QTL Mapping of Fiber Quality and Yield-Related Traits in an Intra-Specific Upland Cotton Using Genotype by Sequencing (GBS)

 , and
, and
Abstract
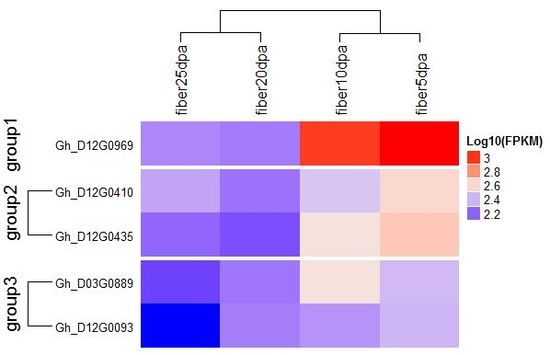
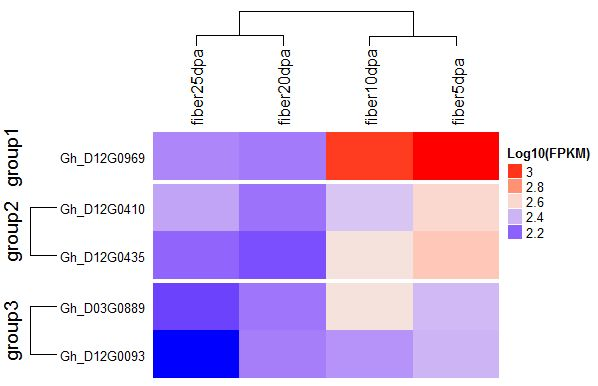
:
1. Introduction
2. Results
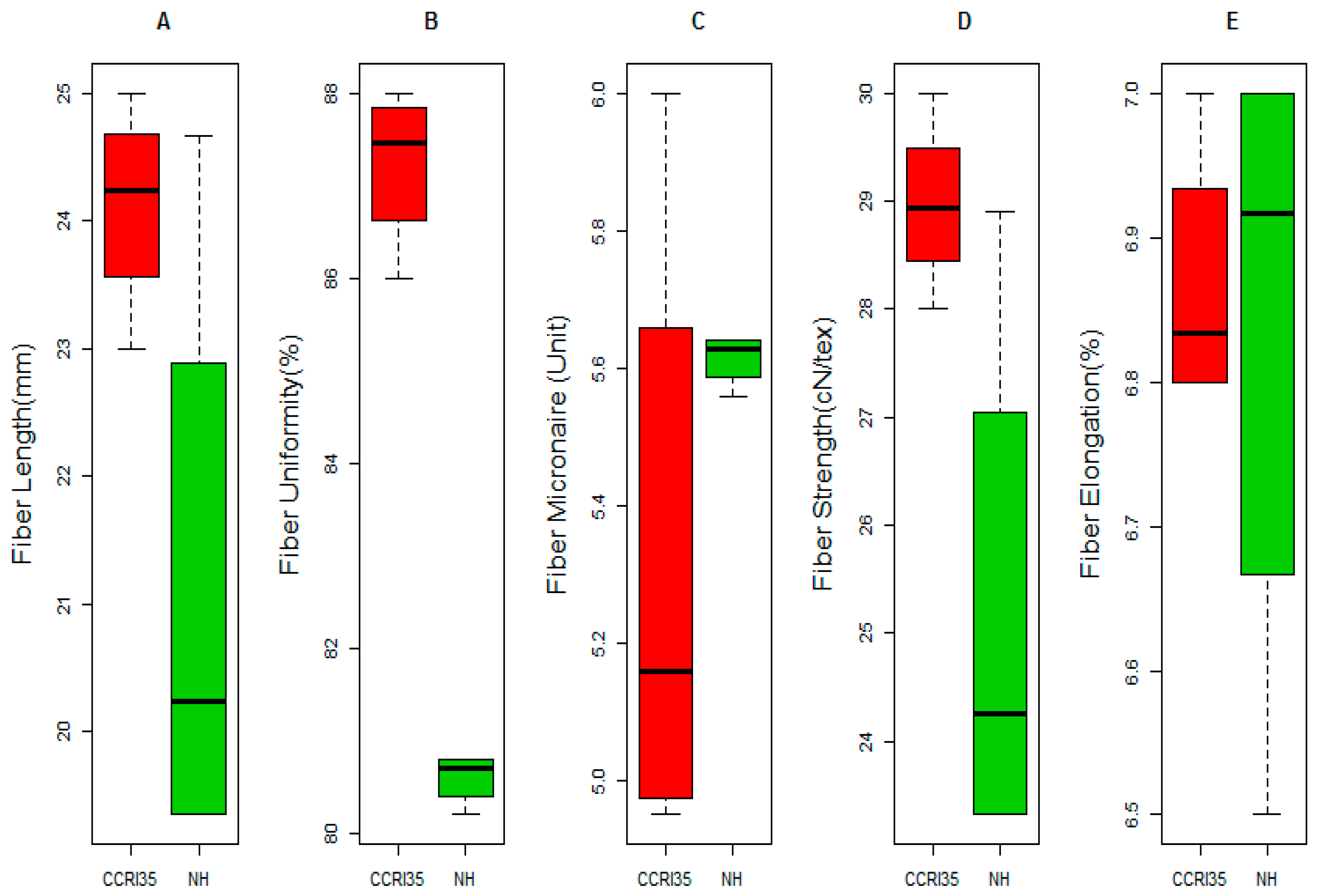
2.1. Phenotypic Variation between the Two Parents
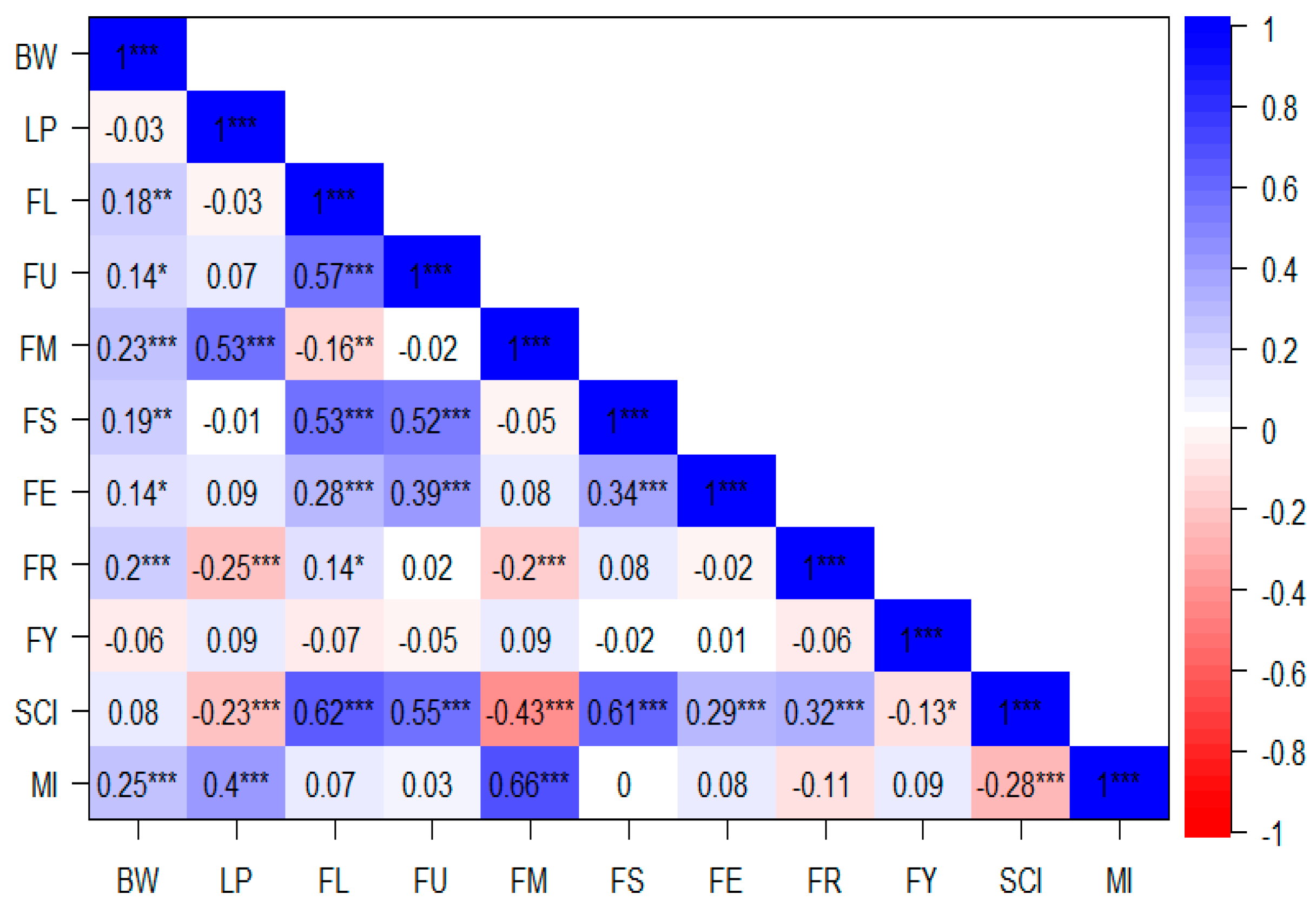
2.2. Correlation Analysis
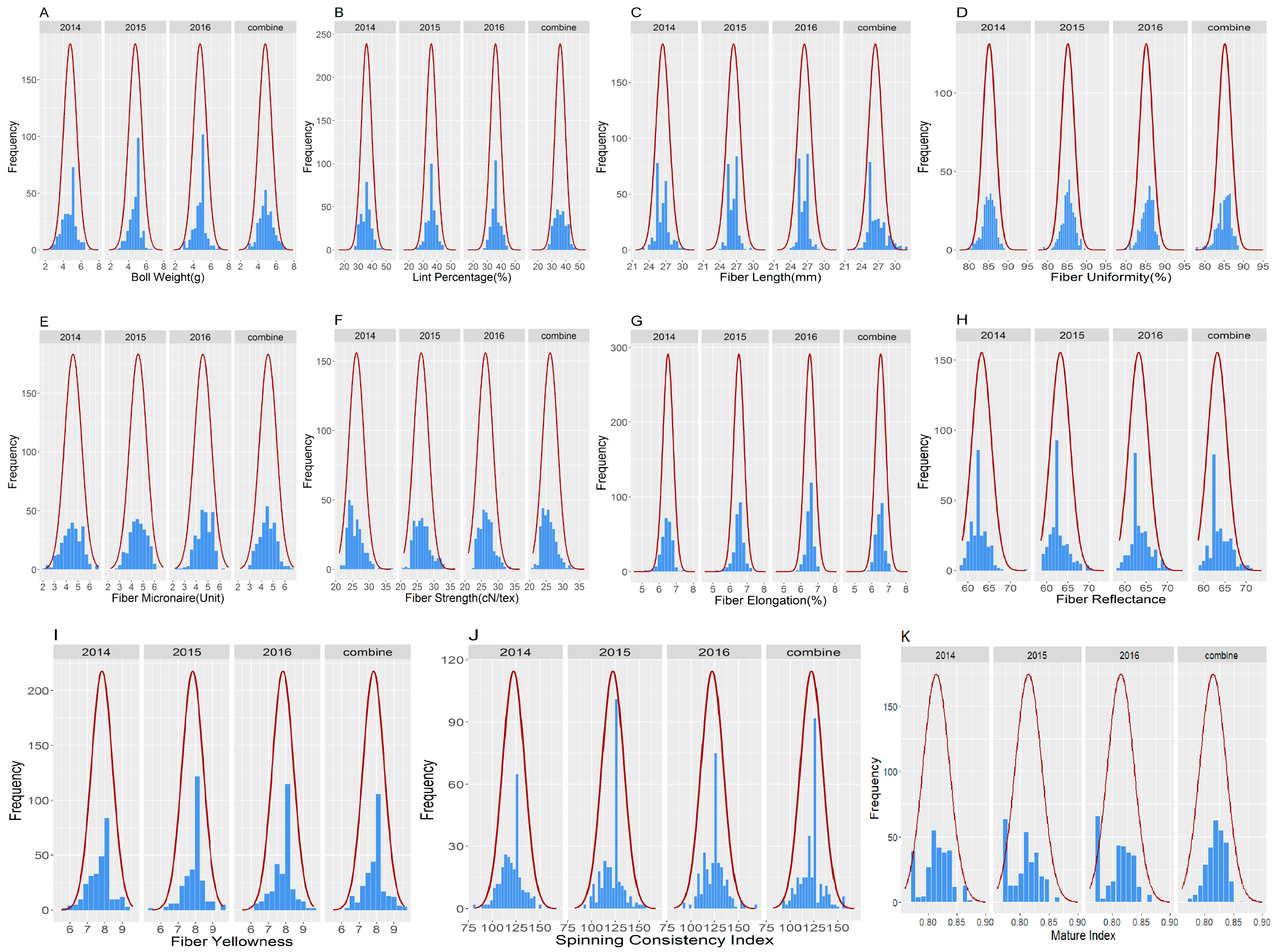
2.3. ANOVA, Broad Sense Heritability and Phenotypic Analysis of Fiber Quality for the Two Parents and the F2:3 Population
2.4. GBS Genotyping, SNP Detection and Annotation
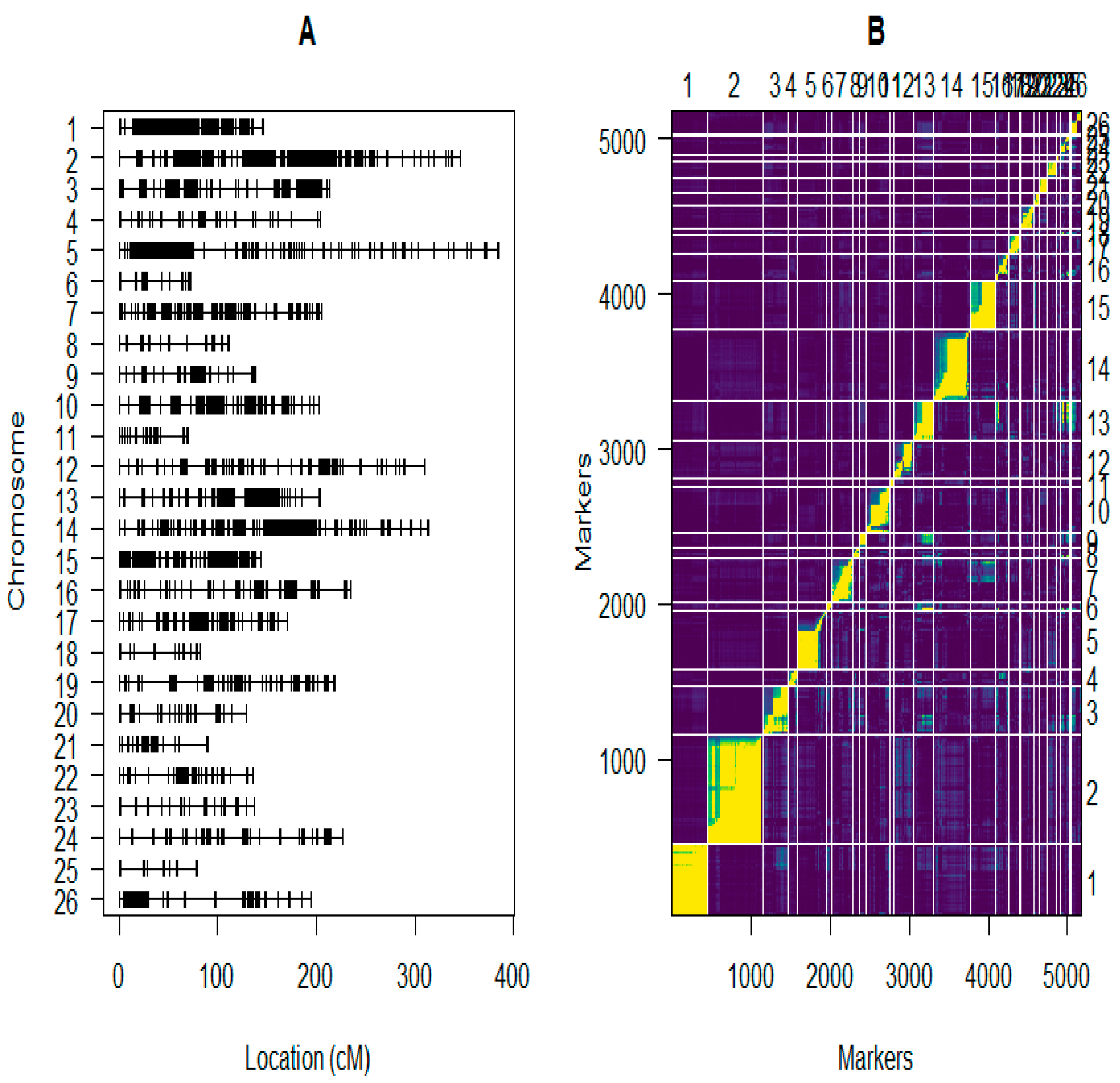
2.5. Construction of the Linkage Maps
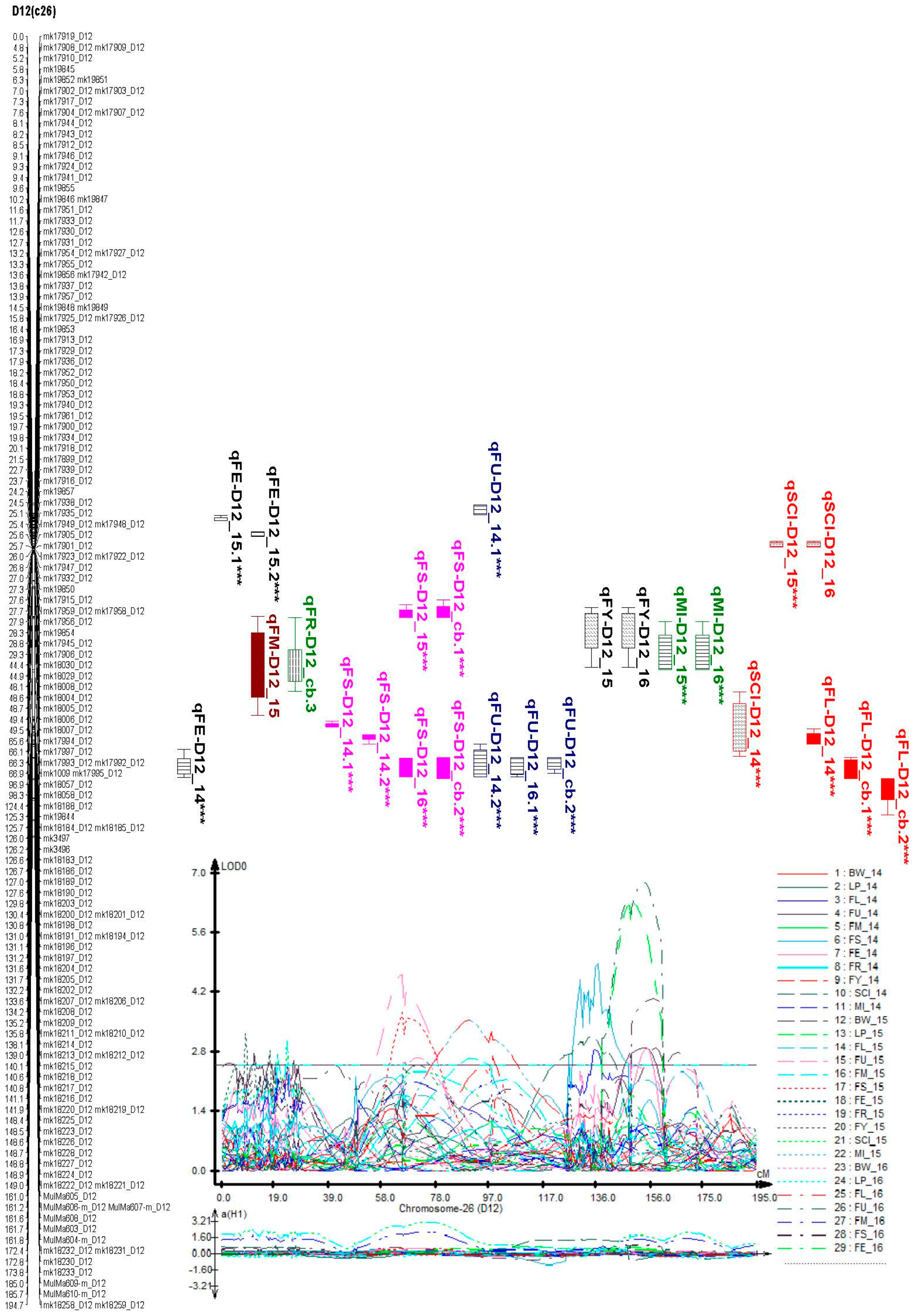
2.6. Identification of Consistent and Clustering QTLs for Yield Related and Fiber Quality Traits
2.7. The Gene Ontology Enrichment Analysis Based on QTL Clusters
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Growth Conditions and Trait Data Collection
4.2. Sample Collection, Library Preparation, Sequencing and SNP Genotyping
4.2.1. DNA Extraction, Quantification and Quality Determination
4.2.2. GBS Library Preparation, Sequencing and SNP Genotyping
4.3. Data Analysis and Linkage Map Construction
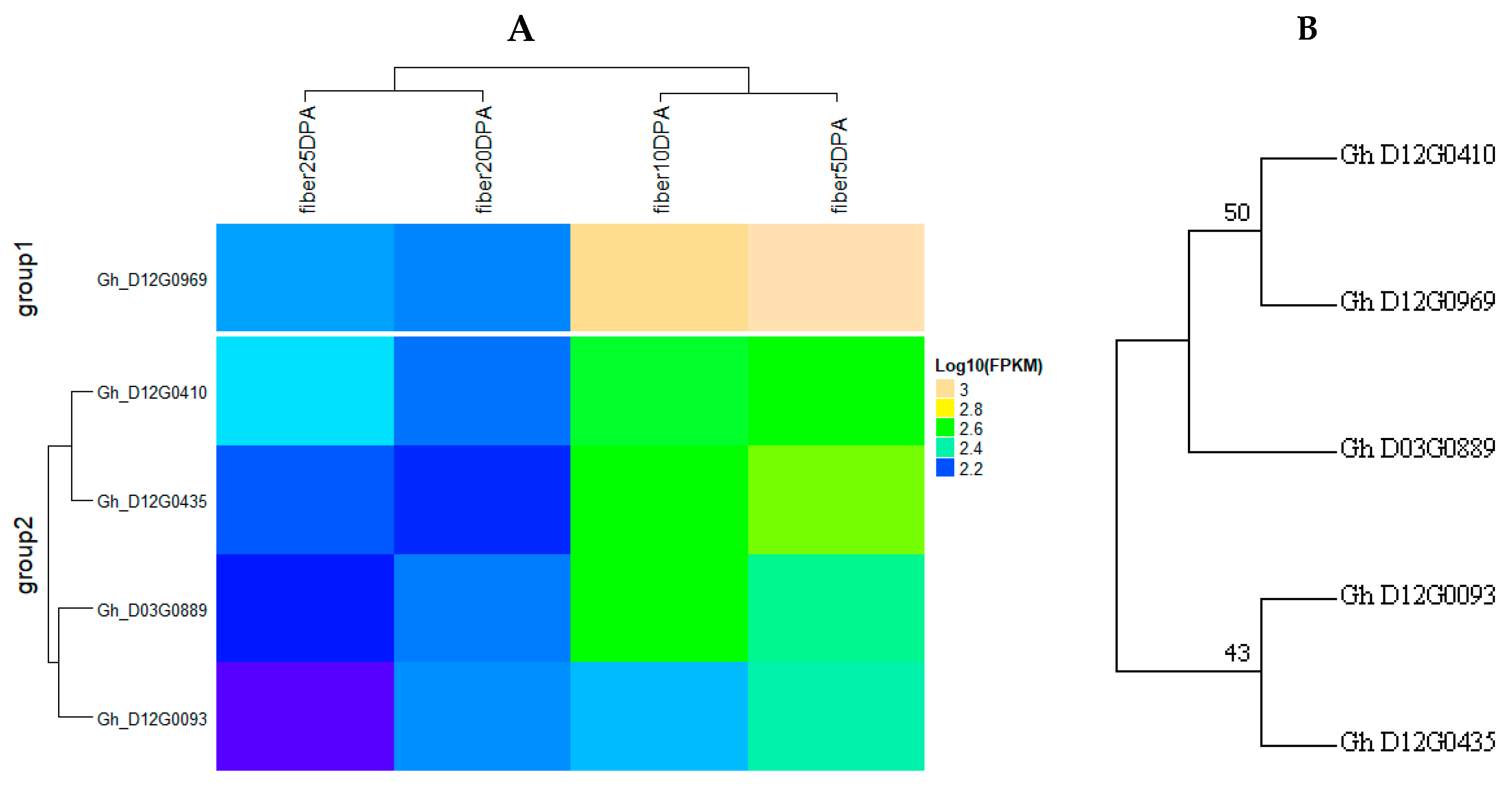
4.4. Gene Mining and Expression Analysis
4.5. The Gene Ontology Enrichment Analysis BaseD on QTL Clusters
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Funding
References
