Identification of Novel Therapeutic Targets for Pulmonary Arterial Hypertension

Abstract
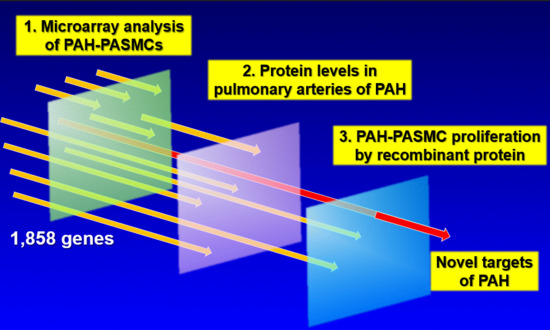
:
1. Introduction
2. Crucial Roles of AMP-Activated Protein Kinase (AMPK) Against PAH
3. Crucial Roles of CyPA and Bsg in the Development of PAH
4. Screening of Inhibitors for CyPA and Bsg
5. Identification of Novel Therapeutic Targets for PAH
6. TAFI as a Novel Therapeutic Target for CTEPH
7. Conclusions
Funding
Conflicts of Interest
References
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2012, 122, 4306–4313. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ. Res. 2014, 115, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Kholdani, C.; Fares, W.H.; Mohsenin, V. Pulmonary hypertension in obstructive sleep apnea: Is it clinically significant? A critical analysis of the association and pathophysiology. Pulm. Circ. 2015, 5, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.-C.; Gibbs, J.S.R. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef]
- Jiang, X.; Yuan, L.; Li, P.; Wang, J.; Wang, P.; Zhang, L.; Sun, B.; Sun, W. Effect of simvastatin on 5-HT and 5-HTT in a rat model of pulmonary artery hypertension. Cell. Physiol. Biochem. 2015, 37, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Grunig, G.; Marsh, L.M.; Esmaeil, N.; Jackson, K.; Gordon, T.; Reibman, J.; Kwapiszewska, G.; Park, S.H. Perspective: Ambient air pollution: Inflammatory response and effects on the lung’s vasculature. Pulm. Circ. 2014, 4, 25–35. [Google Scholar] [CrossRef]
- Irwin, D.C.; Garat, C.V.; Crossno, J.T., Jr.; MacLean, P.S.; Sullivan, T.M.; Erickson, P.F.; Jackman, M.R.; Harral, J.W.; Reusch, J.E.; Klemm, D.J. Obesity-related pulmonary arterial hypertension in rats correlates with increased circulating inflammatory cytokines and lipids and with oxidant damage in the arterial wall but not with hypoxia. Pulm. Circ. 2014, 4, 638–653. [Google Scholar] [CrossRef]
- Perros, F.; Gunther, S.; Ranchoux, B.; Godinas, L.; Antigny, F.; Chaumais, M.C.; Dorfmuller, P.; Hautefort, A.; Raymond, N.; Savale, L.; et al. Mitomycin-induced pulmonary veno-occlusive disease: Evidence from human disease and animal models. Circulation 2015, 132, 834–847. [Google Scholar] [CrossRef]
- Gatzoulis, M.A.; Beghetti, M.; Landzberg, M.J.; Galie, N. Pulmonary arterial hypertension associated with congenital heart disease: Recent advances and future directions. Int. J. Cardiol. 2014, 177, 340–347. [Google Scholar] [CrossRef]
- Chung, L.; Liu, J.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Badesch, D.B.; Miller, D.P.; Nicolls, M.R.; Zamanian, R.T. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: Identifying systemic sclerosis as a unique phenotype. Chest 2010, 138, 1383–1394. [Google Scholar] [CrossRef]
- Rich, J.D.; Rich, S. Clinical diagnosis of pulmonary hypertension. Circulation 2014, 130, 1820–1830. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; West, J.; Loyd, J.E.; Hemnes, A.R. Translational advances in the field of pulmonary hypertension molecular medicine of pulmonary arterial hypertension. From population genetics to precision medicine and gene editing. Am. J. Respir. Crit. Care Med. 2017, 195, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Bakker, W.J.; Harris, I.S.; Mak, T.W. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol. Cell 2007, 28, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullamsetti, S.S.; Savai, R.; Seeger, W.; Goncharova, E.A. Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. Targeting cell growth and proliferation signaling hubs. Am. J. Respir. Crit. Care Med. 2017, 195, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef]
- Boucherat, O.; Provencher, S.; Bonnet, S. Therapeutic Value of ASK1 Inhibition in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 284–286. [Google Scholar] [CrossRef]
- Bourgeois, A.; Omura, J.; Habbout, K.; Bonnet, S.; Boucherat, O. Pulmonary arterial hypertension: New pathophysiological insights and emerging therapeutic targets. Int. J. Biochem. Cell Biol. 2018, 104, 9–13. [Google Scholar] [CrossRef]
