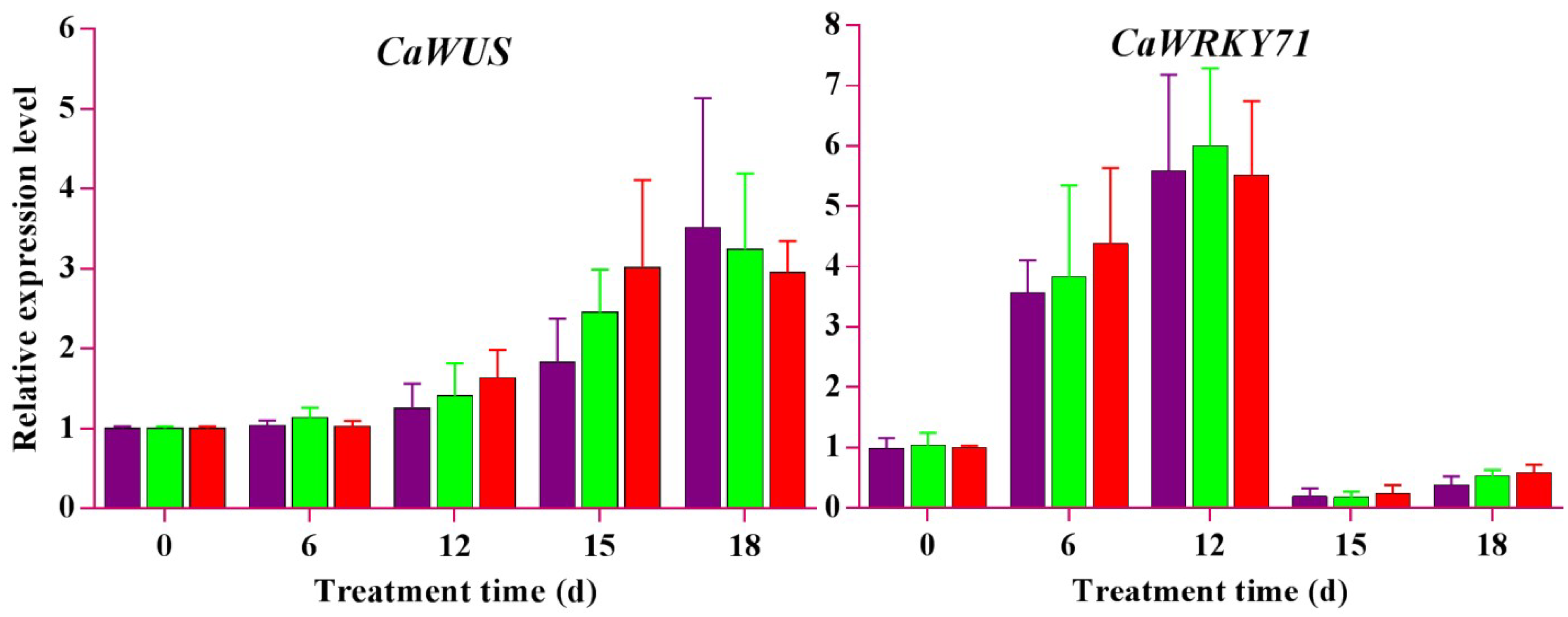
2.8. qRT-PCR Expression Analyses of Candidate Genes
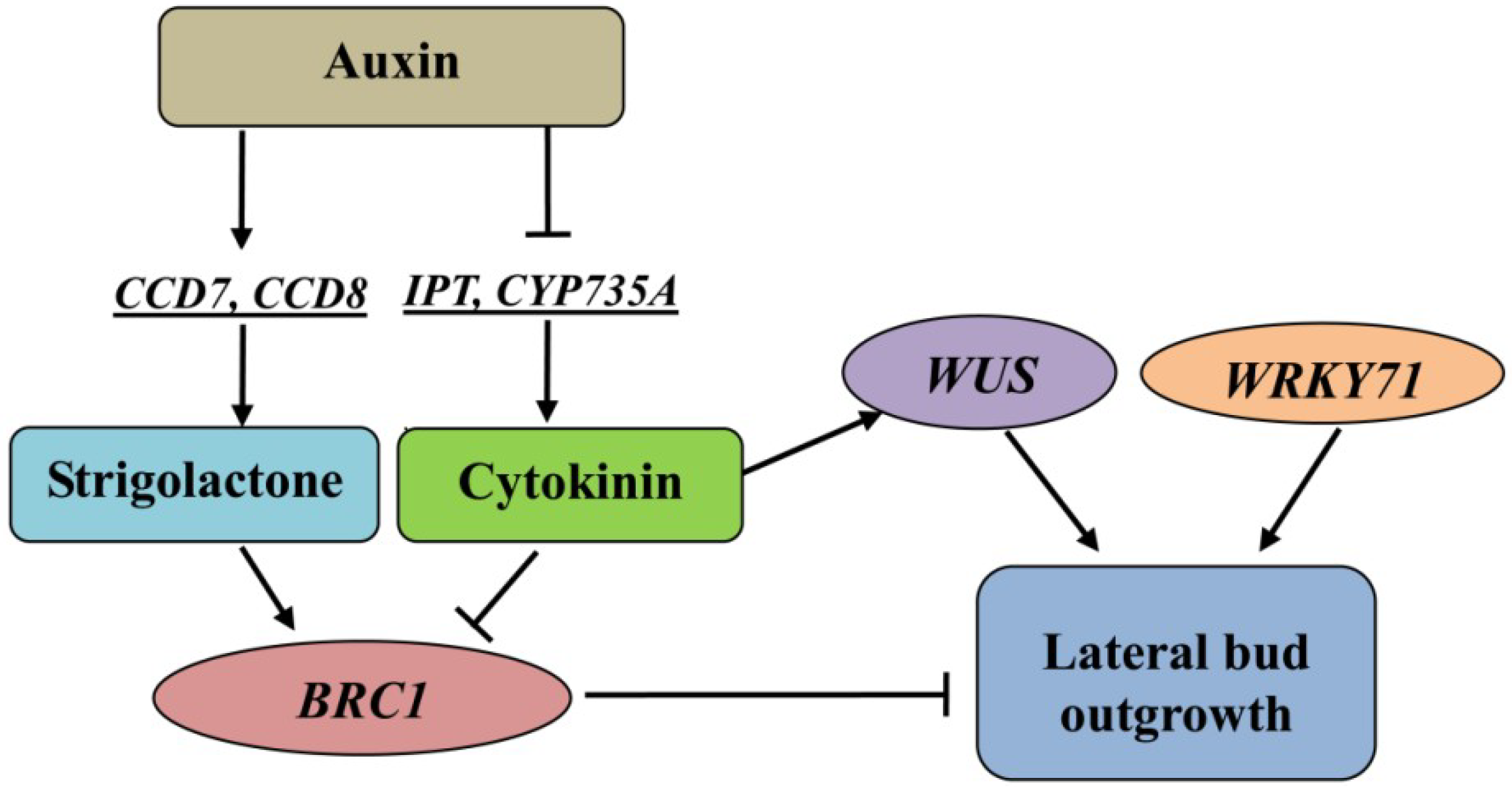
3. Discussion
4. Methods and Materials
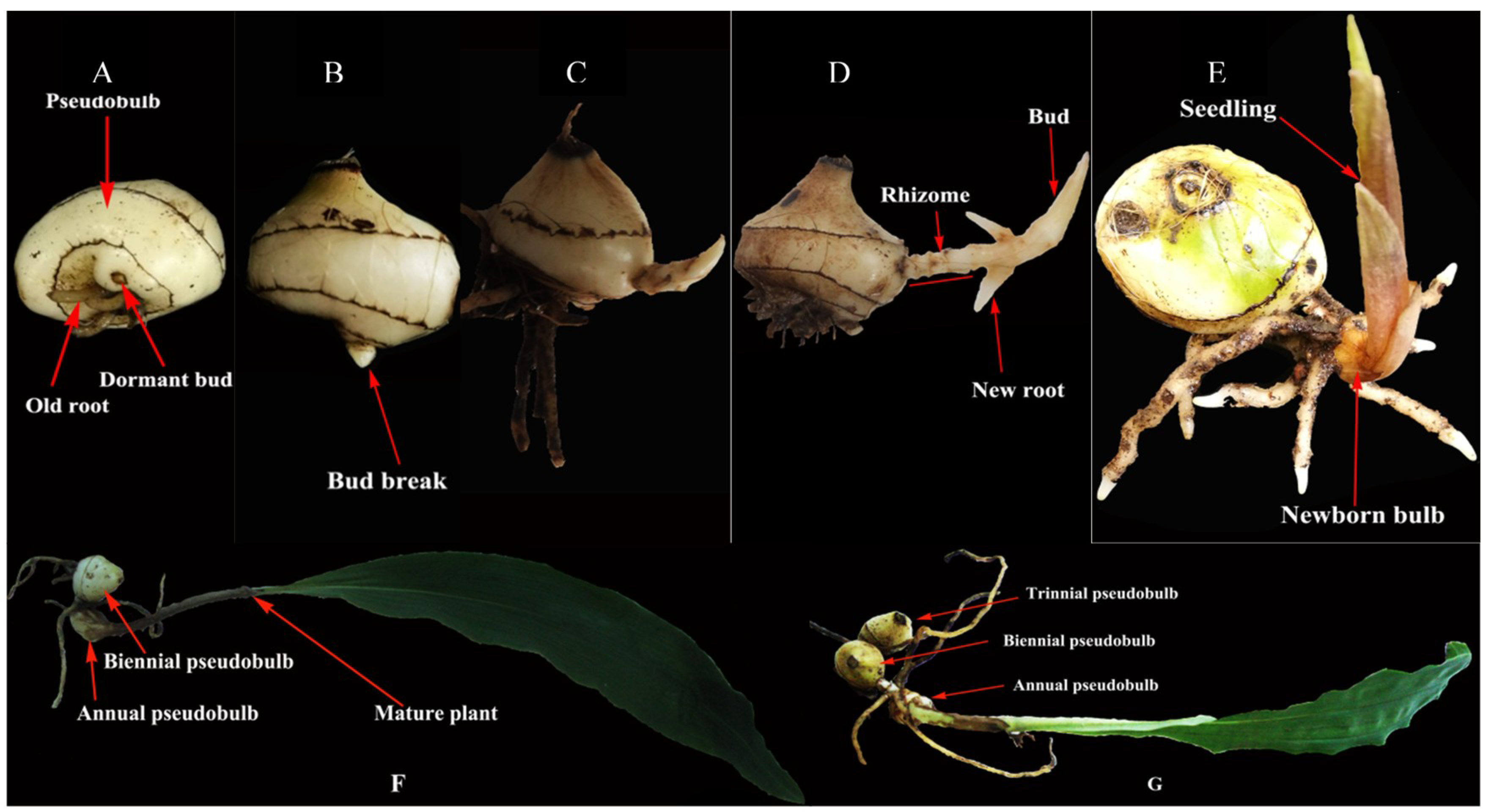
4.1. Plant Materials, Growth Conditions, and Treatments
4.2. RNA Isolation, Quantification, and Qualification
4.3. Library Construction and Sequencing
4.4. De Novo Assembly and Annotation
4.5. Identification of Differentially Expressed Genes
4.6. RNA-Seq Validation and Candidate Gene Expression Analysis Using qRT-PCR
4.7. Measurements of Hormone Contents
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shim, J.S.; Kim, J.H.; Lee, J.Y.; Kim, S.N.; Kwon, H.J. Anti-angiogenic activity of a homoisoflavanone from Cremastra appendiculata. Planta Med. 2004, 70, 171–173. [Google Scholar] [PubMed]
- Ikeda, Y.; Nonaka, H.; Furumai, T.; Iqarashi, Y. Cremastrinea pyrrolizidine alkaloid from Cremastra appendiculata. J. Nat. Prod. 2005, 68, 572–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guan, S.H.; Meng, Y.H.; Zhang, Y.B.; Cheng, C.R.; Shi, Y.Y.; Feng, R.H.; Zeng, F.; Wu, Z.Y.; Zhang, J.X.; et al. Phenanthrenes, 9,10-dihydrophenanthrenes, bibenzyls with their derivatives, and malate or tartrate benzyl ester glucosides from tubers of Cremastra appendiculata. Phytochemistry 2013, 94, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, J.; Zeng, K.W.; Li, P.; Tu, P.F. Three new phenanthrenes form Cremastra appendiculata (D. Don) Makino. Chin. Chem. Lett. 2013, 24, 737–739. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Zeng, K.W.; Jiang, Y.; Tu, P.F. Five new benzylphenanthrenes from Cremastra appendiculata. Fitoterapia 2015, 103, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, J.; Zeng, K.W.; Jiang, Y.; Tu, P.F. Five new biphenanthrenes from Cremastra appendiculata. Molecules 2016, 21, 1089. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.Y.; Chung, M.G. The breeding systems of Cremastra appendiculata and Cymbidium goeringii: High levels of annual fruit failure in two self-compatible orchids. Ann. Bot. Fenn. 2003, 40, 81–85. [Google Scholar]
- Zhang, G.Q.; Liu, K.W.; Li, Z.; Lohaus, R.; Hsiao, Y.Y.; Niu, S.C.; Wang, J.Y.; Lin, Y.C.; Xu, Q.; Chen, L.J.; et al. The apostasia genome and the evolution of orchids. Nature 2017, 549, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Yagame, T.; Funabiki, E.; Nagasawa, E.; Fukiharu, T.; Iwase, K. Identification and symbiotic ability of psathyrellaceae fungi isolated from a photosynthetic orchid, Cremastra appendiculata (Orchidaceae). Am. J. Bot. 2013, 100, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Zhang, M.S.; Wu, Y.Q.; Gao, X.F.; Li, X.L.; Wang, W.Z. The Roles of Auxin in Regulating “Shoot Brancing” of Cremastra appendiculata. J. Plant Growth Regul. 2017, 36, 281–289. [Google Scholar] [CrossRef]
