Receptor Tyrosine Kinase-Targeted Cancer Therapy


Abstract
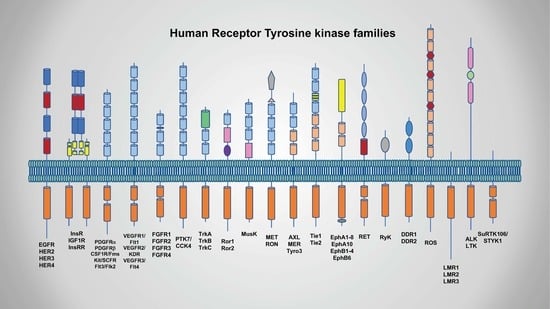
:
1. Introduction
2. Biology of RTKs and Link with Cancer
2.1. Classification and Characterization of RTKs
2.2. Mechanisms of Activation
2.3. EGFR/ERBB Family
2.3.1. EGFR
2.3.2. HER2
2.3.3. HER3
2.4. Anaplastic Lymphoma Kinase (ALK)
2.5. VEGFR, PDGF/kit and FGF
2.6. Hepatocyte Growth Factor (HGF)/Mesenchymal-Epithelial Transition Factor Receptor (MET)
2.7. Insulin/Insulin-Like Growth Factor (IGF) Receptor
3. EGFR Targeted Cancer Therapy, Resistance, & Overcoming Resistance
3.1. Cancer Therapy Targeting EGFR
3.1.1. EGFR TKIs
3.1.2. Anti-EGFR mAbs
3.2. Resistance Mechanisms to EGFR TKIs and Overcoming Resistance
3.2.1. Secondary Mutation of EGFR
3.2.2. Activation of Alternative Pathways
3.2.3. Phenotypic Transformation
3.2.4. Resistance to Apoptotic Cell Death
3.3. Resistance Mechanisms to Anti-EGFR mAbs and Overcoming Resistance
4. HER2 Targeted Cancer Therapy, Resistance, & Overcoming Resistance
4.1. Cancer Therapies Targeting HER2
4.2. Resistance & Overcoming Resistance to Anti-HER2 Therapies
4.2.1. Obstacles in Drug Binding to Her2
4.2.2. Emergence of Bypass Signaling
4.2.3. Failure of Host ADCC Response
5. ALK Targeted Cancer Therapy, Resistance, & Overcoming Resistance
5.1. Targeting of ALK Fusion Protein
5.2. ALK TKI Resistance & Overcoming Resistance
6. VEGF(R) Targeted Cancer Therapy, Resistance, & Overcoming Resistance
6.1. Targeted Therapy to Tyrosine Kinase Domains Including VEGFRs
6.2. Targeted Therapy to VEGF Family and Their Receptors
6.3. Resistance Mechanisms to Anti-VEGF(R) Therapies
6.3.1. FGF(R)
6.3.2. ANG and TIE2
6.3.3. PDGF(R)
6.3.4. HGF and MET
7. Other RTK-Targeted Cancer Therapies: MET/FGF(R)/IGF1R
7.1. MET
7.1.1. MET TKIs
7.1.2. MET mAbs
7.1.3. Resistance to MET Inhibitors
7.2. FGF(R)
7.2.1. Non-Selective FGFR TKIs
7.2.2. Selective FGFR TKIs
7.2.3. FGFR mAbs and FGF Ligand Traps
7.2.4. Resistance to FGFR Inhibitors
7.3. IGF1R
7.3.1. IGF1R-Targeted mAbs and IGF Ligand-Neutralizing mAbs
7.3.2. IGF1R TKIs
7.3.3. Resistance to IGF1R inhibitors
7.4. c-KIT
7.4.1. c-KIT-TKI, Imatinib
7.4.2. Resistance Mechanisms to c-KIT-TKI, Imatinib and Overcoming Resistance
Secondary Mutations in c-KIT
Genomic Amplification of c-KIT
Loss of c-KIT Expression and the Alternative Signaling Activation
8. RTKs on Cancer Therapy
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the erbb signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Engelman, J.A. Erbb receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C.; et al. Targeted disruption of mouse egf receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995, 269, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.J.; Arteaga, C.L.; Muthuswamy, S.K.; Siegel, P.M.; Webster, M.A.; Cardiff, R.D.; Meise, K.S.; Li, F.; Halter, S.A.; Coffey, R.J. Synergistic interaction of the NEU proto-oncogene product and transforming growth factor alpha in the mammary epithelium of transgenic mice. Mol. Cell. Biol. 1996, 16, 5726–5736. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Fukui, Y.; Ueyama, Y.; Tamaoki, N.; Kawamoto, T.; Taniguchi, S.; Shibuya, M. Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c-ERBB) in human brain tumors. Mol. Cell. Biol. 1988, 8, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wen, P.Y.; Mellinghoff, I.K. Targeted molecular therapies against epidermal growth factor receptor: Past experiences and challenges. Neurol. Oncol. 2014, 16 (Suppl. 8), 7–13. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to GEFITINIB. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. Egfr mutations in lung cancer: Correlation with clinical response to GEFITINIB therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.D.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. Somatic mutations of EGFR in colorectal cancers and GLIOBLASTOMAS. N. Engl. J. Med. 2004, 351, 2883. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, C.; Li, F.; Wang, X. Egfr gene copy number as a prognostic marker in colorectal cancer patients treated with cetuximab or panitumumab: A systematic review and meta analysis. PLoS ONE 2013, 8, e56205. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for her2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Corso, S.; Giordano, S. Targeted therapies for gastric cancer: Failures and hopes from clinical trials. Oncotarget 2017, 8, 57654–57669. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. Alk, the chromosome 2 gene locus altered by the t(2;5) in non-hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.; Kadomatsu, K.; Yuasa, S.; Muramatsu, H.; Mamiya, T.; Nabeshima, T.; Fan, Q.W.; Ishiguro, K.; Igakura, T.; Matsubara, S.; et al. Disruption of the midkine gene (MDK) resulted in altered expression of a calcium binding protein in the hippocampus of infant mice and their abnormal behaviour. Genes Cells 1998, 3, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming eml4-alk fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.N.; Bloom, R.; Forys, J.T.; Hiken, J.; Armstrong, J.R.; Branson, J.; McNulty, S.; Velu, P.D.; Pepin, K.; Abel, H.; et al. Genomic heterogeneity of alk fusion breakpoints in non-small-cell lung cancer. Mod. Pathol. 2018, 31, 791–808. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirks, W.G.; Fahnrich, S.; Lis, Y.; Becker, E.; MacLeod, R.A.; Drexler, H.G. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int. J. Cancer 2002, 100, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- DiSalvo, J.; Bayne, M.L.; Conn, G.; Kwok, P.W.; Trivedi, P.G.; Soderman, D.D.; Palisi, T.M.; Sullivan, K.A.; Thomas, K.A. Purification and characterization of a naturally occurring vascular endothelial growth factor.Placenta growth factor heterodimer. J. Biol. Chem. 1995, 270, 7717–7723. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vegf and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Bohman, S.; Dixelius, J.; Berge, T.; Dimberg, A.; Magnusson, P.; Wang, L.; Wikner, C.; Qi, J.H.; Wernstedt, C.; et al. Vegf receptor-2 y951 signaling and a role for the adapter molecule tsad in tumor angiogenesis. EMBO J. 2005, 24, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Warner, A.J.; Lopez-Dee, J.; Knight, E.L.; Feramisco, J.R.; Prigent, S.A. The shc-related adaptor protein, sck, forms a complex with the vascular-endothelial-growth-factor receptor KDR in transfected cells. Biochem. J. 2000, 347, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, K.; Cross, M.J.; Rolny, C.; Hagerkvist, R.; Rahimi, N.; Matsumoto, T.; Claesson-Welsh, L.; Welsh, M. The adaptor protein SHB binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J. Biol. Chem. 2004, 279, 22267–22275. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Wernstedt, C.; Engstrom, U.; Claesson-Welsh, L. Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of sh2 domain-containing molecules. J. Biol. Chem. 1998, 273, 23410–23418. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Wang, G.; Thomas, K.A. Identification of a natural soluble form of the vascular endothelial growth factor receptor, flt-1, and its heterodimerization with KDR. Biochem. Biophys. Res. Commun. 1996, 226, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.D.; Rowinsky, E.K.; Youssoufian, H.; Pytowski, B.; Wu, Y. Vascular endothelial growth factor receptor-1 in human cancer: Concise review and rationale for development of imc-18f1 (human antibody targeting vascular endothelial growth factor receptor-1). Cancer 2010, 116, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Ohashi, A.; Nishida, T.; Isozaki, K.; Kinoshita, K.; Shinomura, Y.; Kitamura, Y. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003, 125, 660–667. [Google Scholar] [CrossRef]
