Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies



Abstract
:1. Introduction
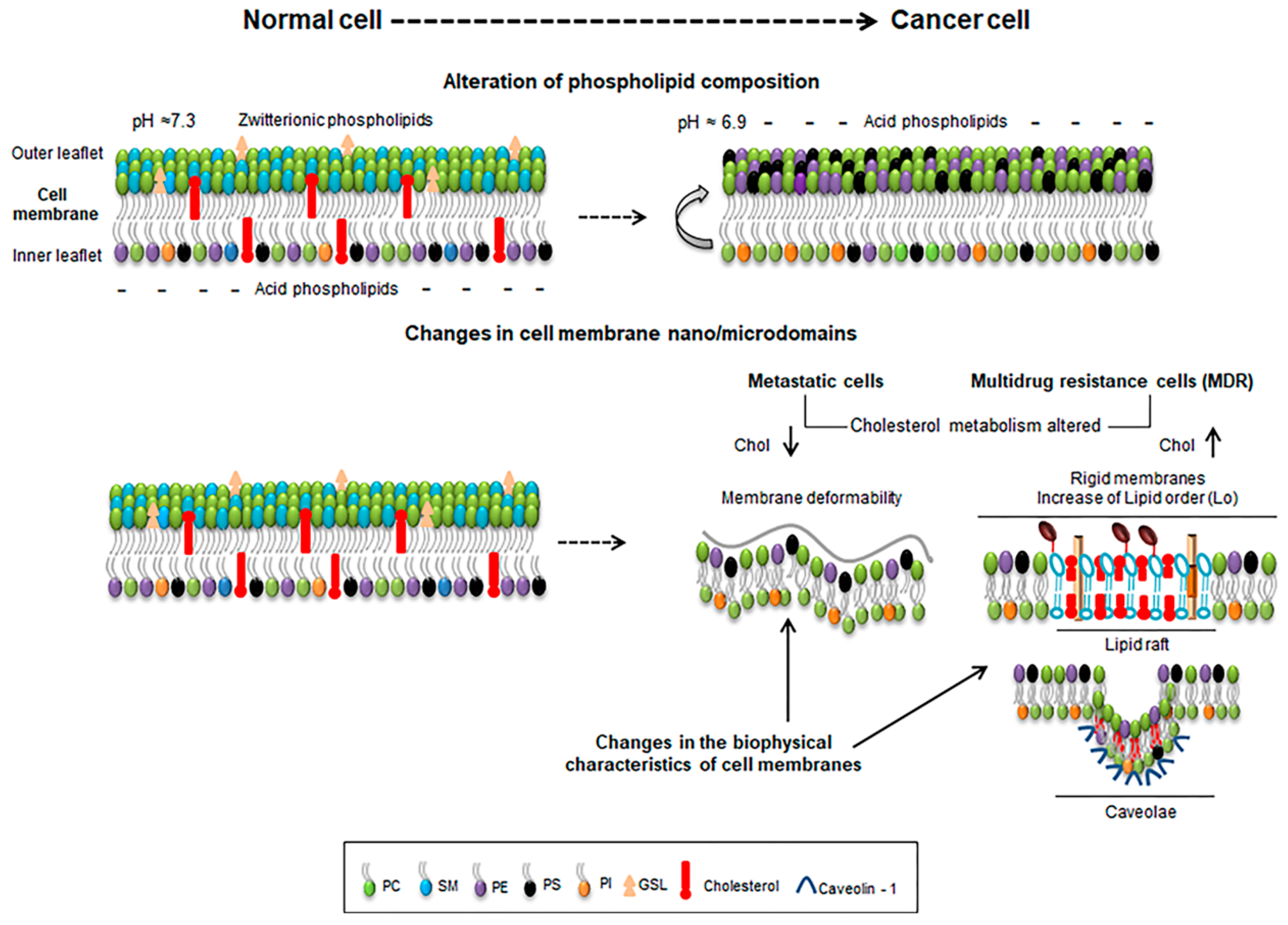
2. Membrane Biophysics of Cancer Cells
3. Therapeutic Strategies and Anticancer Drugs to Modulate Membranes
3.1. Modulation of Invasion and Proliferation Associated Pathways by Lowering Cholesterol Content
3.2. Stabilization of Pro-Apoptotic Membrane Domains
3.3. Lipid Replacement
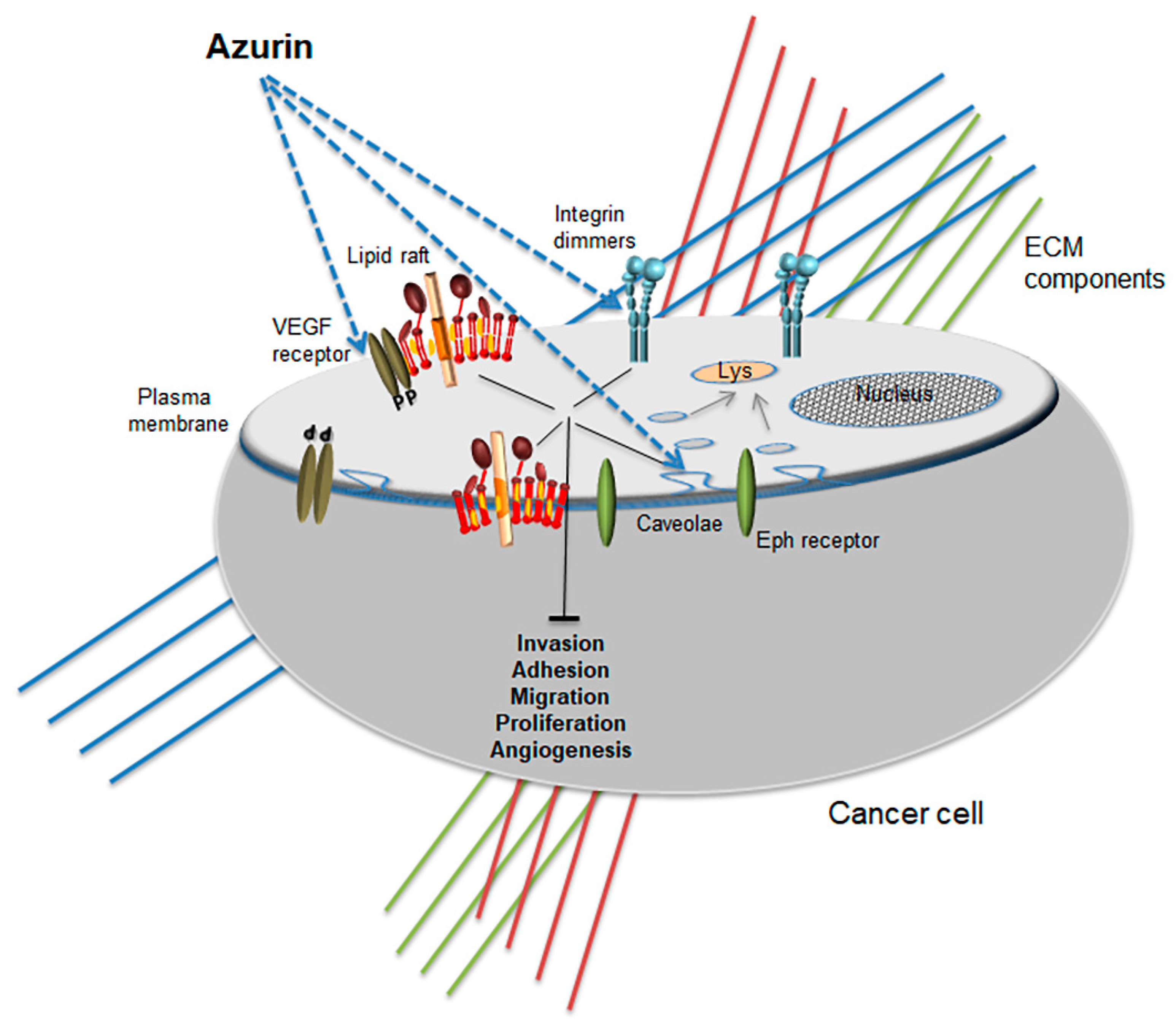
4. The Anticancer Effects of the Bacterial Protein Azurin: A Cell Membrane Targeted Therapy
4.1. Cancer Cells’ Membrane Modulation by Azurin
4.2. Induction of Apoptosis
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of interest
References
- Peetla, C.; Vijayaraghavalu, S.; Labhasetwar, V. Biophysics of cell membrane lipids in cancer drug resistance: Implications for drug transport and drug delivery with nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 1686–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V. Membrane-lipid therapy: A historical perspective of membrane-targeted therapies—From lipid bilayer structure to the pathophysiological regulation of cells. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Marco, P.; Marco, C.; Gálvez, X.; Jiménez-López, J.M.; Carrasco, M.P. Alkylphospholipids: An update on molecular mechanisms and clinical relevance. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Zalba, S.; ten Hagen, T.L.M. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef]
- Escribá, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [Green Version]
- Azordegan, N.; Fraser, V.; Le, K.; Hillyer, L.M.; Ma, D.W.L.; Fischer, G.; Moghadasian, M.H. Carcinogenesis alters fatty acid profile in breast tissue. Mol. Cell. Biochem. 2013, 374, 223–232. [Google Scholar] [CrossRef]
- Ray, U.; Roy, S.S. Aberrant lipid metabolism in cancer cells—The role of oncolipid-activated signaling. FEBS J. 2017, 285, 432–443. [Google Scholar] [CrossRef]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in cancer: The relevance of drug–membrane interaction studies. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2231–2244. [Google Scholar] [CrossRef] [PubMed]
- Hryniewicz-Jankowska, A.; Augoff, K.; Biernatowska, A.; Podkalicka, J.; Sikorski, A.F. Membrane rafts as a novel target in cancer therapy. Biochim. Biophys. Acta 2014, 1845, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.; Mouritsen, O.G.; Anderson, R.G.W. Lipid rafts: At a crossroad between cell biology and physics. Nat. Cell Biol. 2007, 9, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; de Kroon, A.I.P.M. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L. The Fluid-Mosaic Model of Membrane Structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Ran, S.; Stafford, J.H.; Thorpe, P.E. Increased Exposure of Phosphatidylethanolamine on the Surface of Tumor Blood Vessels. Cancer Res. 2002, 62, 6132–6140. [Google Scholar]
- Zwaal, R.F.A.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Stafford, J.H.; Thorpe, P.E. Increased Exposure of Phosphatidylethanolamine on the Surface of Tumor Vascular Endothelium. Neoplasia 2011, 13, 299-IN2. [Google Scholar] [CrossRef]