- Xu, J.; Zha, M.; Li, Y.; Ding, Y.; Chen, L.; Ding, C.; Wang, S. The interaction between nitrogen availability and auxin, cytokinin, and strigolactone in the control of shoot branching in rice (Oryza sativa L.). Plant Cell Rep. 2015, 34, 1647–1662. [Google Scholar] [CrossRef] [PubMed]
- Brewer, P.B.; Dun, E.A.; Gui, R.; Mason, M.G.; Beveridge, C.A. Strigolactone inhibition of branching independent of polar auxin transport. Plant Physiol. 2015, 168, 1820–1829. [Google Scholar] [CrossRef] [PubMed]
- Roman, H.; Girault, T.; Barbier, F.; Péron, T.; Brouard, N.; Pĕnčík, A.; Novák, O.; Vian, A.; Sakr, S.; Lothier, J.; et al. Cytokinins are initial targets of light in the control of bud outgrowth. Plant Physiol. 2016, 172, 489–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldie, T.; Leyser, O. Cytokinin targets auxin transport to promote shoot branching. Plant Physiol. 2018, 177, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.G.; Ross, J.J.; Babst, B.A.; Wienclaw, B.N.; Beveridge, C.A. Sugar demand, not auxin, is the initial regulator of apical dominance. Proc. Natl. Acad. Sci. USA 2014, 111, 6092–6097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furet, P.M.; Lothier, J.; Demotes-Mainard, S.; Travier, S.; Henry, C.; Guérin, V.; Vian, A. Light and nitrogen nutrition regulate apical control in Rosa hybrida L. J. Plant Physiol. 2014, 171, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.E.; Cox, M.C.H.; Ross, J.J.; Kristantini, S.; Beveridge, C.A. Auxin dynamics after decapitation are not correlated with the initial growth of axillary buds. Plant Physiol. 2005, 138, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Zhao, M.L.; Chen, M.S.; Pan, B.Z.; Tao, Y.B.; Xu, Z.F. Comparative transcriptome analysis of axillary buds in response to the shoot branching regulators gibberellin A3 and 6-benzyladenine in Jatropha curcas. Sci. Rep. 2017, 7, 11417–11428. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Takei, K.; Kojima, M.; Sakakibara, H.; Mori, H. Auxin controls local cytokinin biosynthesis in the nodal stem in apical dominance. Plant J. 2006, 45, 1028–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordström, A.; Tarkowski, P.; Tarkowska, D.; Norbaek, R.; Astot, C.; Dolezal, K.; Sandberg, G. Auxin regulation of cytokinin biosynthesis in Arabidopsis thaliana: A factor of potential importance for auxin-cytokinin-regulated development. Proc. Natl. Acad. Sci. USA 2004, 101, 8039–8044. [Google Scholar] [CrossRef] [PubMed]