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Hu, C.; Hua, H.; Li, Z.; Hua, J. Construction of a linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Chin. Sci. Bull. 2013, 58, 3233–3243. [Google Scholar] [CrossRef]
- Kohel, R.J.; Yu, J.; Park, Y.H. Molecular mapping and characterization of traits controlling fiber quality in cotton. Euphytica 2001, 121, 163–172. [Google Scholar] [CrossRef]
- Lokhande, S.; Reddy, K.R. Quantifying Temperature Effects on Cotton Reproductive Efficiency and Fiber Quality. Agron. J. 2014, 106, 1275–1282. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Li, X.B.; Xiao, Y.H. Combining ability and heterosis between high strength lines and transgenic Bt (Bacillus thuringiensis) bollworm-resistant lines in upland cotton (Gossypium hirsutum L.). Agric. Sci. China 2003, 2, 13–18. [Google Scholar]
- Shen, X.; Guo, W.; Lu, Q. Genetic mapping of quantitative trait loci for fiber quality and yield trait by RIL approach in upland cotton. Euphytica 2007, 155, 371–380. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, K.; Li, S.; Yu, S.; Zhai, H.; Wu, M.; Li, X.; Fan, S.; Song, M.; Yang, D.; et al. Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Theor. Appl. Genet. 2013, 126, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Liang, Q.; Wang, Y.; Wang, X.; Wang, K.; Abduweli, A.; Ma, L.; Cai, S.; Hua, J. Identification of stable QTLs controlling fiber traits properties in multi-environment using recombinant inbred lines in Upland cotton (Gossypium hirsutum L.). Euphytica 2015, 205, 877–888. [Google Scholar] [CrossRef]