- Goñi, F.M. The basic structure and dynamics of cell membranes: An update of the Singer-Nicolson model. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1467–1476. [Google Scholar] [CrossRef]
- London, E. How principles of domain formation in model membranes may explain ambiguities concerning lipid raft formation in cells. Biochim. Biophys. Acta-Mol. Cell Res. 2005, 1746, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Subczynski, W.K.; Wisniewska, A. Physical properties of lipid bilayer membranes: Relevance to membrane biological functions. Acta Biochim. Pol. 2000, 47, 613–625. [Google Scholar] [PubMed]
- Hao, M.; Mukherjee, S.; Sun, Y.; Maxfield, F.R. Effects of Cholesterol Depletion and Increased Lipid Unsaturation on the Properties of Endocytic Membranes. J. Biol. Chem. 2004, 279, 14171–14178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preetha, A.; Banerjee, R.; Huilgol, N. Tensiometric profiles and their modulation by cholesterol: Implications in cervical cancer. Cancer Invest. 2007, 25, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, P.; Nabi, I.R. Lipid Rafts, Caveolae, and Their Endocytosis, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; Volume 282, ISBN 9780123812568. [Google Scholar]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, P.J.; Wolf, C. The liquid-ordered phase in membranes. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim. Biophys. Acta 2008, 1785, 182–206. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.N.; Goñi, F.M.; Alonso, A. Compartmentalization of Ceramide Signaling: Physical Foundations and Biological Effects. J. Cell. Physiol. 2000, 184, 285–300. [Google Scholar] [CrossRef]
- Quinn, P.J. Lipid-lipid interactions in bilayer membranes: Married couples and casual liaisons. Prog. Lipid Res. 2012, 51, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Tekpli, X.; Holme, J.A.; Sergent, O.; Lagadic-Gossmann, D. Role for membrane remodeling in cell death: Implication for health and disease. Toxicology 2013, 304, 141–157. [Google Scholar] [CrossRef] [PubMed]
- de Laurentiis, A.; Donovan, L.; Arcaro, A. Lipid rafts and caveolae in signaling by growth factor receptors. Open Biochem. J. 2007, 1, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G. Cellular lipidomics. EMBO J. 2005, 24, 3159–3165. [Google Scholar] [CrossRef] [Green Version]
- Storch, C.H.; Ehehalt, R.; Haefeli, W.E.; Weiss, J. Localization of the Human Breast Cancer Resistance Protein (BCRP/ABCG2) in Lipid Rafts/Caveolae and Modulation of Its Activity by Cholesterol in Vitro. Pharmacology 2007. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Lin, J.; Lu, M.L.; Cells, C.; Solomon, K.R.; Freeman, M.R. Cholesterol-rich Lipid Rafts Mediate Akt-regulated Survival in Prostate Cancer Cells. Cancer Res. 2002, 62, 2227–2231. [Google Scholar] [PubMed]