- Foo, E.; Bullier, E.; Goussot, M.; Foucher, F.; Rameau, C.; Beveridge, C.A. The branching gene RAMOSUS1 mediates interactions among two novel signals and auxin in pea. Plant Cell 2005, 17, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Zhang, S.; Zhang, W.; Li, G.; Chen, Z.; Zhai, W.; Zhao, X.; Pan, X.; Xie, Q.; Zhu, L. The rice HIGH-TILLERING DWARF1 encoding an ortholog of Arabidopsis MAX3 is required for negative regulation of the outgrowth of axillary buds. Plant J. 2006, 48, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Arite, T.; Iwata, H.; Ohshima, K.; Maekawa, M.; Nakajima, M.; Kojima, M.; Sakakibara, H.; Kyozuka, J. DWARF10, an RMS1/DAD1 ortholog, controls lateral bud outgrowth in rice. Plant J. 2007, 51, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.; Stirnberg, P.; Beveridge, C.; Leyser, O. Interactions between auxin and strigolactone in shoot branching control. Plant Physiol. 2009, 151, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Bennett, T.; Sieberer, T.; Willett, B.; Booker, J.; Luschnig, C.; Leyser, O. The Arabidopsis MAX pathway controls shoot branching by regulating auxin transport. Curr. Biol. 2006, 16, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.; Shinohara, N.; Sieberer, T.; Williamson, L.; George, G.; Hepworth, J.; Müller, D.; Domagalska, M.A.; Leyser, O. Strigolactones enhance competition between shoot branches by dampening auxin transport. Development 2010, 137, 2905–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Zhao, L.; Challis, R.; Leyser, O. Strigolactone regulation of shoot branching in chrysanthemum (Dendranthema grandiflorm). J. Exp. Bot. 2010, 61, 3069–3078. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Martínez, J.A.; Poza-Carrión, C.; Cubas, P. Arabidopsis BRANCHED1 acts as an integrator of branching signals within axillary buds. Plant Cell 2007, 19, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Nicolas, M.; Zhang, J.; Yu, H.; Guo, D.; Yuan, R.; Zhang, T.; Yang, J.; Cubas, P.; Qin, G. The TIE1 transcriptional repressor controls shoot branching by directly repressing BRANCHED1 in Arabidopsis. PLoS Genet. 2018, 14, e1007296. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.A.; Raman, S.; Martinez, C.C.; Sinha, N.R.; Schmitz, G.; Theres, K. Trifoliate encodes an MYB transcription factor that modulates leaf and shoot architecture in tomato. Proc. Natl. Acad. Sci. USA 2013, 110, 2401–2406. [Google Scholar] [CrossRef] [PubMed]