- Tan, Z.Y.; Fang, X.M.; Tang, S.Y.; Zhang, J.; Liu, D.J.; Teng, Z.H.; Li, L.; Ni, H.; Zheng, F.; Liu, D.; et al. Genetic map and QTL controlling fiber quality traits in upland cotton (Gossypium hirsutum L.). Euphytica 2015, 203, 615–628. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Jamshed, M.; Jia, F.; Gong, J.; Palanga, K.K.; Shi, Y.; Li, J.; Shang, H.; Liu, A.; Chen, T.; Zhang, Z.; et al. Identification of stable quantitative trait loci (QTLs) for fiber quality traits across multiple environments in Gossypium hirsutum recombinant inbred line population. BMC Genom. 2016, 17, 197. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yuan, D.; Liang, S. Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genom. 2011, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Kurata, N.; Wei, X.; Wang, Z.-X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2012, 30, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Spindel, J.; Wright, M.; Chen, C.; Cobb, J.; Gage, J.; Harrington, S.; Lorieux, M.; Ahmadi, N.; McCouch, S. Bridging the genotyping gap: Using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor. Appl. Genet. 2013, 126, 2699–2716. [Google Scholar] [CrossRef] [PubMed]
- Thomson, M.J.; Zhao, K.; Wright, M.; McNally, K.; Rey, J.; Tung, C.W.; Reynolds, A.; Scheffler, B.; Eizenga, G.; McClung, A.K.H.; et al. High-throughput single nucleotide polymorphism genotyping for breeding applications in rice using the BeadXpress platform. Mol. Breed. 2012, 29, 875–886. [Google Scholar] [CrossRef]