- Michelakis, E.D. Pulmonary arterial hypertension: Yesterday, today, tomorrow. Circ. Res. 2014, 115, 109–114. [Google Scholar] [CrossRef]
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702. [Google Scholar] [CrossRef]
- Yaoita, N.; Satoh, K.; Shimokawa, H. Novel therapeutic targets of pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e97–e102. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Teichert-Kuliszewska, K.; Kutryk, M.J.; Kuliszewski, M.A.; Karoubi, G.; Courtman, D.W.; Zucco, L.; Granton, J.; Stewart, D.J. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: Implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ. Res. 2006, 98, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Weir, E.K.; Wilkins, M.R. Basic science of pulmonary arterial hypertension for clinicians: New concepts and experimental therapies. Circulation 2010, 121, 2045–2066. [Google Scholar] [CrossRef] [PubMed]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Otsuki, T.; Kozu, K.; Numano, K.; Suzuki, K.; et al. Protective roles of endothelial AMP-activated protein kinase against hypoxia-induced pulmonary hypertension in mice. Circ. Res. 2016, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Godo, S.; Saito, H.; Enkhjargal, B.; Shimokawa, H. Dual roles of vascular-derived reactive oxygen species: With a special reference to hydrogen peroxide and cyclophilin A. J. Mol. Cell. Cardiol. 2014, 73, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Kagaya, Y.; Nakano, M.; Ito, Y.; Ohta, J.; Tada, H.; Karibe, A.; Minegishi, N.; Suzuki, N.; Yamamoto, M.; et al. Important role of endogenous erythropoietin system in recruitment of endothelial progenitor cells in hypoxia-induced pulmonary hypertension in mice. Circulation 2006, 113, 1442–1450. [Google Scholar] [CrossRef]
- Tada, H.; Kagaya, Y.; Takeda, M.; Ohta, J.; Asaumi, Y.; Satoh, K.; Ito, K.; Karibe, A.; Shirato, K.; Minegishi, N.; et al. Endogenous erythropoietin system in non-hematopoietic lineage cells plays a protective role in myocardial ischemia/reperfusion. Cardiovasc. Res. 2006, 71, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Satoh, K.; Fukumoto, Y.; Ito, Y.; Kagaya, Y.; Ishii, N.; Sugamura, K.; Shimokawa, H. Important role of erythropoietin receptor to promote VEGF expression and angiogenesis in peripheral ischemia in mice. Circ. Res. 2007, 100, 662–669. [Google Scholar] [CrossRef]
- Satoh, K.; Fukumoto, Y.; Nakano, M.; Kagaya, Y.; Shimokawa, H. Emergence of the erythropoietin/erythropoietin receptor system as a novel cardiovascular therapeutic target. J. Cardiovasc. Pharmacol. 2011, 58, 570–574. [Google Scholar] [CrossRef]
- Kagaya, Y.; Asaumi, Y.; Wang, W.; Takeda, M.; Nakano, M.; Satoh, K.; Fukumoto, Y.; Shimokawa, H. Current perspectives on protective roles of erythropoietin in cardiovascular system: Erythropoietin receptor as a novel therapeutic target. Tohoku J. Exp. Med. 2012, 227, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Fukumoto, Y.; Nakano, M.; Sugimura, K.; Nawata, J.; Demachi, J.; Karibe, A.; Kagaya, Y.; Ishii, N.; Sugamura, K.; et al. Statin ameliorates hypoxia-induced pulmonary hypertension associated with down-regulated stromal cell-derived factor-1. Cardiovasc. Res. 2009, 81, 226–234. [Google Scholar] [CrossRef]
- Fisslthaler, B.; Fleming, I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ. Res. 2009, 105, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Nakajima, S.; Godo, S.; Saito, H.; Ikeda, S.; Shimizu, T.; Enkhjargal, B.; Fukumoto, Y.; Tsukita, S.; Yamada, T.; et al. Rho-kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway in mice. PLoS ONE 2014, 9, e110446. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukumoto, Y.; Tanaka, S.; Satoh, K.; Ikeda, S.; Shimokawa, H. Crucial role of ROCK2 in vascular smooth muscle cells for hypoxia-induced pulmonary hypertension in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2780–2791. [Google Scholar] [CrossRef]