- Kirik, V.; Kölle, K.; Wohlfarth, T.; Miséra, S.; Bäumlein, H. Ectopic expression of a novel MYB gene modifies the architecture of the Arabidopsis inflorescence. Plant J. 1998, 13, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Zhang, J.; Wang, X.; Han, X.; Wei, B.; Wang, J.; Li, B.; Yu, H.; Huang, Q.; Gu, H.; et al. The WRKY transcription factor WRKY71/EXB1 controls shoot branching by transcriptionally regulating RAX genes in Arabidopsis. Plant Cell 2015, 27, 3112–3127. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Qin, G. EXB1/WRKY71 transcription factor regulates both shoot branching and responses to abiotic stresses. Plant Signal Behav. 2016, 11, e1150404–e1150408. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Nie, Q.; Li, Z.; Zhang, J.; Liu, Y.; Long, Y.; Wang, Z.; Wang, G.; Liu, R. Transcriptomic analysis reveals overdominance playing a critical role in nicotine heterosis in Nicotiana tabacum L. BMC Plant Biol. 2018, 18, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Y.; Xin, D.; Chen, W.; Shao, X.; Wang, Y.; Guo, W. RNA-Seq-based transcriptome analysis of dormant flower buds of Chinese cherry (Prunus pseudocerasus). Gene 2015, 555, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, J.N.; Nayak, S.; Jha, S.; Joshi, R.K. Transcriptome profiling of the floral buds and discovery of genes related to sex-differentiation in the dioecious cucurbit Coccinia grandis (L.) Voigt. Gene 2017, 626, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chang, X.; Lin, J.; Chang, Y.; Chen, J.C.; Reid, M.S.; Jiang, C.Z. Transcriptome profiling reveals regulatory mechanisms underlying corolla senescence in petunia. Hortic. Res. 2018, 5, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, X.K.; Zhao, K.; Zheng, T.; Han, Y.; Yuan, C.; Zhang, Q. Transcriptome profiles reveal the crucial roles of hormone and sugar in the bud dormancy of Prunus mume. Sci. Rep. 2018, 8, 5090–5105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, Y.; Lin, N.; Jin, X.; Liao, W.; Zhao, S.; Fu, C.; Yu, L. Transcriptome-wide identification and screening of WRKY factors involved in the regulation of taxol biosynthesis in Taxus chinensis. Sci. Rep. 2018, 8, 5197–5208. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, G.; Tang, N.; Li, Z. Transcriptome analysis reveals molecular signatures of luteoloside accumulation in senescing leaves of Lonicera macranthoides. Int. J. Mol. Sci. 2018, 19, 1012. [Google Scholar] [CrossRef] [PubMed]
- Dun, E.A.; de Saint Germain, A.; Rameau, C.; Beveridge, C.A. Antagonistic action of strigolactone and cytokinin in bud outgrowth control. Plant Physiol. 2012, 158, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Beveridge, C.A. Roles for auxin, cytokinin, and strigolactone in regulating shoot branching. Plant Physiol. 2009, 149, 1929–1944. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, N.; Taylor, C.; Leyser, O. Strigolactone can promote or inhibit shoot branching by triggering rapid depletion of the auxin efflux protein PIN1 from the plasma membrane. PLoS Biol. 2013, 11, e1001474. [Google Scholar] [CrossRef] [PubMed]