- Poland, J.A.; Rife, T.W. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef]
- Wang, S.; Zeng, Z.B. Department of Statistics; North Carolina State University: Raleigh, NC, USA, 2007. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008. [Google Scholar]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Stam, P.; van Ooijen, J.W. JoinMap™ Version 2.0: Software for the Calculation of Genetic Linkage Maps; Plant Research International B.V. and Kyazma B.V.: Wageningen, The Netherlands, 1995; CPRO-DLO: Wageningen, The Netherlands, 1995. [Google Scholar]
- Rong, J.; Abbey, C.; Bowers, J.E. A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genet. Mol. Biol. 2004, 166, 389–417. [Google Scholar] [CrossRef]
- Fang, D.D.; Jenkins, J.N.; Deng, D.D.; McCarty, J.C.; Li, P.; Wu, J. Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (Gossypium hirsutum L.). BMC Genom. 2014, 15, 397. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, H.; Huang, C.; Guo, H.; Li, X.; Zhao, W.; Dai, B.; Yan, Z.; Lin, Z. QTL Mapping for Fiber and Yield Traits in Upland Cotton under Multiple Environments. PLoS ONE 2015, 10, e0130742. [Google Scholar] [CrossRef] [PubMed]
- Oluoch, G.; Zheng, J.; Wang, X.; Khan, M.K.R.; Zhou, Z.; Cai, X.; Wang, C.; Wang, Y.; Li, X.; Wang, H.; et al. QTL mapping for salt tolerance at seedling stage in the interspecific cross of Gossypium tomentosum with Gossypium hirsutum. Euphytica 2016, 209, 223–235. [Google Scholar] [CrossRef]
- Lacape, J.-M.; Llewellyn, D.; Jacobs, J.; Arioli, T.; Becker, D.; Calhoun, S.; Al-Ghazi, Y.; Liu, S.; Palaï, O.; Georges, S.; et al. Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum x G. barbadense RIL population. BMC Plant Biol. 2010, 10, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, J.; Moghaddam, M.; Ghassemi, K.; Nishabouri, M.R. Genetic properties of drought resistance indices. J. Agric. Sci. Technol. 2001, 3, 43–49. [Google Scholar]
- Shao, Q.; Zhang, F.; Tang, S.; Liu, Y.; Fang, X.; Liu, D.; Liu, D.; Zhang, J.; Teng, Z.; Paterson, A.H.; et al. Identifying QTL for fiber quality traits with three upland cotton (Gossypium hirsutum L.) populations. Euphytica 2014, 198, 43–58. [Google Scholar] [CrossRef]
- Lin, Z.; He, D.; Zhang, X.; Nie, Y.; Guo, X.; Feng, C.; Stewart, J.M. Linkage map construction and mapping QTL for cotton fibre quality using SRAP, SSR and RAPD. Plant Breed. 2005, 124, 180–187. [Google Scholar] [CrossRef]
- Shen, X.L.; Guo, W.Z.; Zhu, X.F.; Yuan, Y.L.; Yu, J.Z.; Kohel, R.J.; Zhang, T.Z. Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol. Breed. 2005, 15, 169–181. [Google Scholar] [CrossRef]
- Zhang, Z.-S.; Xiao, Y.-H.; Luo, M.; Li, X.-B.; Luo, X.-Y.; Hou, L.; Li, D.-M.; Pei, Y. Construction of a genetic linkage map and QTL analysis of fiber-related traits in upland cotton (Gossypium hirsutum L.). Euphytica 2005, 144, 91–99. [Google Scholar] [CrossRef]