- Ikeda, S.; Satoh, K.; Kikuchi, N.; Miyata, S.; Suzuki, K.; Omura, J.; Shimizu, T.; Kobayashi, K.; Kobayashi, K.; Fukumoto, Y.; et al. Crucial role of Rho-kinase in pressure overload-induced right ventricular hypertrophy and dysfunction in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Elias-Al-Mamun, M.; Satoh, K.; Tanaka, S.-i.; Shimizu, T.; Nergui, S.; Miyata, S.; Fukumoto, Y.; Shimokawa, H. Combination therapy with fasudil and sildenafil ameliorates monocrotaline-induced pulmonary hypertension and survival in rats. Circ. J. 2014, 78, 967–976. [Google Scholar] [CrossRef]
- Ido, Y.; Carling, D.; Ruderman, N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: Inhibition by the AMP-activated protein kinase activation. Diabetes 2002, 51, 159–167. [Google Scholar] [CrossRef]
- Igata, M.; Motoshima, H.; Tsuruzoe, K.; Kojima, K.; Matsumura, T.; Kondo, T.; Taguchi, T.; Nakamaru, K.; Yano, M.; Kukidome, D.; et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ. Res. 2005, 97, 837–844. [Google Scholar] [CrossRef]
- Enkhjargal, B.; Godo, S.; Sawada, A.; Suvd, N.; Saito, H.; Noda, K.; Satoh, K.; Shimokawa, H. Endothelial AMP-activated protein kinase regulates blood pressure and coronary flow responses through hyperpolarization mechanism in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1505–1513. [Google Scholar] [CrossRef]
- Zou, M.H.; Wu, Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin. Exp. Pharmacol. Physiol. 2008, 35, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Lee, T.S.; Zhu, M.; Gu, C.; Wang, Y.; Zhu, Y.; Shyy, J.Y. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation 2006, 114, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, S.M.; Razavi, H.; Kim, J.; Agrawal, R.; Kundu, R.K.; de Jesus Perez, V.; Zamanian, R.T.; Quertermous, T.; Chun, H.J. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Pirkmajer, S.; Kulkarni, S.S.; Tom, R.Z.; Ross, F.A.; Hawley, S.A.; Hardie, D.G.; Zierath, J.R.; Chibalin, A.V. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes 2015, 64, 360–369. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef]
- Smith, S.C.; Benjamin, E.J.; Bonow, R.O.; Braun, L.T.; Creager, M.A.; Franklin, B.A.; Gibbons, R.J.; Grundy, S.M.; Hiratzka, L.F.; Jones, D.W.; et al. AHA/ACCF Secondary Prevention and Risk Reduction Therapy for Patients With Coronary and Other Atherosclerotic Vascular Disease: 2011 Update: A Guideline from the American Heart Association and American College of Cardiology Foundation. Circulation 2011, 124, 2458–2473. [Google Scholar] [CrossRef]
- Huertas, A.; Perros, F.; Tu, L.; Cohen-Kaminsky, S.; Montani, D.; Dorfmuller, P.; Guignabert, C.; Humbert, M. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: A complex interplay. Circulation 2014, 129, 1332–1340. [Google Scholar] [CrossRef]
- De Frutos, S.; Spangler, R.; Alo, D.; Bosc, L.V. NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha-actin up-regulation. J. Biol. Chem. 2007, 282, 15081–15089. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Matoba, T.; Suzuki, J.; O’Dell, M.R.; Nigro, P.; Cui, Z.; Mohan, A.; Pan, S.; Li, L.; Jin, Z.G.; et al. Cyclophilin A mediates vascular remodeling by promoting inflammation and vascular smooth muscle cell proliferation. Circulation 2008, 117, 3088–3098. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.G.; Melaragno, M.G.; Liao, D.F.; Yan, C.; Haendeler, J.; Suh, Y.A.; Lambeth, J.D.; Berk, B.C. Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ. Res. 2000, 87, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.F.; Jin, Z.G.; Baas, A.S.; Daum, G.; Gygi, S.P.; Aebersold, R.; Berk, B.C. Purification and identification of secreted oxidative stress-induced factors from vascular smooth muscle cells. J. Biol. Chem. 2000, 275, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Fukumoto, Y.; Shimokawa, H. Rho-kinase: Important new therapeutic target in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H287–H296. [Google Scholar] [CrossRef]
- Jin, Z.G.; Lungu, A.O.; Xie, L.; Wang, M.; Wong, C.; Berk, B.C. Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1186–1191. [Google Scholar] [CrossRef]