- Waldie, T.; McCulloch, H.; Leyser, O. Strigolactones and the control of plant development: Lessons from shoot branching. Plant J. 2014, 79, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Moraga, A.; Ahrazem, O.; Pérez-Clemente, R.M.; Gómez-Cadenas, A.; Yoneyama, K.; López-Ráez, J.A.; Molina, R.V.; Gómez-Gómez, L. Apical dominance in saffron and the involvement of the branching enzymes CCD7 and CCD8 in the control of bud sprouting. BMC Plant Biol. 2014, 14, 171–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beveridge, C.A. Long-distance signalling and a mutational analysis of branching in pea. J. Plant Growth Regul. 2000, 32, 193–203. [Google Scholar] [CrossRef]
- Beveridge, C.A.; Symons, G.M.; Turnbull, C.G. Auxin inhibition of decapitation-induced branching is dependent on graft-transmissible signals regulated by genes RMS1 and RMS2. Plant Physiol. 2000, 123, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Brewer, P.B.; Dun, E.A.; Ferguson, B.J.; Rameau, C.; Beveridge, C.A. Strigolactone acts downstream of auxin to regulate bud outgrowth in pea and Arabidopsis. Plant Physiol. 2009, 150, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Thomson, K.S.; Hertel, R.; Müller, S.; Tavares, J.E. 1-N-naphthylphthalamic acid and 2,3,5-triiodobenzoic acid: In-vitro binding to particulate cell fractions and action on auxin transport in corn coleoptiles. Planta 1973, 109, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, J.R.; Garrido, G.; Acosta, M.; Sánchez-Bravo, J. Influence of 2,3,5-Triiodobenzoic Acid and 1-N-Naphthylphthalamic Acid on Indoleacetic Acid Transport in Carnation Cuttings: Relationship with Rooting. J. Plant Growth Regul. 1999, 18, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Teale, W.; Palme, K. Naphthylphthalamic acid and the mechanism of polar auxin transport. J. Exp. Bot. 2018, 69, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Skoog, F.; Thimann, K.V. Further Experiments on the Inhibition of the Development of Lateral Buds by Growth Hormone. Proc. Natl. Acad. Sci. USA 1934, 19, 714–716. [Google Scholar] [CrossRef]