- Chen, Q.; Pan, Z.; Zhao, M.; Wang, Q.; Qiao, C.; Miao, L.; Ding, X. High cholesterol in lipid rafts reduces the sensitivity to EGFR-TKI therapy in non-small cell lung cancer. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.G. The caveolae membrane system. Annu. Rev. Biochem. 1998, 67, 199–225. [Google Scholar] [CrossRef]
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef]
- Zeisig, R.; Koklič, T.; Wiesner, B.; Fichtner, I.; Sentjurč, M. Increase in fluidity in the membrane of MT3 breast cancer cells correlates with enhanced cell adhesion in vitro and increased lung metastasis in NOD/SCID mice. Arch. Biochem. Biophys. 2007, 459, 98–106. [Google Scholar] [CrossRef]
- Sok, M.; Sentjurc, M.; Schara, M.; Stare, J.; Rott, T. Cell membrane fluidity and prognosis of lung cancer. Ann. Thorac. Surg. 2002, 73, 1567–1571. [Google Scholar] [CrossRef]
- Ramu, A.; Glaubiger, D.; Magrath, I.T.; Cells, P. Plasma Membrane Lipid Structural Order in Doxorubicin-sensitive and -resistant P388 Cells Plasma Membrane Lipid Structural Order in Doxorubicin-sensitive. Cancer Res. 1983, 43, 5533–5537. [Google Scholar] [PubMed]
- May, G.L.; Wright, L.C.; Dyne, M.; Mackinnon, W.B.; Fox, R.M.; Mountford, C.E. Plasma membrane lipid composition of vinblastine sensitive and resistant human leukaemic lymphoblasts. Int. J. Cancer 1988, 42, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Peetla, C.; Bhave, R.; Vijayaraghavalu, S.; Stine, A.; Kooijman, E.; Labhasetwar, V. Biophysical Characterization of and Doxorubicin Interactions with Membrane Lipids. Mol. Pharm. 2010, 7, 1264–1276. [Google Scholar] [CrossRef]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Jodoin, J.; Gingras, D.; Béliveau, R. P-glycoprotein is localized in caveolae in resistant cells and in brain capillaries. FEBS Lett. 2000, 466, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, N.; Schumann-Gillett, A.; Mark, A.E.; O’Mara, M.L. Understanding the accumulation of P-glycoprotein substrates within cells: The effect of cholesterol on membrane partitioning. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 776–782. [Google Scholar] [CrossRef]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 1–19. [Google Scholar] [CrossRef]
- Alves, A.C.; Magarkar, A.; Horta, M.; Lima, J.L.F.C.; Bunker, A.; Nunes, C.; Reis, S. Influence of doxorubicin on model cell membrane properties: Insights from in vitro and in silico studies. Sci. Rep. 2017, 7, 6343. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Kartal Yandim, M.; Apohan, E.; Baran, Y. Therapeutic potential of targeting ceramide/glucosylceramide pathway in cancer. Cancer Chemother. Pharmacol. 2013, 71, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V. Membrane-lipid therapy: A new approach in molecular medicine. Trends Mol. Med. 2006, 12, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Suzui, M.; Weinstein, I.B. Effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression, and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clin. Cancer Res. 2001, 7, 4220–4229. [Google Scholar] [PubMed]