- Nigro, P.; Satoh, K.; O’Dell, M.R.; Soe, N.N.; Cui, Z.; Mohan, A.; Abe, J.; Alexis, J.D.; Sparks, J.D.; Berk, B.C. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J. Exp. Med. 2011, 208, 53–66. [Google Scholar] [CrossRef]
- Khromykh, L.M.; Kulikova, N.L.; Anfalova, T.V.; Muranova, T.A.; Abramov, V.M.; Vasiliev, A.M.; Khlebnikov, V.S.; Kazansky, D.B. Cyclophilin A produced by thymocytes regulates the migration of murine bone marrow cells. Cell. Immunol. 2007, 249, 46–53. [Google Scholar] [CrossRef]
- Yurchenko, V.; Zybarth, G.; O’Connor, M.; Dai, W.W.; Franchin, G.; Hao, T.; Guo, H.; Hung, H.C.; Toole, B.; Gallay, P.; et al. Active site residues of cyclophilin A are crucial for its signaling activity via CD147. J. Biol. Chem. 2002, 277, 22959–22965. [Google Scholar] [CrossRef]
- Miller, L.H.; Ackerman, H.C.; Su, X.Z.; Wellems, T.E. Malaria biology and disease pathogenesis: Insights for new treatments. Nat. Med. 2013, 19, 156–167. [Google Scholar] [CrossRef]
- Satoh, K.; Satoh, T.; Kikuchi, N.; Omura, J.; Kurosawa, R.; Suzuki, K.; Sugimura, K.; Aoki, T.; Nochioka, K.; Tatebe, S.; et al. Basigin mediates pulmonary hypertension by promoting inflammation and vascular smooth muscle cell proliferation. Circ. Res. 2014, 115, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Seizer, P.; Schonberger, T.; Schott, M.; Lang, M.R.; Langer, H.F.; Bigalke, B.; Kramer, B.F.; Borst, O.; Daub, K.; Heidenreich, O.; et al. EMMPRIN and its ligand cyclophilin A regulate MT1-MMP, MMP-9 and M-CSF during foam cell formation. Atherosclerosis 2010, 209, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Seizer, P.; Ochmann, C.; Schonberger, T.; Zach, S.; Rose, M.; Borst, O.; Klingel, K.; Kandolf, R.; MacDonald, H.R.; Nowak, R.A.; et al. Disrupting the EMMPRIN (CD147)-cyclophilin A interaction reduces infarct size and preserves systolic function after myocardial ischemia and reperfusion. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, B.; Valente, A.J.; Prabhu, S.D.; Shanmugam, P.; Delafontaine, P.; Chandrasekar, B. EMMPRIN activates multiple transcription factors in cardiomyocytes, and induces interleukin-18 expression via Rac1-dependent PI3K/Akt/IKK/NF-kappaB andMKK7/JNK/AP-1 signaling. J. Mol. Cell. Cardiol. 2010, 49, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, B.; Valente, A.J.; Reddy, V.S.; Siwik, D.A.; Chandrasekar, B. Resveratrol blocks interleukin-18-EMMPRIN cross-regulation and smooth muscle cell migration. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H874–H886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, J.; Jin, Z.G.; Meoli, D.F.; Matoba, T.; Berk, B.C. Cyclophilin A is secreted by a vesicular pathway in vascular smooth muscle cells. Circ. Res. 2006, 98, 811–817. [Google Scholar] [CrossRef]
- Satoh, K.; Nigro, P.; Matoba, T.; O’Dell, M.R.; Cui, Z.; Shi, X.; Mohan, A.; Yan, C.; Abe, J.; Illig, K.A.; et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat. Med. 2009, 15, 649–656. [Google Scholar] [CrossRef]
- Shimokawa, H.; Takeshita, A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1767–1775. [Google Scholar] [CrossRef]
- Abe, K.; Shimokawa, H.; Morikawa, K.; Uwatoku, T.; Oi, K.; Matsumoto, Y.; Hattori, T.; Nakashima, Y.; Kaibuchi, K.; Sueishi, K.; et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ. Res. 2004, 94, 385–393. [Google Scholar] [CrossRef]
- Do e, Z.; Fukumoto, Y.; Takaki, A.; Tawara, S.; Ohashi, J.; Nakano, M.; Tada, T.; Saji, K.; Sugimura, K.; Fujita, H.; et al. Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ. J. 2009, 73, 1731–1739. [Google Scholar] [CrossRef]
- Satoh, K.; Fukumoto, Y.; Sugimura, K.; Miura, Y.; Aoki, T.; Nochioka, K.; Tatebe, S.; Miyamichi-Yamamoto, S.; Shimizu, T.; Osaki, S.; et al. Plasma cyclophilin A is a novel biomarker for coronary artery disease. Circ. J. 2013, 77, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, Y.; Yamada, N.; Matsubara, H.; Mizoguchi, M.; Uchino, K.; Yao, A.; Kihara, Y.; Kawano, M.; Watanabe, H.; Takeda, Y.; et al. Double-blind, placebo-controlled clinical trial with a Rho-kinase inhibitor in pulmonary arterial hypertension. Circ. J. 2013, 77, 2619–2625. [Google Scholar] [CrossRef]