- Leibfried, A.; To, J.P.C.; Busch, W.; Stehling, S.; Kehle, A.; Demar, M.; Kieber, J.J.; Lohmann, J.U. WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature 2005, 438, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Chickarmane, V.S.; Gordon, S.P.; Tarr, P.T.; Heisler, M.G.; Meyerowitz, E.M. Cytokinin signaling as a positional cue for patterning the apical-basal axis of the growing Arabidopsis shoot meristem. Proc. Natl. Acad. Sci. USA 2012, 109, 4002–4007. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.P.; Chickarmane, V.S.; Ohno, C.; Meyerowitz, E.M. Multiple feedback loops through cytokinin signaling control stem cell number within the Arabidopsis shoot meristem. Proc. Natl. Acad. Sci. USA 2009, 106, 16529–16534. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Liu, Z.; Qiao, M.; Li, J.; Li, S.; Xiang, F. ARR12 promotes de novo shoot regeneration in Arabidopsis thaliana via activation of WUSCHEL expression. J. Integr. Plant Biol. 2017, 59, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Chen, H.; Huang, L.; ÓNeil, R.C.; Shiokhirev, M.N.; Ecker, J.R. A B-ARR-mediated cytokinin transcriptional network directs hormone cross-regulation and shoot development. Nat. Commun. 2018, 9, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.F.; Jürgens, G.; Laux, T. The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef]
- Brand, U.; Fletcher, J.C.; Hobe, M.; Meyerowitz, E.M.; Simon, R. Dependence of stem cell fate in Arabidopsis on a feedback loop regulated by CLV3 activity. Science 2000, 289, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Somssich, M.; Je, B.I.; Simon, R.; Jackson, D. CLAVATA-WUSCHEL signaling in the shoot meristem. Development 2016, 143, 3238–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kurepa, J.; Smalle, J. AXR1 promotes the Arabidopsis cytokinin response by facilitating ARR5 proteolysis. Plant J. 2013, 74, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, B.; Sheen, J. Cytokinin and auxin interplay in root stem-cell specification during early embryogenesis. Nature 2008, 453, 1094–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, E.L.; Alder, A.; Hunn, S.; Ferguson, J.; Lehtonen, M.T.; Scheler, B.; Kerres, K.L.; Wiedemann, G.; Safavi-Rizi, V.; Nordzieke, S.; et al. Strigolactone biosynthesis is evolutionarily conserved, regulated by phosphate starvation and contributes to resistance against phytopathogenic fungi in a moss Physcomitrella patens. New Phytol. 2017, 216, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, R.; Qian, Q.; Yan, M.; Meng, X.; Fu, Z.; Yan, C.; Jiang, B.; Su, Z.; Li, J.; et al. DWARF27, an iron-containing protein required for the biosynthesis of strigolactones, regulates rice tiller bud outgrowth. Plant Cell 2009, 21, 1512–1525. [Google Scholar] [CrossRef] [PubMed]
- Alder, A.; Jamil, M.; Marzorati, M.; Bruno, M.; Vermathen, M.; Bigler, P.; Ghisla, S.; Bouwmeester, H.; Beyer, P.; Al-Babili, S. The path from b-carotene to carlactone, a strigolactone-like plant hormone. Science 2012, 335, 1348–1351. [Google Scholar] [CrossRef] [PubMed]