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. Pdgfra activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Hirota, S.; Taniguchi, M.; Hashimoto, K.; Isozaki, K.; Nakamura, H.; Kanakura, Y.; Tanaka, T.; Takabayashi, A.; Matsuda, H.; et al. Familial gastrointestinal stromal tumours with germline mutation of the kit gene. Nat. Genet. 1998, 19, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Nishida, T.; Isozaki, K.; Taniguchi, M.; Nishikawa, K.; Ohashi, A.; Takabayashi, A.; Obayashi, T.; Okuno, T.; Kinoshita, K.; et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of kit gene. Gastroenterology 2002, 122, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting fgfr signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Dutt, A.; Ramos, A.H.; Hammerman, P.S.; Mermel, C.; Cho, J.; Sharifnia, T.; Chande, A.; Tanaka, K.E.; Stransky, N.; Greulich, H.; et al. Inhibitor-sensitive fgfr1 amplification in human non-small cell lung cancer. PLoS ONE 2011, 6, e20351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis-Filho, J.S.; Simpson, P.T.; Turner, N.C.; Lambros, M.B.; Jones, C.; Mackay, A.; Grigoriadis, A.; Sarrio, D.; Savage, K.; Dexter, T.; et al. Fgfr1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin. Cancer Res. 2006, 12, 6652–6662. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Arao, T.; Hamaguchi, T.; Shimada, Y.; Kato, K.; Oda, I.; Taniguchi, H.; Koizumi, F.; Yanagihara, K.; Sasaki, H.; et al. Fgfr2 gene amplification and clinicopathological features in gastric cancer. Br. J. Cancer 2012, 106, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, A.O.; Patey, S.J.; Kan, S.H.; van den Ouweland, A.M.; Hamel, B.C. Fgfs, their receptors, and human limb malformations: Clinical and molecular correlations. Am. J. Med. Genet. 2002, 112, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.C.; Robertson, S.C.; Kanemitsu, M.Y.; Meyer, A.N.; Tynan, J.A.; Donoghue, D.J. Transformation and stat activation by derivatives of fgfr1, fgfr3, and fgfr4. Oncogene 2000, 19, 3309–3320. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of fgfr and tacc genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Ooi, A.; Kobayashi, M.; Mai, M.; Yanagihara, K.; Nakanishi, I. Amplification of C-MYC, K-SAM, and c-met in gastric cancers: Detection by fluorescence in situ hybridization. Lab. Investig. J. Tech. Methods Pathol. 1998, 78, 1143–1153. [Google Scholar]
- Tong, C.Y.; Hui, A.B.; Yin, X.L.; Pang, J.C.; Zhu, X.L.; Poon, W.S.; Ng, H.K. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. J. Neurosurg. 2004, 100, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. Met amplification occurs with or without t790m mutations in egfr mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Ferro, S.; Prat, M.; Bongarzone, I.; Pilotti, S.; Belfiore, A.; Costantino, A.; Vigneri, R.; Pierotti, M.A.; et al. Overexpression of the c-met/hgf receptor gene in human thyroid carcinomas. Oncogene 1992, 7, 2549–2553. [Google Scholar] [PubMed]
- Hiscox, S.E.; Hallett, M.B.; Puntis, M.C.; Nakamura, T.; Jiang, W.G. Expression of the hgf/sf receptor, c-met, and its ligand in human colorectal cancers. Cancer Investig. 1997, 15, 513–521. [Google Scholar] [CrossRef]
- Furukawa, T.; Duguid, W.P.; Kobari, M.; Matsuno, S.; Tsao, M.S. Hepatocyte growth factor and met receptor expression in human pancreatic carcinogenesis. Am. J. Pathol. 1995, 147, 889–895. [Google Scholar] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Katsaros, D.; Crepaldi, T.; Gaglia, P.; Zola, P.; Sismondi, P.; Comoglio, P.M. Overexpression of the met/hgf receptor in ovarian cancer. Int. J. Cancer 1994, 58, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Prechtel, D.; Resau, J.H.; Gauger, K.; Welk, A.; Lindemann, K.; Salanti, G.; Richter, T.; Knudsen, B.; Vande Woude, G.F.; et al. C-met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of her2/neu. Int. J. Cancer 2005, 113, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Junker, K.; Nakaigawa, N.; Kinjerski, T.; Weirich, G.; Miller, M.; Lubensky, I.; Neumann, H.P.; Brauch, H.; Decker, J.; et al. Novel mutations of the met proto-oncogene in papillary renal carcinomas. Oncogene 1999, 18, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.S.; Rosen, O.M. The role of insulin receptor autophosphorylation in signal transduction. J. Biol. Chem. 1991, 266, 22653–22660. [Google Scholar] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Ando, A.; Yonezawa, K.; Gout, I.; Nakata, T.; Ueda, H.; Hara, K.; Kitamura, Y.; Noda, Y.; Takenawa, T.; Hirokawa, N.; et al. A complex of grb2-dynamin binds to tyrosine-phosphorylated insulin receptor substrate-1 after insulin treatment. EMBO J. 1994, 13, 3033–3038. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Burgess, A.W.; Clayton, A.H.; Scott, A.M. Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer Res. 2012, 72, 2924–2930. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sooro, M.A.; Zhang, N.; Zhang, P. Targeting egfr-mediated autophagy as a potential strategy for cancer therapy. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohba, M.; Ohmori, T. Molecular-targeted therapies for epidermal growth factor receptor and its resistance mechanisms. Int. J. Mol. Sci. 2017, 18, 2420. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; Navaratnam, S.; Pitz, M.W.; Maniate, J.M.; Wiechec, E.; Baust, H.; Gingerich, J.; Skliris, G.P.; Murphy, L.C.; Los, M. Targeting the EGFR pathway for cancer therapy. Curr. Med. Chem. 2006, 13, 3483–3492. [Google Scholar] [CrossRef] [PubMed]
- Metro, G.; Finocchiaro, G.; Cappuzzo, F. Anti-cancer therapy with egfr inhibitors: Factors of prognostic and predictive significance. Ann. Oncol. 2006, 17 (Suppl. 2), 42–45. [Google Scholar] [CrossRef]