- Shimizu, M.; Deguchi, A.; Lim, J.T.E.; Moriwaki, H.; Kopelovich, L.; Weinstein, I.B. (−)-Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor-2 signaling pathways in human colon cancer cells. Clin. cancer Res. 2005, 11, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Nagao, T.; Ingolfsson, H.I.; Maxfield, F.R.; Andersen, O.S.; Kopelovich, L.; Weinstein, I.B. The inhibitory effect of (−)-epigallocatechin gallate on activation of the epidermal growth factor receptor is associated with altered lipid order in HT29 colon cancer cells. Cancer Res. 2007, 67, 6493–6501. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Wakasaki, T.; Toh, S.; Shimizu, M.; Adachi, S. Chemoprevention of head and neck cancer by green tea extract: EGCG-the role of EGFR signaling and “lipid raft”. J. Oncol. 2011, 2011. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Tighiouart, M.; Lee, J.E.; Shin, H.J.; Khuri, R.; Yang, C.S.; Chen, Z.G.; Shin, D.M. Synergistic inhibition of head and neck tumor growth by green tea (−)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int. J. Cancer 2008, 123, 1005–1014. [Google Scholar] [CrossRef]
- Eddy, S.F.; Kane, S.E.; Sonenshein, G.E. Trastuzumab-resistant HER2-driven breast cancer cells are sensitive to epigallocatechin-3 gallate. Cancer Res. 2007, 67, 9018–9023. [Google Scholar] [CrossRef]
- Grunt, T.W. Interacting Cancer Machineries: Cell Signaling, Lipid Metabolism, and Epigenetics. Trends Endocrinol. MeTable 2018, 29, 86–98. [Google Scholar] [CrossRef]
- Di Vizio, D.; Adam, R.M.; Kim, J.; Kim, R.; Sotgia, F.; Williams, T.; Demichelis, F.; Solomon, K.R.; Loda, M.; Rubin, M.A.; et al. Caveolin-1 interacts with a lipid raft-associated population of fatty acid synthase. Cell Cycle 2008, 7, 2257–2267. [Google Scholar] [CrossRef] [Green Version]
- Arkenau, H.T.; Voskoboynik, M.; Infante, J.; Brenner, A.; Patel, M.; Borazanci, E.; Falchook, G.; Molife, L.R.; Pant, S.; Dean, E.; et al. Evidence of activity of a new mechanism of action (MoA): A first-in-human study of the first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640, as monotherapy or in combination. Eur. J. Cancer 2015, 51, S724. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Clendening, J.W.; Goard, C.A.; Penn, L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: Potential therapeutic relevance. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lan, T.; Hou, J.; Zhang, J.; An, Y.; Tie, L.; Pan, Y.; Liu, J.; Li, X. Atorvastatin sensitizes human non-small cell lung carcinomas to carboplatin via suppression of AKT activation and upregulation of TIMP-1. Int. J. Biochem. Cell Biol. 2012, 44, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Shen, H.-M.; Shui, G.; Wenk, M.R.; Ong, C.-N. Emodin Inhibits Tumor Cell Adhesion through Disruption of the Membrane Lipid Raft-Associated Integrin Signaling Pathway. Cancer Res. 2006, 66, 5807–5815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Shen, H.M.; Ong, C.N. Emodin inhibits tumor cell migration through suppression of the phosphatidylinositol 3-kinase-Cdc42/Rac1 pathway. Cell. Mol. Life Sci. 2005, 62, 1167–1175. [Google Scholar] [CrossRef]