- Hennig, L.; Gruissem, W.; Grossniklaus, U.; Köhler, C. Transcriptional programs of early reproductive stages in Arabidopsis. Plant Physiol. 2004, 135, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Doebley, J.; Stec, A.; Hubbard, L. The evolution of apical dominance in maize. Nature 1997, 386, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Braun, N.; de Saint Germain, A.; Pillot, J.P.; Boutet-Mercey, S.; Dalmais, M.; Antoniadi, I.; Li, X.; Maia-Grondard, A.; Signor, C.L.; Bouteiller, N.; et al. The pea TCP transcription factor PsBRC1 acts downstream of strigolactones to control shoot branching. Plant Physiol. 2012, 158, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Kebrom, T.H.; Burson, B.L.; Finlayson, S.A. Phytochrome B represses Teosinte Branched1 expression and induces sorghum axillary bud outgrowth in response to light signals. Plant Physiol. 2006, 140, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Minakuchi, K.; Kameoka, H.; Yasuno, N.; Umehara, M.; Luo, L.; Kobayashi, K.; Hanada, A.; Ueno, K.; Asami, T.; Yamaguchi, S.; et al. FINE CULM1 (FC1) works downstream of strigolactones to inhibit the outgrowth of axillary buds in rice. Plant Cell Physiol. 2010, 51, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Martín-Trillo, M.; Cubas, B. TCP genes: A family snapshot ten years later. Trends Plant Sci. 2009, 15, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Seale, M.; Bennett, T.; Leyser, O. BRC1 expression regulates bud activation potential but is not necessary or sufficient for bud growth inhibition in Arabidopsis. Development 2017, 144, 1661–1673. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, P.; Ge, L.; Yong, J.W.H.; Tan, S.N. Analytical methods for cytokinins. Trends Anal. Chem. 2009, 28, 323–335. [Google Scholar] [CrossRef]
- Ma, Z.; Ge, L.; Lee, A.S.Y.; Yong, J.W.H.; Tan, S.N.; Ong, E.S. Simultaneous analysis of different classes of phytohormones in coconut (Cocos nucifera L.) water using high-performance liquid chromatography and liquid chromatography-tandem mass spectrometry after solid-phase extraction. Anal. Chim. Acta 2008, 610, 274–281. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases (Gbp) | Q20 (%) | GC (%) | Mapped Rate (%) |
|---|---|---|---|---|---|---|
| G1_1 | 48,451,288 | 46,640,074 | 7.00 | 96.62 | 48.16 | 77.97 |
| G1_2 | 53,695,306 | 51,981,244 | 7.80 | 97.19 | 49.33 | 79.33 |
| G1_3 | 56,169,226 | 53,749,702 | 8.06 | 96.46 | 47.67 | 76.83 |
| D2_1 | 56,176,712 | 53,803,124 | 8.07 | 97.54 | 47.01 | 74.20 |