- Giaccone, G.; Gonzalez-Larriba, J.L.; van Oosterom, A.T.; Alfonso, R.; Smit, E.F.; Martens, M.; Peters, G.J.; van der Vijgh, W.J.; Smith, R.; Averbuch, S.; et al. Combination therapy with gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, gemcitabine and cisplatin in patients with advanced solid tumors. Ann. Oncol. 2004, 15, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrelli, F.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Barni, S. Cetuximab and panitumumab in kras wild-type colorectal cancer: A meta-analysis. Int. J. Colorectal Dis. 2011, 26, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Lima, C.M.; Soares, H.P.; Raez, L.E.; Singal, R. Egfr targeting of solid tumors. Cancer Control 2007, 14, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, C.; Baldessari, C.; Napolitano, M.; Orsi, G.; Grizzi, G.; Bertolini, F.; Barbieri, F.; Cascinu, S. Resistance to egfr inhibitors in non-small cell lung cancer: Clinical management and future perspectives. Crit. Rev. Oncol. Hematol. 2018, 123, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. Egfr antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Xie, C.; Yu, X.; Liu, J. Egfr tki as first-line treatment for patients with advanced egfr mutation-positive non-small-cell lung cancer. Oncotarget 2017, 8, 75712–75726. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yan, H. Skin toxicity with anti-egfr monoclonal antibody in cancer patients: A meta-analysis of 65 randomized controlled trials. Cancer Chemother. Pharmacol. 2018, 82, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Gorden, K.J.; Mesbah, P.; Kolesar, J.M. Egfr inhibitors as first-line therapy in egfr mutation-positive patients with nsclc. J. Oncol. Pharm. Pract. 2012, 18, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.B. Nondermatologic adverse events associated with anti-egfr therapy. Oncology 2006, 20, 35–40. [Google Scholar] [PubMed]
- Vogel, W.H.; Jennifer, P. Management strategies for adverse events associated with egfr tkis in non-small cell lung cancer. J. Adv. Pract. Oncol. 2016, 7, 723–735. [Google Scholar] [PubMed]
- Diaz-Serrano, A.; Gella, P.; Jimenez, E.; Zugazagoitia, J.; Paz-Ares Rodriguez, L. Targeting egfr in lung cancer: Current standards and developments. Drugs 2018, 79, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Sim, E.H.; Yang, I.A.; Wood-Baker, R.; Bowman, R.V.; Fong, K.M. Gefitinib for advanced non-small cell lung cancer. Cochrane Database Syst. Rev. 2018, 1, Cd006847. [Google Scholar] [CrossRef] [PubMed]
- Landi, L.; Cappuzzo, F. Experience with erlotinib in the treatment of non-small cell lung cancer. Ther. Adv. Respir. Dis. 2015, 9, 146–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genova, C.; Rijavec, E.; Barletta, G.; Burrafato, G.; Biello, F.; Dal Bello, M.G.; Coco, S.; Truini, A.; Alama, A.; Boccardo, F.; et al. Afatinib for the treatment of advanced non-small-cell lung cancer. Expert Opin. Pharmacother. 2014, 15, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Khanal, R.; Sharma, A.; Yan, F.; Sharma, N. Afatinib and lung cancer. Expert Rev. Anticancer Ther. 2014, 14, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Afatinib: A review in advanced non-small cell lung cancer. Targeted Oncol. 2016, 11, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Wirth, S.M. Afatinib in non-small cell lung cancer. J. Adv. Pract. Oncol. 2015, 6, 448–455. [Google Scholar] [PubMed]
- Brzezniak, C.; Carter, C.A.; Giaccone, G. Dacomitinib, a new therapy for the treatment of non-small cell lung cancer. Expert Opin. Pharmacother. 2013, 14, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.G.; Vallee, A.; Theoleyre, S. Egfr t790m resistance mutation in non small-cell lung carcinoma. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 444, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to egfr targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. Egfr-tkis resistance via egfr-independent signaling pathways. Mol. Cancer 2018, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Soejima, K.; Yasuda, H.; Hirano, T. Osimertinib for egfr t790m mutation-positive non-small cell lung cancer. Expert Rev. Clin. Pharmacol. 2017, 10, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cang, S.; Liu, D. Third-generation inhibitors targeting egfr t790m mutation in advanced non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, Z.; Li, Y.; Zhou, Q. Development of epidermal growth factor receptor tyrosine kinase inhibitors against egfr t790m. Mutation in non small-cell lung carcinoma. Open Med. 2016, 11, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, H.; Katakami, N.; Okamoto, I.; Kato, T.; Kim, Y.H.; Imamura, F.; Shinkai, M.; Hodge, R.A.; Uchida, H.; Hida, T. Osimertinib in japanese patients with egfr t790m mutation-positive advanced non-small-cell lung cancer: Aura3 trial. Cancer Sci. 2018, 109, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated egfr-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
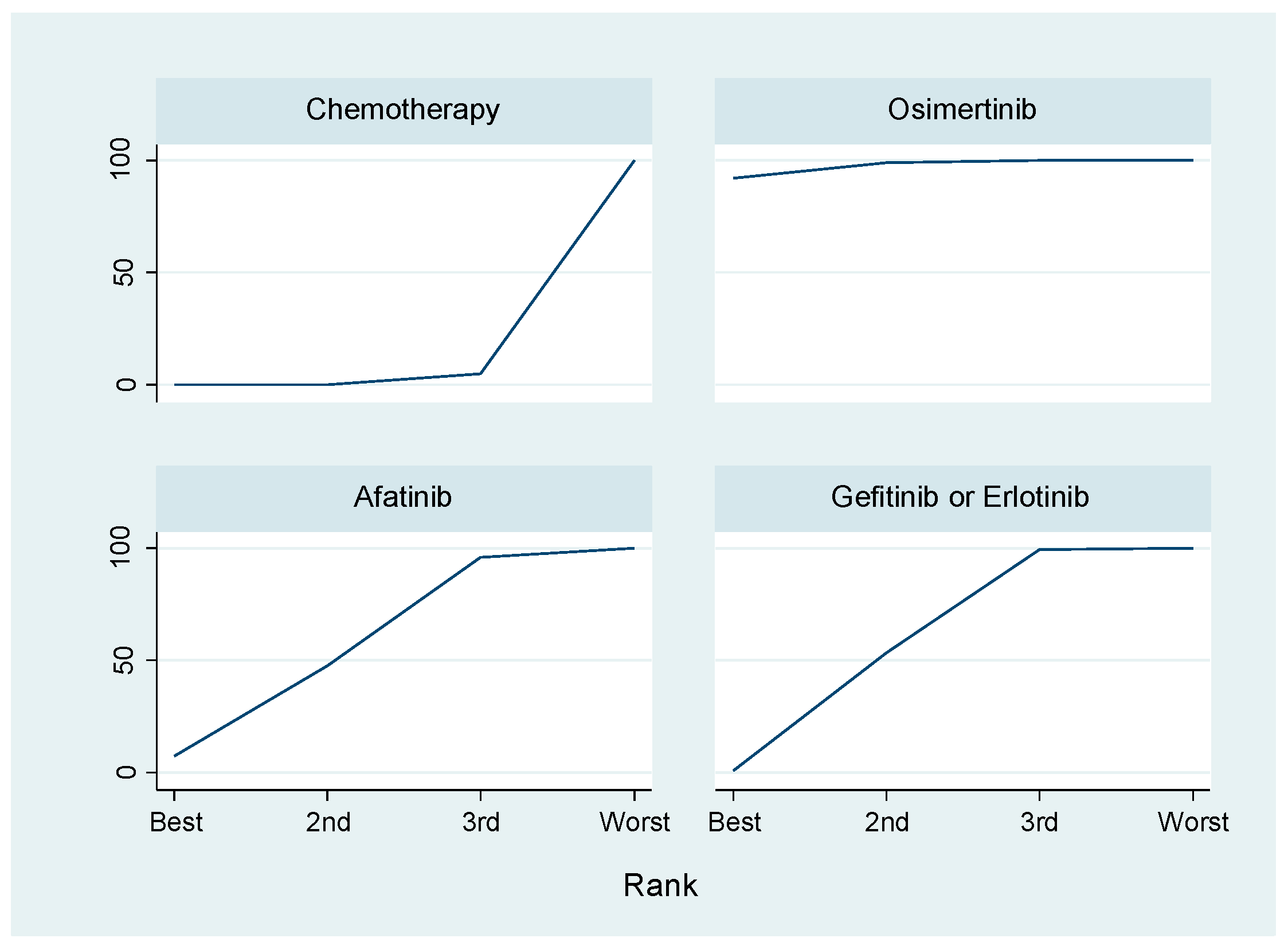
- Lin, J.Z.; Ma, S.K.; Wu, S.X.; Yu, S.H.; Li, X.Y. A network meta-analysis of nonsmall-cell lung cancer patients with an activating egfr mutation: Should osimertinib be the first-line treatment? Medicine 2018, 97, e11569. [Google Scholar] [CrossRef] [PubMed]
- White, I.R. Network meta-analysis. Stata J. 2015, 15, 951–985. [Google Scholar]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of asian patients with advanced non-small-cell lung cancer harbouring egfr mutations (lux-lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced egfr mutation-positive non-small-cell lung cancer (optimal, ctong-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (wjtog3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Alorabi, M.; Shonka, N.A.; Ganti, A.K. Egfr monoclonal antibodies in locally advanced head and neck squamous cell carcinoma: What is their current role? Crit. Rev. Oncol. Hematol. 2016, 99, 170–179. [Google Scholar] [CrossRef] [PubMed]
- van Helden, E.J.; Menke-van der Houven van Oordt, C.W.; Heymans, M.W.; Ket, J.C.F.; van den Oord, R.; Verheul, H.M.W. Optimal use of anti-egfr monoclonal antibodies for patients with advanced colorectal cancer: A meta-analysis. Cancer Metastasis Rev. 2017, 36, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-folfox4 treatment and ras mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fu, L. Mechanisms of resistance to egfr tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to egfr-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.A.; O’Kane, G.M.; Vincent, M.D.; Leighl, N.B. Third-generation tyrosine kinase inhibitors targeting epidermal growth factor receptor mutations in non-small cell lung cancer. Front. Oncol. 2017, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Franchina, T.; Ricciardi, G.R.R.; Smiroldo, V.; Picciotto, M.; Zanghi, M.; Rolfo, C.; Adamo, V. Third generation egfr tkis in egfr-mutated nsclc: Where are we now and where are we going. Crit. Rev. Oncol. Hematol. 2017, 117, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Kumarakulasinghe, N.B.; Huang, Y.Q.; Ang, Y.L.E.; Choo, J.R.; Goh, B.C.; Soo, R.A. Third generation egfr tkis: Current data and future directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or platinum-pemetrexed in egfr t790m-positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yu, L.; Zhang, Z.; Ren, X.; Smaill, J.B.; Ding, K. Targeting EGFR(l858r/t790m) and EGFR(l858r/t790m/c797s) resistance mutations in NSCLC: Current developments in medicinal chemistry. Med. Res. Rev. 2018. [Google Scholar] [CrossRef]