- Ok, S.; Kim, S.-M.; Kim, C.; Nam, D.; Shim, B.S.; Kim, S.-H.; Ahn, K.S.; Choi, S.-H.; Ahn, K.S. Emodin inhibits invasion and migration of prostate and lung cancer cells by downregulating the expression of chemokine receptor CXCR4. Immunopharmacol. Immunotoxicol. 2012, 34, 768–778. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Lipid rafts and Fas/CD95 signaling in cancer chemotherapy. Recent Pat. Anticancer Drug Discov. 2011, 6, 274–283. [Google Scholar] [CrossRef]
- Estella-Hermoso de Mendoza, A.; Préat, V.; Mollinedo, F.; Blanco-Prieto, M.J. In vitro and in vivo efficacy of edelfosine-loaded lipid nanoparticles against glioma. J. Control. Release 2011, 156, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; Campanero, M.A.; Blanco-Prieto, M.J. Lipid raft-targeted therapy in multiple myeloma. Oncogene 2010, 29, 3748–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crul, M.; Rosing, H.; de Klerk, G.J.; Dubbelman, R.; Traiser, M.; Reichert, S.; Knebel, N.G.; Schellens, J.H.M.; Beijnen, J.H.; ten Bokkel Huinink, W.W. Phase I and pharmacological study of daily oral administration of perifosine (D-21266) in patients with advanced solid tumours. Eur. J. Cancer 2002, 38, 1615–1621. [Google Scholar] [CrossRef]
- Vink, S.R.; Schellens, J.H.M.; Van Blitterswijk, W.J.; Verheij, M. Tumor and normal tissue pharmacokinetics of perifosine, an oral anti-cancer alkylphospholipid. Invest. New Drugs 2005, 23, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Chen, G.; Wang, J.; Wang, F. Perifosine induces cell cycle arrest and apoptosis in human hepatocellular carcinoma cell lines by blockade of Akt phosphorylation. Cytotechnology 2010, 62, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2009, 109, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomé, C.H.; dos Santos, G.A.; Ferreira, G.A.; Scheucher, P.S.; Izumi, C.; Leopoldino, A.M.; Simão, A.M.; Ciancaglini, P.; de Oliveira, K.T.; Chin, A.; et al. Linker for Activation of T-cell Family Member2 (LAT2) a Lipid Raft Adaptor Protein for AKT Signaling, Is an Early Mediator of Alkylphospholipid Anti-leukemic Activity. Mol. Cell. Proteomics 2012, 11, 1898–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomide, A.B.; Thomé, C.H.; Dos Santos, G.A.; Ferreira, G.A.; Faça, V.M.; Rego, E.M.; Greene, L.J.; Stabeli, R.G.; Ciancaglini, P.; Itri, R. Disrupting membrane raft domains by alkylphospholipids. Biochim. Biophys. Acta-Biomembr. 2013, 1828, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. J. Cell. Biochem. 2004, 91, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Aires, V.; Colin, D.J.; Limagne, E.; Scagliarini, A.; Cotte, A.K.; Ghiringhelli, F. Importance of lipid microdomains, rafts, in absorption, delivery, and biological effects of resveratrol. Ann. N. Y. Acad. Sci. 2013, 1290, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Itoh, M.; Taguchi, Y.; Yamaoka, S.; Umehara, H.; Ichikawa, S.I.; Hirabayashi, Y.; Holleran, W.M.; Okazaki, T. Ceramide reduction and transcriptional up-regulation of glucosylceramide synthase through doxorubicin-activated Sp1 in drug-resistant HL-60/ADR cells. Cancer Res. 2004, 64, 6271–6279. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric oxide generated by tumor-associated macrophages is responsible for cancer resistance to cisplatin and correlated with syntaxin 4 and acid sphingomyelinase inhibition. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Kok, J.W.; Sietsma, H. Sphingolipid metabolism enzymes as targets for anticancer therapy. Curr. Drug Targets 2004, 5, 375–382. [Google Scholar] [CrossRef]
- Lladó, V.; López, D.J.; Ibarguren, M.; Alonso, M.; Soriano, J.B.; Escribá, P.V.; Busquets, X. Regulation of the cancer cell membrane lipid composition by NaCHOleate: Effects on cell signaling and therapeutical relevance in glioma. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Sparg, S.G.; Light, M.E.; van Staden, J. Biological activities and distribution of plant saponins. J. Ethnopharmacol. 2004, 94, 219–243. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Niesler, N.; Trautner, A.; Sama, S.; Jerz, G.; Panjideh, H.; Weng, A. Glycosylated Triterpenoids as Endosomal Escape Enhancers in Targeted Tumor Therapies. Biomedicines 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Goto, M.; Punj, V.; Zaborina, O.; Chen, M.L.; Kimbara, K.; Majumdar, D.; Cunningham, E.; Das Gupta, T.K.; Chakrabarty, A.M. Bacterial redox protein azurin, tumor suppressor protein p53, and regression of cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 14098–14103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Goto, M.; Punj, V.; Zaborina, O.; Kimbara, K.; Das Gupta, T.K.; Chakrabarty, A.M. The Bacterial Redox Protein Azurin Induces Apoptosis in J774 Macrophages through Complex Formation and Stabilization of the Tumor Suppressor Protein p53. Infect. Immun. 2002, 70, 7054–7062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Hiraoka, Y.; Ikehata, M.; Kimbara, K.; Avner, B.S.; Das Gupta, T.K.; Chakrabarty, A.M. Apoptosis or growth arrest: Modulation of tumor suppressor p53’s specificity by bacterial redox protein azurin. Proc. Natl. Acad. Sci. USA 2004, 101, 4770–4775. [Google Scholar] [CrossRef]
- Punj, V.; Bhattacharyya, S.; Saint-dic, D.; Vasu, C.; Cunningham, E.A.; Graves, J. Bacterial cupredoxin azurin as an inducer of apoptosis and regression in human breast cancer. Oncogene 2004, 23, 2367. [Google Scholar] [CrossRef]
- Bernardes, N.; Ribeiro, A.S.; Abreu, S.; Mota, B.; Matos, R.G.; Arraiano, C.M.; Seruca, R.; Paredes, J.; Fialho, A.M. The bacterial protein azurin impairs invasion and FAK/Src signaling in P-cadherin-overexpressing breast cancer models. PLoS ONE 2013, 19, e69023. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, N.; Ribeiro, A.S.; Abreu, S.; Vieira, A.F.; Carreto, L.; Santos, M.; Seruca, R.; Paredes, J.; Fialho, A.M. High-throughput molecular profiling of a P-cadherin overexpressing breast cancer model reveals new targets for the anti-cancer bacterial protein azurin. Int. J. Biochem. Cell Biol. 2014, 50, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, N.; Abreu, S.; Carvalho, F.A.; Fernandes, F.; Santos, N.C.; Fialho, A.M. Modulation of membrane properties of lung cancer cells by azurin enhances the sensitivity to EGFR-targeted therapy and decreased β1 integrin-mediated adhesion. Cell Cycle 2016, 15, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albergaria, A.; Ribeiro, A.S.; Vieira, A.F.; Sousa, B.; Nobre, A.R.; Seruca, R.; Schmitt, F.; Paredes, J. P-cadherin role in normal breast development and cancer. Int. J. Dev. Biol. 2011, 55, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, A.F.; Ribeiro, A.S.; Dionísio, M.R.; Sousa, B.; Nobre, A.R.; Albergaria, A.; Santiago-Gómez, A.; Mendes, N.; Gerhard, R.; Schmitt, F.; et al. P-cadherin signals through the laminin receptor α6β4 integrin to induce stem cell and invasive properties in basal-like breast cancer cells. Oncotarget 2014, 5, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, N.; Garizo, A.R.; Pinto, S.N.; Caniço, B.; Perdigão, C.; Fernandes, F.; Fialho, A.M. Azurin interaction with the lipid raft components ganglioside GM-1 and caveolin-1 increases membrane fluidity and sensitivity to anti-cancer drugs. Cell Cycle 2018, 17. [Google Scholar] [CrossRef]
- Apiyo, D.; Wittung-Stafshede, P. Unique complex between bacterial azurin and tumor-suppressor protein p53. Biochem. Biophys. Res. Commun. 2005, 332, 965–968. [Google Scholar] [CrossRef]
- Taranta, M.; Bizzarri, A.R.; Cannistraro, S. Probing the interaction between p53 and the bacterial protein azurin by single molecule force spectroscopy. J. Mol. Recognit. 2008, 21, 63–70. [Google Scholar] [CrossRef]
- Taranta, M.; Bizzarri, A.R.; Cannistraro, S. Modeling the interaction between the N-terminal domain of the tumor suppressor p53 and azurin. J. Mol. Recognit. 2009, 22, 215–222. [Google Scholar] [CrossRef]
- Yamada, T.; Das Gupta, T.K.; Beattie, C.W. P28-Mediated activation of p53 in G2-M phase of the cell cycle enhances the efficacy of DNA damaging and antimitotic chemotherapy. Cancer Res. 2016, 76, 2354–2365. [Google Scholar] [CrossRef]
- Mehta, R.R.; Hawthorne, M.; Peng, X.; Shilkaitis, A.; Mehta, R.G.; Beattie, C.W.; Das Gupta, T.K. A 28-amino-acid peptide fragment of the cupredoxin azurin prevents carcinogen-induced mouse mammary lesions. Cancer Prev. Res. 2010, 3, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mehta, R.R.; Lekmine, F.; Christov, K.; King, M.L.; Majumdar, D.; Shilkaitis, A.; Green, A.; Bratescu, L.; Beattie, C.W.; et al. A peptide fragment of azurin induces a p53-mediated cell cycle arrest in human breast cancer cells. Mol. Cancer Ther. 2009, 8, 2947–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Rae Bressler, L.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lulla, R.R.; Goldman, S.; Yamada, T.; Beattie, C.W.; Bressler, L.; Pacini, M.; Pollack, I.F.; Fisher, P.G.; Packer, R.J.; Dunkel, I.J.; et al. Phase I trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: A Pediatric Brain Tumor Consortium Study. Neuro. Oncol. 