- Guo, H.; Majmudar, G.; Jensen, T.C.; Biswas, C.; Toole, B.P.; Gordon, M.K. Characterization of the gene for human EMMPRIN, a tumor cell surface inducer of matrix metalloproteinases. Gene 1998, 220, 99–108. [Google Scholar] [CrossRef]
- Satoh, K.; Shimokawa, H.; Berk, B.C. Cyclophilin A: Promising new target in cardiovascular therapy. Circ. J. 2010, 74, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Koyasu, S. Regulation of MAPK signaling pathways through immunophilin-ligand complex. Curr. Top. Med. Chem. 2003, 3, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Dromparis, P.; Paulin, R.; Sutendra, G.; Qi, A.C.; Bonnet, S.; Michelakis, E.D. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ. Res. 2013, 113, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L. Potassium channel diversity in the pulmonary arteries and pulmonary veins: Implications for regulation of the pulmonary vasculature in health and during pulmonary hypertension. Pharmacol. Ther. 2007, 115, 56–69. [Google Scholar] [CrossRef]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: Implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006, 13, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Roger, V.L. Epidemiology of heart failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Vachiery, J.L.; Adir, Y.; Barbera, J.A.; Champion, H.; Coghlan, J.G.; Cottin, V.; De Marco, T.; Galie, N.; Ghio, S.; Gibbs, J.S.; et al. Pulmonary hypertension due to left heart diseases. J. Am. Coll. Cardiol. 2013, 62, D100–D108. [Google Scholar] [CrossRef]
- Delgado, J.F.; Conde, E.; Sanchez, V.; Lopez-Rios, F.; Gomez-Sanchez, M.A.; Escribano, P.; Sotelo, T.; Gomez de la Camara, A.; Cortina, J.; de la Calzada, C.S. Pulmonary vascular remodeling in pulmonary hypertension due to chronic heart failure. Eur. J. Heart Fail. 2005, 7, 1011–1016. [Google Scholar] [CrossRef] [Green Version]
- Ghio, S.; Gavazzi, A.; Campana, C.; Inserra, C.; Klersy, C.; Sebastiani, R.; Arbustini, E.; Recusani, F.; Tavazzi, L. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J. Am. Coll. Cardiol. 2001, 37, 183–188. [Google Scholar] [CrossRef]
- Lai, Y.C.; Tabima, D.M.; Dube, J.J.; Hughan, K.S.; Vanderpool, R.R.; Goncharov, D.A.; St Croix, C.M.; Garcia-Ocana, A.; Goncharova, E.A.; Tofovic, S.P.; et al. SIRT3-AMP-activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction. Circulation 2016, 133, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Aguero, J.; Ishikawa, K.; Hadri, L.; Santos-Gallego, C.G.; Fish, K.M.; Kohlbrenner, E.; Hammoudi, N.; Kho, C.; Lee, A.; Ibanez, B.; et al. Intratracheal gene delivery of SERCA2a ameliorates chronic post-capillary pulmonary hypertension: A large animal model. J. Am. Coll. Cardiol. 2016, 67, 2032–2046. [Google Scholar] [CrossRef]
- Sunamura, S.; Satoh, K.; Kurosawa, R.; Ohtsuki, T.; Kikuchi, N.; Elias-Al-Mamun, M.; Shimizu, T.; Ikeda, S.; Suzuki, K.; Satoh, T.; et al. Different roles of myocardial ROCK1 and ROCK2 in cardiac dysfunction and postcapillary pulmonary hypertension in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E7129–E7138. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Nigro, P.; Zeidan, A.; Soe, N.N.; Jaffre, F.; Oikawa, M.; O’Dell, M.R.; Cui, Z.; Menon, P.; Lu, Y.; et al. Cyclophilin A promotes cardiac hypertrophy in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1116–1123. [Google Scholar] [CrossRef]
- Suzuki, K.; Satoh, K.; Ikeda, S.; Sunamura, S.; Otsuki, T.; Satoh, T.; Kikuchi, N.; Omura, J.; Kurosawa, R.; Nogi, M.; et al. Basigin promotes cardiac fibrosis and failure in response to chronic pressure overload in mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K. Cyclophilin A in cardiovascular homeostasis and diseases. Tohoku J. Exp. Med. 2015, 235, 1–15. [Google Scholar] [CrossRef]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Xue, C.; Sowden, M.; Berk, B.C. Extracellular cyclophilin A, especially acetylated, causes pulmonary hypertension by stimulating endothelial apoptosis, redox stress, and inflammation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1138–1146. [Google Scholar] [CrossRef]