- Wu, J.Y.; Shih, J.Y. Effectiveness of tyrosine kinase inhibitors on uncommon e709x epidermal growth factor receptor mutations in non-small-cell lung cancer. OncoTargets Ther. 2016, 9, 6137–6145. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Narayan, R.N.; Horton, L.; Patel, T.R.; Habib, A.A. The role of EGFR-met interactions in the pathogenesis of glioblastoma and resistance to treatment. Current Cancer Drug Targets 2017, 17, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Agwa, E.S.; Ma, P.C. Targeting the met receptor tyrosine kinase in non-small cell lung cancer: Emerging role of tivantinib. Cancer Manag. Res. 2014, 6, 397–404. [Google Scholar] [PubMed]
- Abdelaziz, A.; Vaishampayan, U. Cabozantinib for the treatment of kidney cancer. Expert Rev. Anticancer Ther. 2017, 17, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Kishino, Y.; Murata, Y.; Kusumoto, S.; Ishida, H.; Shirai, T.; Hirose, T.; et al. Acquired resistance mechanisms to combination met-TKI/EGFR-TKI exposure in met-amplified EGFR-TKI-resistant lung adenocarcinoma harboring an activating EGFR mutation. Mol. Cancer Ther. 2016, 15, 3040–3054. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohba, M.; Arata, S.; Ohmori, T. Establishing dual resistance to EGFR-TKI and met-TKI in lung adenocarcinoma cells in vitro with a 2-step dose-escalation procedure. J. Vis. Exp. 2017, 126, e55967. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Murata, Y.; Kusumoto, S.; Ando, K.; Ishida, H.; Ohnishi, T.; Sasaki, Y. Distinct afatinib resistance mechanisms identified in lung adenocarcinoma harboring an EGFR mutation. Mol. Cancer Res. 2017, 15, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Ohmori, T.; Inoue, F.; Kadofuku, T.; Hosaka, T.; Ishida, H.; Shirai, T.; Okuda, K.; Hirose, T.; Horichi, N.; et al. Enhancement of sensitivity to tumor necrosis factor alpha in non-small cell lung cancer cells with acquired resistance to GEFITINIB. Clin. Cancer Res. 2005, 11, 8872–8879. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. Fas and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.L.; Tan, S.Z.; Liu, G.; Tsao, M.S. Known and putative mechanisms of resistance to egfr targeted therapies in NSCLC patients with EGFR mutations—A review. Transl. Lung Cancer Res. 2015, 4, 67–81. [Google Scholar] [PubMed]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type kras is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Weickhardt, A.J.; Price, T.J.; Chong, G.; Gebski, V.; Pavlakis, N.; Johns, T.G.; Azad, A.; Skrinos, E.; Fluck, K.; Dobrovic, A.; et al. Dual targeting of the epidermal growth factor receptor using the combination of cetuximab and erlotinib: Preclinical evaluation and results of the phase ii dux study in chemotherapy-refractory, advanced colorectal cancer. J. Clin. Oncol. 2012, 30, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Eng, C.; Nowara, E.; Swieboda-Sadlej, A.; Tebbutt, N.C.; Mitchell, E.; Davidenko, I.; Stephenson, J.; Elez, E.; Prenen, H.; et al. Randomized phase IB/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin. Cancer Res. 2014, 20, 4240–4250. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.D.; Figari, I.; Fendly, B.; Wong, W.L.; Carter, P.; Gorman, C.; Shepard, H.M. Differential responses of human tumor cell lines to anti-p185her2 monoclonal antibodies. Cancer Immunol. Immunother. 1993, 37, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. Pten activation contributes to tumor inhibition by trastuzumab, and loss of pten predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. Targeting the function of the her2 oncogene in human cancer therapeutics. Oncogene 2007, 26, 6577–6592. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Fan, X.; Meng, W.; Deng, H.; Zhang, N.; An, Z. Engagement of immune effector cells by trastuzumab induces her2/erbb2 downregulation in cancer cells through stat1 activation. Breast Cancer Res. 2014, 16, R33. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J. Emerging approaches for treating her2-positive metastatic breast cancer beyond trastuzumab. Ann. Oncol. 2013, 24, 2492–2500. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effects of the novel, reversible epidermal growth factor receptor/erbb-2 tyrosine kinase inhibitor, gw2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar] [PubMed]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of hki-272, an orally active, irreversible inhibitor of the her-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef] [PubMed]
- Christianson, T.A.; Doherty, J.K.; Lin, Y.J.; Ramsey, E.E.; Holmes, R.; Keenan, E.J.; Clinton, G.M. Nh2-terminally truncated her-2/neu protein: Relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res. 1998, 58, 5123–5129. [Google Scholar] [PubMed]
- Molina, M.A.; Saez, R.; Ramsey, E.E.; Garcia-Barchino, M.J.; Rojo, F.; Evans, A.J.; Albanell, J.; Keenan, E.J.; Lluch, A.; Garcia-Conde, J.; et al. Nh(2)-terminal truncated her-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin. Cancer Res. 2002, 8, 347–353. [Google Scholar] [PubMed]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95her2, a truncated form of the her2 receptor, and response to anti-her2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.; Hunter, C.; Bignell, G.; Edkins, S.; Davies, H.; Teague, J.; Stevens, C.; O’Meara, S.; Smith, R.; Parker, A.; et al. Lung cancer: Intragenic erbb2 kinase mutations in tumours. Nature 2004, 431, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.; Lingen, M.W.; Martin, L.E.; Harris, P.L.; Brannigan, B.W.; Haserlat, S.M.; Okimoto, R.A.; Sgroi, D.C.; Dahiya, S.; Muir, B.; et al. Response of some head and neck cancers to epidermal growth factor receptor tyrosine kinase inhibitors may be linked to mutation of erbb2 rather than EGFR. Clin. Cancer Res. 2005, 11, 8105–8108. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Soung, Y.H.; Seo, S.H.; Kim, S.Y.; Park, C.H.; Wang, Y.P.; Park, K.; Nam, S.W.; Park, W.S.; Kim, S.H.; et al. Somatic mutations of erbb2 kinase domain in gastric, colorectal, and breast carcinomas. Clin. Cancer Res. 2006, 12, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Trowe, T.; Boukouvala, S.; Calkins, K.; Cutler, R.E., Jr.; Fong, R.; Funke, R.; Gendreau, S.B.; Kim, Y.D.; Miller, N.; Woolfrey, J.R.; et al. Exel-7647 inhibits mutant forms of ERBB2 associated with lapatinib resistance and neoplastic transformation. Clin. Cancer Res. 2008, 14, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Brewer, M.R.; Sheehan, J.H.; Koch, J.P.; Sliwoski, G.R.; Nagy, R.; Lanman, R.; Berger, M.F.; Hyman, D.M.; Solit, D.B.; et al. An acquired HER2(T798I) gatekeeper mutation induces resistance to neratinib in a patient with her2 mutant-driven breast cancer. Cancer Discov. 2017, 7, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Wu, Y.; Scaltriti, M.; Meric-Bernstam, F.; Hunt, K.K.; Dawood, S.; Esteva, F.J.; Buzdar, A.U.; Chen, H.; Eksambi, S.; et al. Loss of her2 amplification following trastuzumab-based neoadjuvant systemic therapy and survival outcomes. Clin. Cancer Res. 2009, 15, 7381–7388. [Google Scholar] [CrossRef] [PubMed]