| D2_2 | 55,346,934 | 53,003,636 | 7.95 | 96.72 | 47.48 | 77.02 |
| D2_3 | 50,324,224 | 48,136,950 | 7.22 | 97.54 | 46.71 | 71.16 |
| TD2_1 | 46,568,478 | 45,272,406 | 6.79 | 97.39 | 47.49 | 72.62 |
| TD2_2 | 48,746,182 | 46,913,750 | 7.04 | 96.18 | 46.33 | 69.36 |
| TD2_3 | 45,843,728 | 44,538,914 | 6.68 | 96.91 | 46.57 | 71.04 |
| TG2_1 | 49,637,968 | 48,035,784 | 7.21 | 97.71 | 49.57 | 73.10 |
| TG2_2 | 54,961,460 | 53,625,420 | 8.04 | 96.58 | 48.75 | 74.09 |
| TG2_3 | 52,872,172 | 51,352,168 | 7.7 | 96.25 | 49.1 | 74.15 |
| Summary | 618,793,678 | 597,053,172 | ||||
| Genes | 239,732 | |||||
| Mean length | 921 bp | |||||
| N50 length | 1282 bp | |||||
| GO Accession | GO Term | No. of Background Genes in This GO Term | NO. of Differentially Expressed Genes in This GO Term | Corrected p Value |
|---|---|---|---|---|
| Biological process | ||||
| GO:0008207 | C21-steroid hormone metabolic process | 302 | 33 | 9.94 × 10−3 |
| GO:0034754 | cellular hormone metabolic process | 359 | 35 | 2.18 × 10−2 |
| GO:0042445 | hormone metabolic process | 406 | 36 | 4.12 × 10−2 |
| GO:0010817 | regulation of hormone levels | 443 | 37 | 3.58 × 10−2 |
| GO:0009755 | hormone-mediated signaling pathway | 190 | 15 | 3.53 × 10−2 |
| GO:0003707 | steroid hormone receptor activity | 65 | 8 | 3.56 × 10−2 |
| GO:0032870 | cellular response to hormone stimulus | 196 | 15 | 3.98 × 10−2 |
| GO:0009725 | response to hormone | 486 | 29 | 4.79 × 10−2 |
| GO:0016116 | carotenoid metabolic process | 282 | 33 | 6.20 × 10−3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, X.; Zhang, M.; Li, X.; Ye, R.; Wang, X. Transcriptome Profiles Reveal the Crucial Roles of Auxin and Cytokinin in the “Shoot Branching” of Cremastra appendiculata. Int. J. Mol. Sci. 2018, 19, 3354. https://doi.org/10.3390/ijms19113354
Lv X, Zhang M, Li X, Ye R, Wang X. Transcriptome Profiles Reveal the Crucial Roles of Auxin and Cytokinin in the “Shoot Branching” of Cremastra appendiculata. International Journal of Molecular Sciences. 2018; 19(11):3354. https://doi.org/10.3390/ijms19113354
Chicago/Turabian StyleLv, Xiang, Mingsheng Zhang, Xiaolan Li, Ruihua Ye, and Xiaohong Wang. 2018. "Transcriptome Profiles Reveal the Crucial Roles of Auxin and Cytokinin in the “Shoot Branching” of Cremastra appendiculata" International Journal of Molecular Sciences 19, no. 11: 3354. https://doi.org/10.3390/ijms19113354
APA StyleLv, X., Zhang, M., Li, X., Ye, R., & Wang, X. (2018). Transcriptome Profiles Reveal the Crucial Roles of Auxin and Cytokinin in the “Shoot Branching” of Cremastra appendiculata. International Journal of Molecular Sciences, 19(11), 3354. https://doi.org/10.3390/ijms19113354