- Higashi, M.; Shimokawa, H.; Hattori, T.; Hiroki, J.; Mukai, Y.; Morikawa, K.; Ichiki, T.; Takahashi, S.; Takeshita, A. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: Effect on endothelial NAD(P)H oxidase system. Circ. Res. 2003, 93, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Fukumoto, Y.; Suzuki, J.; Saji, K.; Nawata, J.; Tawara, S.; Shinozaki, T.; Kagaya, Y.; Shimokawa, H. Long-term inhibition of Rho-kinase ameliorates diastolic heart failure in hypertensive rats. J. Cardiovasc. Pharmacol. 2008, 51, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Guan, P.; Zhang, J.P.; Chang, Y.Z.; Gu, L.J.; Hao, F.K.; Shi, Z.H.; Wang, F.Y.; Chu, L. Preventive effects of fasudil on adriamycin-induced cardiomyopathy: Possible involvement of inhibition of RhoA/ROCK pathway. Food Chem. Toxicol. 2011, 49, 2975–2982. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Guan, P.; Zhang, J.P.; Li, Y.Q.; Chang, Y.Z.; Shi, Z.H.; Wang, F.Y.; Chu, L. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses isoproterenol-induced heart failure in rats via JNK and ERK1/2 pathways. J. Cell. Biochem. 2011, 112, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Gao, J.; Bi, X.; He, H.; Shi, X.; Weng, S.; Chen, Y.; Yang, Y.; Ye, Y.; Fu, G. The Rho-kinase inhibitor, fasudil, ameliorates diabetes-induced cardiac dysfunction by improving calcium clearance and actin remodeling. J. Mol. Med. 2017, 95, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Ellawindy, A.; Satoh, K.; Sunamura, S.; Kikuchi, N.; Suzuki, K.; Minami, T.; Ikeda, S.; Tanaka, S.; Shimizu, T.; Enkhjargal, B.; et al. Rho-kinase inhibition during early cardiac development causes arrhythmogenic right ventricular cardiomyopathy in mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2172–2184. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Narang, N.; Chen, P.; Yu, B.; Knapp, M.; Janardanan, J.; Blair, J.; Liao, J.K. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017, 2, e93187. [Google Scholar] [CrossRef]
- Lee, J.H.; Zheng, Y.; von Bornstadt, D.; Wei, Y.; Balcioglu, A.; Daneshmand, A.; Yalcin, N.; Yu, E.; Herisson, F.; Atalay, Y.B.; et al. Selective ROCK2 inhibition in focal cerebral ischemia. Ann. Clin. Transl. Neurol. 2014, 1, 2–14. [Google Scholar]
- Zanin-Zhorov, A.; Weiss, J.M.; Nyuydzefe, M.S.; Chen, W.; Scher, J.U.; Mo, R.; Depoil, D.; Rao, N.; Liu, B.; Wei, J.; et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 16814–16819. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.; Paz, K.; Du, J.; Reichenbach, D.K.; Taylor, P.A.; Panoskaltsis-Mortari, A.; Vulic, A.; Luznik, L.; MacDonald, K.K.; Hill, G.R.; et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a STAT3-dependent mechanism. Blood 2016, 127, 2144–2154. [Google Scholar] [CrossRef]
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-kinase in the cardiovascular system. Circ. Res. 2016, 118, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Borlaug, B.A. Pulmonary hypertension due to left heart disease. Circulation 2012, 126, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Cascao, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A spectrum of treatment opportunities in chronic diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Hirotani, S.; Otsu, K.; Nishida, K.; Higuchi, Y.; Morita, T.; Nakayama, H.; Yamaguchi, O.; Mano, T.; Matsumura, Y.; Ueno, H.; et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation 2002, 105, 509–515. [Google Scholar] [CrossRef]
- Farkas, D.; Alhussaini, A.A.; Kraskauskas, D.; Kraskauskiene, V.; Cool, C.D.; Nicolls, M.R.; Natarajan, R.; Farkas, L. Nuclear factor kappaB inhibition reduces lung vascular lumen obliteration in severe pulmonary hypertension in rats. Am. J. Respir. Cell Mol. Biol. 2014, 51, 413–425. [Google Scholar] [CrossRef]