- Garrett, J.T.; Olivares, M.G.; Rinehart, C.; Granja-Ingram, N.D.; Sanchez, V.; Chakrabarty, A.; Dave, B.; Cook, R.S.; Pao, W.; McKinely, E.; et al. Transcriptional and posttranslational up-regulation of her3 (erbb3) compensates for inhibition of the her2 tyrosine kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 5021–5026. [Google Scholar] [CrossRef] [PubMed]
- Wehrman, T.S.; Raab, W.J.; Casipit, C.L.; Doyonnas, R.; Pomerantz, J.H.; Blau, H.M. A system for quantifying dynamic protein interactions defines a role for herceptin in modulating erbb2 interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 19063–19068. [Google Scholar] [CrossRef] [PubMed]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., III; Sweeney, C. Met receptor contributes to trastuzumab resistance of her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Kim, H.; Liska, D.; Gao, S.; Christensen, J.G.; Weiser, M.R. Met activation mediates resistance to lapatinib inhibition of her2-amplified gastric cancer cells. Mol. Cancer Ther. 2012, 11, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in her2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.N.; You, F.; Schnitt, S.J.; Witkiewicz, A.; Lu, X.; Sgroi, D.; Ryan, P.D.; Come, S.E.; Burstein, H.J.; Lesnikoski, B.A.; et al. Predictors of resistance to preoperative trastuzumab and vinorelbine for her2-positive early breast cancer. Clin. Cancer Res. 2007, 13, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L. Her3 and mutant EGFR meet met. Nat. Med. 2007, 13, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.Y.; Hong, J.Y.; Lee, H.J.; Park, H.J.; Lee, S.K. Targeting the degradation of axl receptor tyrosine kinase to overcome resistance in gefitinib-resistant non-small cell lung cancer. Oncotarget 2015, 6, 10146–10160. [Google Scholar] [CrossRef] [PubMed]
- Jameson, M.J.; Beckler, A.D.; Taniguchi, L.E.; Allak, A.; Vanwagner, L.B.; Lee, N.G.; Thomsen, W.C.; Hubbard, M.A.; Thomas, C.Y. Activation of the insulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous carcinoma cells. Mol. Cancer Ther. 2011, 10, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Jones, D.R.; Rodriguez-Viciana, P.; Gonzalez-Garcia, A.; Leonardo, E.; Wennstrom, S.; von Kobbe, C.; Toran, J.L.; L, R.B.; Calvo, V.; et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J. 1998, 17, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philp, A.J.; Campbell, I.G.; Leet, C.; Vincan, E.; Rockman, S.P.; Whitehead, R.H.; Thomas, R.J.; Phillips, W.A. The phosphatidylinositol 3’-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001, 61, 7426–7429. [Google Scholar] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Elster, N.; Cremona, M.; Morgan, C.; Toomey, S.; Carr, A.; O’Grady, A.; Hennessy, B.T.; Eustace, A.J. A preclinical evaluation of the pi3k alpha/delta dominant inhibitor bay 80-6946 in her2-positive breast cancer models with acquired resistance to the her2-targeted therapies trastuzumab and lapatinib. Breast Cancer Res. Treat. 2015, 149, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by SRC family kinases. Annu. Rev. Cell Dev. Biology 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Bonham, K. Src gene expression in human cancer: The role of transcriptional activation. Biochem. Cell Biol. 2004, 82, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Rexer, B.N.; Ham, A.J.; Rinehart, C.; Hill, S.; Granja-Ingram Nde, M.; Gonzalez-Angulo, A.M.; Mills, G.B.; Dave, B.; Chang, J.C.; Liebler, D.C.; et al. Phosphoproteomic mass spectrometry profiling links src family kinases to escape from her2 tyrosine kinase inhibition. Oncogene 2011, 30, 4163–4174. [Google Scholar] [CrossRef] [PubMed]
- Warmerdam, P.A.; van de Winkel, J.G.; Vlug, A.; Westerdaal, N.A.; Capel, P.J. A single amino acid in the second ig-like domain of the human fc gamma receptor ii is critical for human igg2 binding. J. Immunol. 1991, 147, 1338–1343. [Google Scholar] [PubMed]
- Koene, H.R.; Kleijer, M.; Algra, J.; Roos, D.; von dem Borne, A.E.; de Haas, M. Fc gammariiia-158v/f polymorphism influences the binding of igg by natural killer cell fc gammariiia, independently of the fc gammariiia-48l/r/h phenotype. Blood 1997, 90, 1109–1114. [Google Scholar] [PubMed]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin g fragment c receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with her-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Roca, L.; Dieras, V.; Roche, H.; Lappartient, E.; Kerbrat, P.; Cany, L.; Chieze, S.; Canon, J.L.; Spielmann, M.; Penault-Llorca, F.; et al. Correlation of her2, fcgr2a, and fcgr3a gene polymorphisms with trastuzumab related cardiac toxicity and efficacy in a subgroup of patients from unicancer-pacs 04 trial. Breast Cancer Res. Treat. 2013, 139, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, C.; Mogushi, K.; Morioka, M.S.; Yamamoto, H.; Tamura, K.; Fujiwara, Y.; Tanaka, H. Fc-gamma receptor polymorphism and gene expression of peripheral blood mononuclear cells in patients with her2-positive metastatic breast cancer receiving single-agent trastuzumab. Breast Cancer 2016, 23, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, alk, to a nucleolar protein gene, npm, in non-hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Mohanty, J.; Tome, F.; Puleo, D.E.; Plotnikov, A.N.; Ahmed, M.; Kaur, N.; Poliakov, A.; Cinnaiyan, A.M.; Lax, I.; et al. Identification of a biologically active fragment of ALK and LTK-ligand 2 (augmentor-alpha). Proc. Natl. Acad. Sci. USA 2018, 115, 8340–8345. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor alpha and beta (fam150) are ligands of the receptor tyrosine kinases alk and ltk: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor eml4-alk. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in alk-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
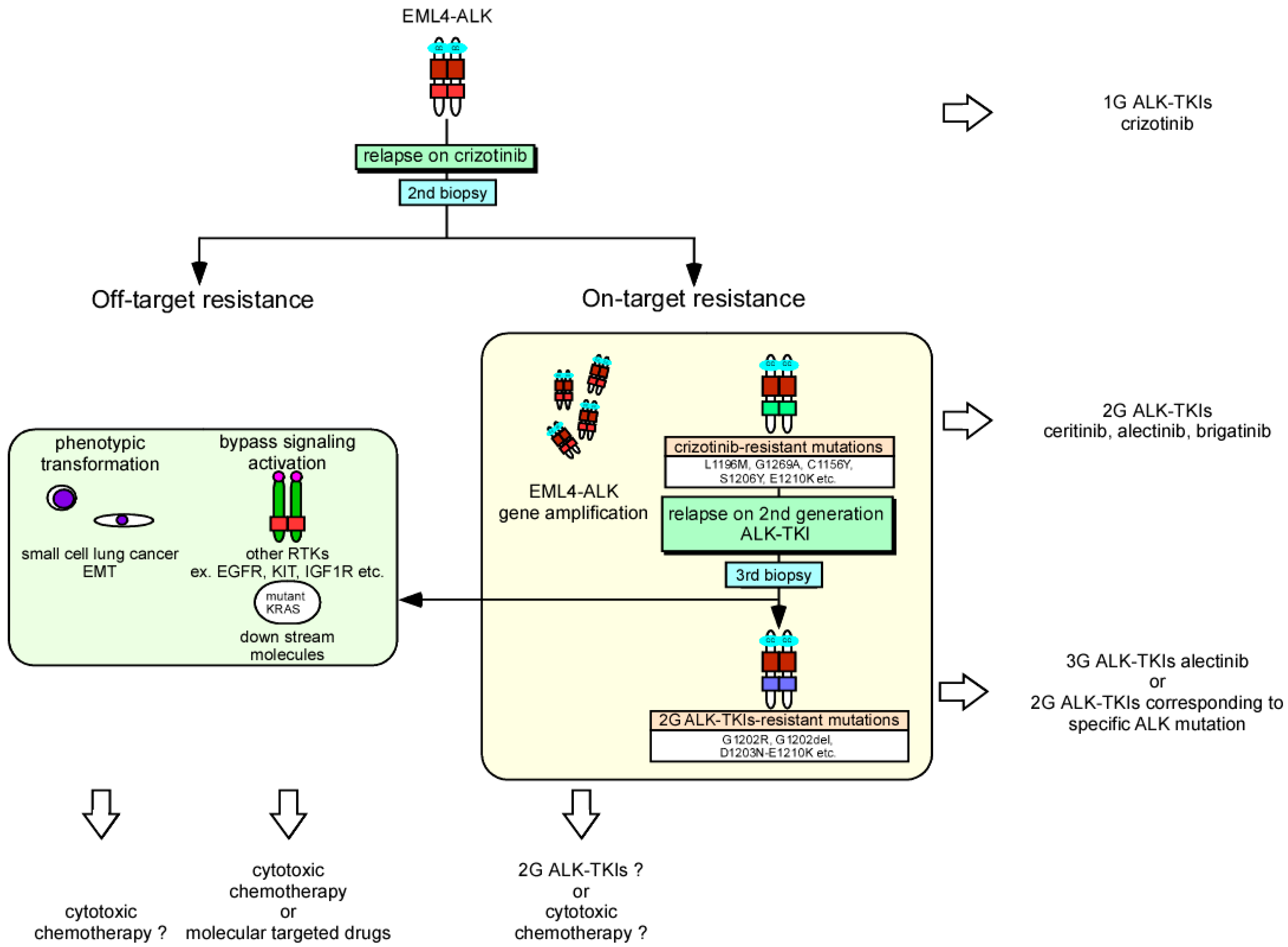