- Zhang, Z.-S.; Hu, M.-C.; Zhang, J.; Liu, D.-J.; Zheng, J.; Zhang, K.; Wang, W.; Wan, Q. Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol. Breed. 2009, 24, 49–61. [Google Scholar] [CrossRef]
- Wang, B.H.; Guo, W.Z.; Zhu, X.F.; Wu, Y.T.; Huang, N.T.; Zhang, T. QTL mapping of fiber quality in an elite hybrid derived-RIL population of upland cotton. Euphytica 2006, 152, 367–378. [Google Scholar] [CrossRef]
- He, D.H.; Lin, Z.X.; Zhang, X.L.; Nie, Y.C.; Guo, X.P.; Zhang, Y.X.; Li, W. QTL mapping for economic traits based on a dense genetic map of cotton with PCR-based markers using the interspecific cross of Gossypium hirsutum × Gossypium barbadense. Euphytica 2007, 153, 181–197. [Google Scholar] [CrossRef]
- Rong, J.; Feltus, F.A.; Waghmare, V.N.; Pierce, G.J.; Chee, P.W.; Draye, X.; Saranga, Y.; Wright, R.J.; Wilkins, T.A.; May, O.L.; et al. Meta-analysis of polyploidy cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 2007, 176, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ding, Y.; Zhou, B.; Guo, W.; Lv, Y.; Zhu, X.; Zhang, T. QTL mapping in A-genome diploid Asiatic cotton and their congruence analysis with AD-genome tetraploid cotton in genus Gossypium. J. Genet. Genom. 2008, 35, 751–762. [Google Scholar] [CrossRef]
- Sun, F.-D.; Zhang, J.-H.; Wang, S.-F.; Gong, W.-K.; Shi, Y.-Z.; Liu, A.-Y.; Li, J.-W.; Gong, J.-W.; Shang, H.-H.; Yuan, Y.-L. QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol. Breed. 2012, 30, 569–582. [Google Scholar] [CrossRef]