- Yuan, W.; Ge, H.; He, B. Pro-inflammatory activities induced by CyPA-EMMPRIN interaction in monocytes. Atherosclerosis 2010, 213, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, N.; Satoh, K.; Kurosawa, R.; Yaoita, N.; Elias-Al-Mamun, M.; Siddique, M.A.H.; Omura, J.; Satoh, T.; Nogi, M.; Sunamura, S.; et al. Selenoprotein P Promotes the Development of Pulmonary Arterial Hypertension: A Possible Novel Therapeutic Target. Circulation 2018. [Google Scholar] [CrossRef]
- Hill, K.E.; Wu, S.; Motley, A.K.; Stevenson, T.D.; Winfrey, V.P.; Capecchi, M.R.; Atkins, J.F.; Burk, R.F. Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J. Biol. Chem. 2012, 287, 40414–40424. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005, 25, 215–235. [Google Scholar] [CrossRef]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 2003, 278, 13640–13646. [Google Scholar] [CrossRef]
- Saito, Y.; Takahashi, K. Characterization of selenoprotein P as a selenium supply protein. Eur. J. Biochem 2002, 269, 5746–5751. [Google Scholar] [CrossRef] [Green Version]
- Tujebajeva, R.M.; Harney, J.W.; Berry, M.J. Selenoprotein P expression, purification, and immunochemical characterization. J. Biol. Chem. 2000, 275, 6288–6294. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metabolism 2010, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Strauss, E.; Oszkinis, G.; Staniszewski, R. SEPP1 gene variants and abdominal aortic aneurysm: Gene association in relation to metabolic risk factors and peripheral arterial disease coexistence. Sci. Rep. 2014, 4, 7061. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, F.P.; Raman, A.V.; Rueli, R.H.; Bellinger, M.T.; Dewing, A.S.; Seale, L.A.; Andres, M.A.; Uyehara-Lock, J.H.; White, L.R.; Ross, G.W.; et al. Changes in selenoprotein P in substantia nigra and putamen in Parkinson’s disease. J. Parkinsons Dis. 2012, 2, 115–126. [Google Scholar]
- Barrett, C.W.; Reddy, V.K.; Short, S.P.; Motley, A.K.; Lintel, M.K.; Bradley, A.M.; Freeman, T.; Vallance, J.; Ning, W.; Parang, B.; et al. Selenoprotein P influences colitis-induced tumorigenesis by mediating stemness and oxidative damage. J. Clin. Investig. 2015, 125, 2646–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef]
- Saito, Y.; Sato, N.; Hirashima, M.; Takebe, G.; Nagasawa, S.; Takahashi, K. Domain structure of bi-functional selenoprotein P. Biochem. J. 2004, 381, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Hoe, H.S.; Harris, D.C.; Rebeck, G.W. Multiple pathways of apolipoprotein E signaling in primary neurons. J. Neurochem. 2005, 93, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeper, M.M.; McLaughlin, V.V.; Dalaan, A.M.; Satoh, T.; Galiè, N. Treatment of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 323–336. [Google Scholar] [CrossRef]
- Takayama, H.; Misu, H.; Iwama, H.; Chikamoto, K.; Saito, Y.; Murao, K.; Teraguchi, A.; Lan, F.; Kikuchi, A.; Saito, R.; et al. Metformin suppresses expression of the selenoprotein P gene via an AMP-activated kinase (AMPK)/FoxO3a pathway in H4IIEC3 hepatocytes. J. Biol. Chem. 2014, 289, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Kalogris, C.; Garulli, C.; Pietrella, L.; Gambini, V.; Pucciarelli, S.; Lucci, C.; Tilio, M.; Zabaleta, M.E.; Bartolacci, C.; Andreani, C.; et al. Sanguinarine suppresses basal-like breast cancer growth through dihydrofolate reductase inhibition. Biochem. Pharmacol. 2014, 90, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Yaoita, N.; Satoh, K.; Satoh, T.; Sugimura, K.; Tatebe, S.; Yamamoto, S.; Aoki, T.; Miura, M.; Miyata, S.; Kawamura, T.; et al. Thrombin-activatable fibrinolysis inhibitor in chronic thromboembolic pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Satoh, K.; Yaoita, N.; Kikuchi, N.; Omura, J.; Kurosawa, R.; Numano, K.; Al-Mamun, E.; Siddique, M.A.; Sunamura, S.; et al. Activated TAFI promotes the development of chronic thromboembolic pulmonary hypertension: A possible novel therapeutic target. Circ. Res. 2017, 120, 1246–1262. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, T.; Tekinalp, G.; Yigit, S.; Kirazli, S.; Korkmaz, A.; Gurgey, A. Thrombin activatable fibrinolysis inhibitor activity (TAFIa) levels in neonates with meconium-stained amniotic fluid. J. Matern.-Fetal Neonatal Med. 2008, 21, 123–128. [Google Scholar] [CrossRef]