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in alk-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretic, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Cho, B.C.; Kim, H.R.; Lee, H.J.; Shim, H.S. A case of alk-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to crizotinib. J. Thorac. Oncol. 2016, 11, e55–e58. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation alk inhibitors in alk-rearranged lung cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, W.S.; Choi, Y.J.; Choi, C.M.; Rho, J.K.; Lee, J.C. Epithelial-mesenchymal transition leads to crizotinib resistance in h2228 lung cancer cells with eml4-alk translocation. Mol. Oncol. 2013, 7, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Dardaei, L.; Wang, H.Q.; Singh, M.; Fordjour, P.; Shaw, K.X.; Yoda, S.; Kerr, G.; Yu, K.; Liang, J.; Cao, Y.; et al. Shp2 inhibition restores sensitivity in alk-rearranged non-small-cell lung cancer resistant to alk inhibitors. Nat. Med. 2018, 24, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with alk gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision medicine takes on drug resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Sakashita, T.; Yanagitani, N.; Ninomiya, H.; Horiike, A.; Friboulet, L.; Gainor, J.F.; Motoi, N.; Dobashi, A.; Sakata, S.; et al. P-glycoprotein mediates ceritinib resistance in anaplastic lymphoma kinase-rearranged non-small cell lung cancer. EBioMedicine 2016, 3, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced alk-rearranged non-small-cell lung cancer (ascend-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Perol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus crizotinib in untreated alk-positive non-small-cell lung cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- FDA. Highlights of Prescribing Information. Available online: https://www.Accessdata.Fda.Gov/drugsatfda_docs/label/2017/208434s003lbl.Pdf (accessed on 21 May 2018).
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with alk-positive non-small-cell lung cancer (j-alex): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Kim, D.W.; Tiseo, M.; Ahn, M.J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: A randomized, multicenter phase ii trial. J. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with alk or ros1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. Targeting angiogenesis in cancer therapy: Moving beyond vascular endothelial growth factor. Oncologist 2015, 20, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Ilic, I.; Jankovic, S.; Ilic, M. Bevacizumab combined with chemotherapy improves survival for patients with metastatic colorectal cancer: Evidence from meta analysis. PLoS ONE 2016, 11, e0161912. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The aurelia open-label randomized phase iii trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (rainbow): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Tabernero, J.; Takayuki, Y.; Cohn, A.L. Ramucirumab versus placebo in combination with second-line folfiri in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (raise): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [PubMed]
- Garon, E.B.; Ciuleanu, T.E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.; et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage iv non-small-cell lung cancer after disease progression on platinum-based therapy (revel): A multicentre, double-blind, randomised phase 3 trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausová, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase iii randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of vegf signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Grünwald, V.; Ravaud, A.; Ou, Y.C.; Castellano, D.; Lin, C.C.; Gschwend, J.E.; Harzstark, A.; Beall, S.; Pirotta, N.; et al. Phase ii results of dovitinib (tki258) in patients with metastatic renal cell cancer. Clin. Cancer Res. 2014, 20, 3012–3022. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Porta, C.; Vogelzang, N.J.; Sternberg, C.N.; Szczylik, C.; Zolnierek, J.; Kollmannsberger, C.; Rha, S.Y.; Bjarnason, G.A.; Melichar, B.; et al. Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 286–296. [Google Scholar] [CrossRef]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Wyser Rmili, C.; Leow, C.C.; De Palma, M. Role of angiopoietin-2 in adaptive tumor resistance to vegf signaling blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Biel, N.M.; Siemann, D.W. Targeting the angiopoietin-2/tie-2 axis in conjunction with vegf signal interference. Cancer Lett 2016, 380, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Mamer, S.B.; Chen, S.; Weddell, J.C.; Palasz, A.; Wittenkeller, A.; Kumar, M.; Imoukhuede, P.I. Discovery of high-affinity pdgf-vegfr interactions: Redefining rtk dynamics. Sci. Rep. 2017, 7, 16439. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. Vegf inhibits tumor cell invasion and mesenchymal transition through a met/vegfr2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Hammers, H.J.; Donskov, F.; Roth, B.J.; Peltola, K.; et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (meteor): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef]
- Sattler, M.; Reddy, M.M.; Hasina, R.; Gangadhar, T.; Salgia, R. The role of the c-met pathway in lung cancer and the potential for targeted therapy. Ther. Adv. Med. Oncol. 2011, 3, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.T.; Tebbutt, N.C.; Davidenko, I.; Murad, A.M.; Al-Batran, S.E.; Ilson, D.H.; Tjulandin, S.; Gotovkin, E.; Karaszewska, B.; Bondarenko, I.; et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced met-positive gastric or gastro-oesophageal junction cancer (rilomet-1): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1467–1482. [Google Scholar] [CrossRef]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results from the phase iii randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage iiib or iv non-small-cell lung cancer: Metlung. J. Clin. Oncol. 2017, 35, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Bang, Y.J.; Lordick, F.; Alsina, M.; Chen, M.; Hack, S.P.; Bruey, J.M.; Smith, D.; McCaffery, I.; Shames, D.S.; et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in her2-negative, met-positive gastroesophageal adenocarcinoma: The metgastric randomized clinical trial. JAMA Oncol. 2017, 3, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced alk-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Cella, D.; Escudier, B.; Tannir, N.M.; Powles, T.; Donskov, F.; Peltola, K.; Schmidinger, M.; Heng, D.Y.C.; Mainwaring, P.N.; Hammers, H.J.; et al. Quality of life outcomes for cabozantinib versus everolimus in patients with metastatic renal cell carcinoma: Meteor phase iii randomized trial. J. Clin. Oncol. 2018, 36, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; De Bono, J.; Sternberg, C.; Le Moulec, S.; Oudard, S.; De Giorgi, U.; Krainer, M.; Bergman, A.; Hoelzer, W.; De Wit, R.; et al. Phase iii study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: Comet-1. J. Clin. Oncol. 2016, 34, 3005–3013. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Cepero, V.; Sierra, J.R.; Corso, S.; Ghiso, E.; Casorzo, L.; Perera, T.; Comoglio, P.M.; Giordano, S. Met and kras gene amplification mediates acquired resistance to met tyrosine kinase inhibitors. Cancer Res. 2010, 70, 7580–7590. [Google Scholar] [CrossRef] [PubMed]