2016, 18, 1319–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Gorman, G.S.; Coward, L.U.; Noker, P.E.; Mccormick, D.; Horn, T.L.; Harder, J.B.; Muzzio, M. Preclinical pharmacokinetics, metabolism, and toxicity of azurin-p28 (NSC745104) a peptide inhibitor of p53 ubiquitination. Cancer Chemother. Pharmacol. 2011, 28, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.N.; Mehta, R.R.; Yamada, T.; Lekmine, F.; Christov, K.; Chakrabarty, A.M.; Green, A.; Bratescu, L.; Shilkaitis, A.; Beattie, C.W.; et al. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res. 2009, 69, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.R.; Yamada, T.; Taylor, B.N.; Christov, K.; King, M.L.; Majumdar, D.; Lekmine, F.; Tiruppathi, C.; Shilkaitis, A.; Bratescu, L.; et al. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis 2011, 14, 355–369. [Google Scholar] [CrossRef]
- Giansanti, F.; Panella, G.; Leboffe, L.; Antonini, G. Lactoferrin from milk: Nutraceutical and pharmacological properties. Pharmaceuticals 2016, 9, 61. [Google Scholar] [CrossRef]
- Sun, X.; Jiang, R.; Przepiorski, A.; Reddy, S.; Palmano, K.P.; Krissansen, G.W. “Iron-saturated” bovine lactoferrin improves the chemotherapeutic effects of tamoxifen in the treatment of basal-like breast cancer in mice. BMC Cancer 2012, 12, 1–12. [Google Scholar] [CrossRef]
- Fang, B.; Guo, H.Y.; Zhang, M.; Jiang, L.; Ren, F.Z. The six amino acid antimicrobial peptide bLFcin6 penetrates cells and delivers siRNA. FEBS J. 2013, 280, 1007–1017. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Acting Directly at the Membrane | Ref | ||
| i) Lowering cholesterol levels | Cholesterol binding agents: filipin |  | [6] |
| Cholesterol chemical depletion: MβCD |  | [38] | |
| ii) Stabilization of pro-apoptotic domains | ALPs Edelfosine |  | [70,71,72,73,74,75,76,77,78] |
| Miltefosine |  | ||
| perifosine |  | ||
| ODPC |  | ||
| Acting Intracellularly in lipid metabolism pathways | |||
| i) Fatty acid synthesis inhibition | TVB-2640 | unavailable | [62] |
| ii) Statins (inhibition of mevalonate pathway) | Lovastatin |  | [65] |
| Atorvastatin |  | [66] | |
| iii) Cer metabolism (activation of SMase) | Ara-C; |  | [83] |
| Decitabine; |  | [1,6] | |
| glucosylceramide analogue (PDMP) |  | [52,84] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardes, N.; Fialho, A.M. Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies. Int. J. Mol. Sci. 2018, 19, 3871. https://doi.org/10.3390/ijms19123871
Bernardes N, Fialho AM. Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies. International Journal of Molecular Sciences. 2018; 19(12):3871. https://doi.org/10.3390/ijms19123871
Chicago/Turabian StyleBernardes, Nuno, and Arsenio M. Fialho. 2018. "Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies" International Journal of Molecular Sciences 19, no. 12: 3871. https://doi.org/10.3390/ijms19123871
APA StyleBernardes, N., & Fialho, A. M. (2018). Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies. International Journal of Molecular Sciences, 19(12), 3871. https://doi.org/10.3390/ijms19123871