- Bajzar, L.; Nesheim, M.; Morser, J.; Tracy, P.B. Both cellular and soluble forms of thrombomodulin inhibit fibrinolysis by potentiating the activation of thrombin-activable fibrinolysis inhibitor. J. Biol. Chem. 1998, 273, 2792–2798. [Google Scholar] [CrossRef]
- Sugimura, K.; Fukumoto, Y.; Miura, Y.; Nochioka, K.; Miura, M.; Tatebe, S.; Aoki, T.; Satoh, K.; Yamamoto, S.; Yaoita, N.; et al. Three-dimensional-optical coherence tomography imaging of chronic thromboembolic pulmonary hypertension. Eur. Heart J. 2013, 34, 2121. [Google Scholar] [CrossRef]
- Aoki, T.; Sugimura, K.; Nochioka, K.; Miura, M.; Tatebe, S.; Yamamoto, S.; Yaoita, N.; Suzuki, H.; Sato, H.; Kozu, K.; et al. Effects of balloon pulmonary angioplasty on oxygenation in patients with chronic thromboembolic pulmonary hypertension-Importance of intrapulmonary shunt. Circ. J. 2016, 80, 2227–2234. [Google Scholar] [CrossRef]
- Sato, H.; Ota, H.; Sugimura, K.; Aoki, T.; Tatebe, S.; Miura, M.; Yamamoto, S.; Yaoita, N.; Suzuki, H.; Satoh, K.; et al. Balloon pulmonary angioplasty improves biventricular functions and pulmonary flow in chronic thromboembolic pulmonary hypertension. Circ. J. 2016, 80, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, S.; Sugimura, K.; Aoki, T.; Miura, M.; Nochioka, K.; Yaoita, N.; Suzuki, H.; Sato, H.; Yamamoto, S.; Satoh, K.; et al. Multiple beneficial effects of balloon pulmonary angioplasty in patients with chronic thromboembolic pulmonary hypertension. Circ. J. 2016, 80, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, S.; Fukumoto, Y.; Sugimura, K.; Nakano, M.; Miyamichi, S.; Satoh, K.; Oikawa, M.; Shimokawa, H. Optical coherence tomography as a novel diagnostic tool for distal type chronic thromboembolic pulmonary hypertension. Circ. J. 2010, 74, 1742–1744. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Fukumoto, Y.; Saji, K.; Sugimura, K.; Demachi, J.; Nawata, J.; Shimokawa, H. Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessel. 2010, 25, 144–149. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| New Candidate Molecules | Novel Targets | References |
|---|---|---|
| Fasudil | Rho-kinase (human, inharation) | [134] |
| Fasudil | Rho-kinase (human, oral) | [72] |
| Fasudil | Rho-kinase (rodents) | [37] |
| Metformin | AMPK (mouse) | [25] |
| PPARα agonist (Fenofibrate, WY14643) | TAFI (mouse) | [126] |
| TAFIa inhibitor (carboxypeptidase inhibitor) | TAFI (mouse) | [126] |
| Sanguinarin | Selenoprotein P (rat) | [107] |
| Celastrol | Cyclophilin A and Basigin (mouse) | [85] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Sunamura, S.; Siddique, M.A.H.; Omura, J.; Yaoita, N.; Shimokawa, H. Identification of Novel Therapeutic Targets for Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 4081. https://doi.org/10.3390/ijms19124081
Satoh K, Kikuchi N, Satoh T, Kurosawa R, Sunamura S, Siddique MAH, Omura J, Yaoita N, Shimokawa H. Identification of Novel Therapeutic Targets for Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2018; 19(12):4081. https://doi.org/10.3390/ijms19124081
Chicago/Turabian StyleSatoh, Kimio, Nobuhiro Kikuchi, Taijyu Satoh, Ryo Kurosawa, Shinichiro Sunamura, Mohammad Abdul Hai Siddique, Junichi Omura, Nobuhiro Yaoita, and Hiroaki Shimokawa. 2018. "Identification of Novel Therapeutic Targets for Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 19, no. 12: 4081. https://doi.org/10.3390/ijms19124081
APA StyleSatoh, K., Kikuchi, N., Satoh, T., Kurosawa, R., Sunamura, S., Siddique, M. A. H., Omura, J., Yaoita, N., & Shimokawa, H. (2018). Identification of Novel Therapeutic Targets for Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 19(12), 4081. https://doi.org/10.3390/ijms19124081