- Bahcall, M.; Sim, T.; Paweletz, C.P.; Patel, J.D.; Alden, R.S.; Kuang, Y.; Sacher, A.G.; Kim, N.D.; Lydon, C.A.; Awad, M.M.; et al. Acquired metd1228v mutation and resistance to met inhibition in lung cancer. Cancer Discov. 2016, 6, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Corso, S.; Comoglio, P.M.; Giordano, S. Increase of met gene copy number confers resistance to a monovalent met antibody and establishes drug dependence. Mol. Oncol. 2014, 8, 1561–1574. [Google Scholar] [CrossRef] [PubMed]
- Corso, S.; Ghiso, E.; Cepero, V.; Sierra, J.R.; Migliore, C.; Bertotti, A.; Trusolino, L.; Comoglio, P.M.; Giordano, S. Activation of her family members in gastric carcinoma cells mediates resistance to met inhibition. Mol. Cancer 2010, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Migliore, C.; Morando, E.; Ghiso, E.; Anastasi, S.; Leoni, V.P.; Apicella, M.; Cora, D.; Sapino, A.; Pietrantonio, F.; De Braud, F.; et al. Mir-205 mediates adaptive resistance to met inhibition via errfi1 targeting and raised egfr signaling. EMBO Mol. Med. 2018, 10, e8746. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.H.; Chuah, C.; Guerci-Bresler, A.; Rosti, G.; Simpson, D.; Assouline, S.; Etienne, G.; Nicolini, F.E.; le Coutre, P.; Clark, R.E.; et al. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 612–621. [Google Scholar] [CrossRef]
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A phase ib open-label multicenter study of azd4547 in patients with advanced squamous cell lung cancers. Clin. Cancer Res. 2017, 23, 5366–5373. [Google Scholar] [CrossRef] [PubMed]
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.P.; Hidalgo, M.; Schellens, J.H.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of bgj398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: Results of a global phase i, dose-escalation and dose-expansion study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [PubMed]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase i dose-escalation study of jnj-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.; Andrews, S.; Blockley, L.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in fgfr3 as a mechanism of acquired resistance. Oncogene 2013, 32, 3059–3070. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Chen, H.; Wortmann, A.; Loch, D.; Gartside, M.G.; Dehkhoda, F.; Blais, S.P.; Neubert, T.A.; Mohammadi, M.; Pollock, P.M. The n550k/h mutations in fgfr2 confer differential resistance to pd173074, dovitinib, and ponatinib atp-competitive inhibitors. Neoplasia 2013, 15, 975–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation fgfr kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araujo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. Fgfr a promising druggable target in cancer: Molecular biology and new drugs. Crit Rev Oncol Hematol 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Sell, C.; Dumenil, G.; Deveaud, C.; Miura, M.; Coppola, D.; DeAngelis, T.; Rubin, R.; Efstratiadis, A.; Baserga, R. Effect of a null mutation of the insulin-like growth factor i receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell. Biol. 1994, 14, 3604–3612. [Google Scholar] [CrossRef] [PubMed]
- Zha, J.; O’Brien, C.; Savage, H.; Huw, L.Y.; Zhong, F.; Berry, L.; Lewis Phillips, G.D.; Luis, E.; Cavet, G.; Hu, X.; et al. Molecular predictors of response to a humanized anti-insulin-like growth factor-i receptor monoclonal antibody in breast and colorectal cancer. Mol. Cancer Ther. 2009, 8, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Asmane, I.; Watkin, E.; Alberti, L.; Duc, A.; Marec-Berard, P.; Ray-Coquard, I.; Cassier, P.; Decouvelaere, A.V.; Ranchere, D.; Kurtz, J.E.; et al. Insulin-like growth factor type 1 receptor (igf-1r) exclusive nuclear staining: A predictive biomarker for igf-1r monoclonal antibody (ab) therapy in sarcomas. Eur. J. Cancer 2012, 48, 3027–3035. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, A.; Hixon, M.L.; Karp, D.D.; Li, D.; Green, S.; Dolled-Filhart, M.; Paz-Ares, L.G.; Novello, S.; Blakely, J.; Langer, C.J.; et al. Pre-treatment levels of circulating free igf-1 identify nsclc patients who derive clinical benefit from figitumumab. Br. J. Cancer 2011, 104, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Novello, S.; Park, K.; Krzakowski, M.; Karp, D.D.; Mok, T.; Benner, R.J.; Scranton, J.R.; Olszanski, A.J.; Jassem, J. Randomized, phase iii trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2014, 32, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Bondarenko, I.; Blackhall, F.; Barlesi, F.; Hsia, T.C.; Jassem, J.; Milanowski, J.; Popat, S.; Sanchez-Torres, J.M.; Novello, S.; et al. Randomized, phase iii trial of figitumumab in combination with erlotinib versus erlotinib alone in patients with nonadenocarcinoma nonsmall-cell lung cancer. Ann. Oncol. 2015, 26, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Azevedo, S.; Okusaka, T.; Van Laethem, J.L.; Lipton, L.R.; Riess, H.; Szczylik, C.; Moore, M.J.; Peeters, M.; Bodoky, G.; et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: The gamma trial. Ann. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Berruti, A.; Baudin, E.; Demeure, M.J.; Gilbert, J.; Haak, H.; Kroiss, M.; Quinn, D.I.; Hesseltine, E.; Ronchi, C.L.; et al. Linsitinib (osi-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: A double-blind, randomised, phase 3 study. Lancet Oncol. 2015, 16, 426–435. [Google Scholar] [CrossRef]
- Leighl, N.B.; Rizvi, N.A.; de Lima, L.G., Jr.; Arpornwirat, W.; Rudin, C.M.; Chiappori, A.A.; Ahn, M.J.; Chow, L.Q.; Bazhenova, L.; Dechaphunkul, A.; et al. Phase 2 study of erlotinib in combination with linsitinib (osi-906) or placebo in chemotherapy-naive patients with non-small-cell lung cancer and activating epidermal growth factor receptor mutations. Clin. Lung Cancer 2017, 18, 34–42.e2. [Google Scholar] [CrossRef] [PubMed]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to igf-1r targeted therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Hodi, F.S.; Fisher, D.E. From genes to drugs: Targeted strategies for melanoma. Nat. Rev. Cancer 2012, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Kindler, T.; Breitenbuecher, F.; Marx, A.; Beck, J.; Hess, G.; Weinkauf, B.; Duyster, J.; Peschel, C.; Kirkpatrick, C.J.; Theobald, M.; et al. Efficacy and safety of imatinib in adult patients with c-kit-positive acute myeloid leukemia. Blood 2004, 103, 3644–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardanani, A.; Tefferi, A. Systemic mastocytosis in adults: A review on prognosis and treatment based on 342 mayo clinic patients and current literature. Curr. Opin. Hematol. 2010, 17, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Gramza, A.W.; Corless, C.L.; Heinrich, M.C. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin. Cancer Res. 2009, 15, 7510–7518. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.; Roberts, P.J.; Heinz, D.; et al. Long-term results from a randomized phase ii trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing kit. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Besmer, P.; Guo, T.; Arkun, K.; Hom, G.; Koryotowski, B.; Leversha, M.A.; Jeffrey, P.D.; Desantis, D.; Singer, S.; et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res. 2005, 11, 4182–4190. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Garrett, C.R.; Schoffski, P.; Shah, M.H.; Verweij, J.; Leyvraz, S.; Hurwitz, H.I.; Pousa, A.L.; Le Cesne, A.; Goldstein, D.; et al. Complete longitudinal analyses of the randomized, placebo-controlled, phase iii trial of sunitinib in patients with gastrointestinal stromal tumor following imatinib failure. Clin. Cancer Res. 2012, 18, 3170–3179. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (grid): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Tamborini, E.; Pricl, S.; Negri, T.; Lagonigro, M.S.; Miselli, F.; Greco, A.; Gronchi, A.; Casali, P.G.; Ferrone, M.; Fermeglia, M.; et al. Functional analyses and molecular modeling of two c-kit mutations responsible for imatinib secondary resistance in gist patients. Oncogene 2006, 25, 6140–6146. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Holden, J.A.; Choi, H.; Zhu, J.; Wu, E.F.; Jones, K.A.; Ward, J.H.; Andtbacka, R.H.; Randall, R.L.; Scaife, C.L.; et al. Evolution from heterozygous to homozygous kit mutation in gastrointestinal stromal tumor correlates with the mechanism of mitotic nondisjunction and significant tumor progression. Mod. Pathol. 2008, 21, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel v600e braf mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosom. Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurama, K.; Noma, K.; Takaoka, M.; Tomono, Y.; Watanabe, N.; Hatakeyama, S.; Ohmori, O.; Hirota, S.; Motoki, T.; Shirakawa, Y.; et al. Inhibition of focal adhesion kinase as a potential therapeutic strategy for imatinib-resistant gastrointestinal stromal tumor. Mol. Cancer Ther. 2009, 8, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarn, C.; Rink, L.; Merkel, E.; Flieder, D.; Pathak, H.; Koumbi, D.; Testa, J.R.; Eisenberg, B.; von Mehren, M.; Godwin, A.K. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 8387–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Huynh, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. Fgfr-mediated reactivation of mapk signaling attenuates antitumor effects of imatinib in gastrointestinal stromal tumors. Cancer Discov. 2015, 5, 438–451. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| MDs | SE | 95% CIs | p-Value | ||
|---|---|---|---|---|---|
| Chemotherapy | 14.15 | 4.43 | 5.46–22.83 | <0.001 | |
| Osimertinib vs. | Gefitinib/Erlotinib | 7.94 | 3.62 | 0.83–15.15 | 0.029 |
| Afatinib | 8.53 | 5.6 | −2.62–19.33 | 0.136 |
| Treatment | SUCRA | PrBest | MeanRank |
|---|---|---|---|
| Chemotherapy | 2.7 | 0.1 | 3.9 |
| Gefitininb/Erlotinib | 48.4 | 0.6 | 2.5 |
| Afatinib | 50.6 | 4.4 | 2.5 |
| Osimertinib | 98.3 | 95.0 | 1.1 |
| Drugs | Sorafenib | Sunitinib | Regorafenib | Pazopanib | Axitinib | Cabozantinib | Vandetanib |
|---|---|---|---|---|---|---|---|
| Targets | VEGFR, PDGFR, c-KIT, FLT-3, RET | VEGFR, PDGFR, c-KIT, FLT-3, RET | VEGFR, PDGFR, FGFR, TIE2, c-KIT, FLT-3, RET | VEGFR, PDGFR, FGFR, c-KIT | VEGFR, PDGFR, c-KIT | VEGFR, c-MET, RET, TIE2, FLT-3, RET, AXL | VEGFR, EGFR, RET, TIE2, SRC |
| Clinical Indications | HCC a, RCC b, Thyroid carcinoma | GIST c, RCC | mCRC d, GIST | RCC, STS e | RCC | MTC f | MTC |
| Drug | Bevacizumab | Ramucirumab | Aflibercept |
|---|---|---|---|
| Target | VEGF-A | VEGFR2 | VEGFR |
| Clinical Indications | Glioblastoma, mCRC 1, NSCLC 2, Ovarian cancer | Gastric cancer, mCRC 1, NSCLC 2 | mCRC 1 |
| Agents | Phase | Disease Characteristics | Comparison | Clinical Trial ID |
|---|---|---|---|---|
| HGF antibodies | ||||
| Rilotumumab | III | MET-positive G/GEJ cancer a | Chemo ± Rilotumumab | NCT01697072 [211] |
| MET antibodies | ||||
| onartuzumab | III | MET-positive NSCLC b | Erlotinib ± onartumumab | NCT01456325 [212] |
| onartuzumab | III | HER2(-)/MET(+)-GEC c | mFOLFOX6 ± onartumumab | NCT01662869 [213] |
| MET TKI | ||||
| Crizotinib | III | ALK (+)-NSCLC | Chemo vs. crizotinib | NCT00932893 [214] |
| Crizotinib | III | ALK (+)-NSCLC | Chemo vs. crizotinib | NCT01154140 [177] |
| Crizotinib | III | ALK (+)-NSCLC | Alectinib vs. crizotinib | NCT02075840 [189] |
| Cabozantinib | III | HCC d | Cabozantinib vs. Placebo | NCT01908426 [215] |
| Cabozantinib | III | RCC e | Cabozantinib vs. Everolimus | NCT01865747 [216] |
| Cabozantinib | III | mCRPC f | Cabozantinib vs. Prednisone | NCT01605227 [217] |
| Agents | Phase | Disease Characteristics | Comparison | Clinical Trial ID |
|---|---|---|---|---|
| Non-selective | ||||
| Dovitinib | III | RCC a | Dovitinib vs. Sorafenib | NCT01223027 [204] |
| Ponatinib | III | CML b | Ponatinib vs. Imatinib | NCT01650805 [224] |
| Agents | Phase | Disease Characteristics | Comparison | Clinical Trial ID |
|---|---|---|---|---|
| IGF-1R mAbs | ||||
| figitumumab | III | NSCLC a | chemo ± figitumumab | NCT00596830 [237] |
| figitumumab | III | NSCLC | erlotinib ± figitumumab | NCT00673049 [238] |
| ganitumab | III | Pancreatic adenocarcinoma | gemcitabine ± ganitumab | NCT01231347 [239] |
| IGF-1R TKI | ||||
| linsitinib | III | Adrenocortical carcinoma | linsitinib vs. Placebo | NCT00924989 [240] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. https://doi.org/10.3390/ijms19113491
Yamaoka T, Kusumoto S, Ando K, Ohba M, Ohmori T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. International Journal of Molecular Sciences. 2018; 19(11):3491. https://doi.org/10.3390/ijms19113491
Chicago/Turabian StyleYamaoka, Toshimitsu, Sojiro Kusumoto, Koichi Ando, Motoi Ohba, and Tohru Ohmori. 2018. "Receptor Tyrosine Kinase-Targeted Cancer Therapy" International Journal of Molecular Sciences 19, no. 11: 3491. https://doi.org/10.3390/ijms19113491
APA StyleYamaoka, T., Kusumoto, S., Ando, K., Ohba, M., & Ohmori, T. (2018). Receptor Tyrosine Kinase-Targeted Cancer Therapy. International Journal of Molecular Sciences, 19(11), 3491. https://doi.org/10.3390/ijms19113491