- Zhang, S.W.; Zhu, X.F.; Feng, L.C.; Gao, X.; Yang, B.; Zhang, T.Z.; Zhou, B.L. Mapping of fiber quality QTLs reveals useful variation and footprints of cotton domestication using introgression lines. Sci. Rep. 2016, 6, 31954. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, C.; Zhao, W.; Dai, B.; Shen, C.; Zhang, B.; Li, D.; Lin, Z. Identification of QTL for Fiber Quality and Yield Traits Using Two Immortalized Backcross Populations in Upland Cotton. PLoS ONE 2016, 11, e0166970. [Google Scholar] [CrossRef] [PubMed]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.C.; Wilkins, T.A. Cotton (Gossypium hirsutum) Seed Trichomes Expand via Diffuse Growing Mechanism. Can. J. Bot. Rev. Can. Bot. 1995, 73, 746–757. [Google Scholar] [CrossRef]
- Guan, X.; Lee, J.J.; Pang, M.; Shi, X.; Stelly, D.M.; Chen, Z.J. Activation of Arabidopsis Seed Hair Development by Cotton Fiber-Related Genes. PLoS ONE 2011, 6, e21301. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Li, Q.; Fan, X.; Yang, W.; Xue, Y. The R2R3 MYB Transcription Factor GhMYB109 Is Required for Cotton Fiber Development. Genetics 2008, 180, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Wu, M.; Pei, W.; Li, H.; Li, X.; Zhang, J.; Yu, J.; Yu, S. Quantitative phosphoproteomic profiling of fiber differentiation and initiation in a fiberless mutant of cotton. BMC Genom. 2014, 15, 466. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, B.; Hovav, R.; Rapp, R.; Verma, N.; Udall, J.A.; Wendel, J.F. Global Analysis of Gene Expression in Cotton Fibers from Wild and Domesticated Gossypium barbadense. Evol. Dev. 2008, 10, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; He, S.; Gong, W.; Sun, J.; Pan, Z.; Xu, F.; Lu, Y.; Du, X. Comprehensive analysis of differentially expressed genes and transcriptional regulation induced by salt stress in two contrasting cotton genotypes. BMC Genom. 2014, 15, 760. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Xu, F.; Sun, J.; Peng, Z.; He, S.; Pan, Z.; Du, X. iTRAQ-Based Comparative Proteomic Analysis of Seedling Leaves of Two Upland Cotton Genotypes Differing in Salt Tolerance. Front. Plant Sci. 2017, 8, 2113. [Google Scholar] [CrossRef] [PubMed]
- Socquet-Juglard, D.; Christen, D.; Devenes, G.; Gessler, C.; Duffy, B.; Patocchi, A. Mapping Architectural, Phenological, and Fruit Quality QTLs in Apricot. Plant Mol. Biol. Rep. 2013, 31, 387–397. [Google Scholar] [CrossRef]
- Paterson, A.H.; Brubaker, C.; Wendel, J.F. A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol. Biol. Rep. 1993, 11, 122–127. [Google Scholar] [CrossRef]
- Krizman, M.; Jakse, J.; Baricevic, D.; Javornik, B.; Prosek, M. Robust CTAB-activated charcoal protocol for plant DNA extraction. Acta Agric. Slov. 2006, 87, 427–433. [Google Scholar]
- Wilfinger, W.W.; Chomczynski, M.K. P 260/280 and 260/230 Ratios NanoDrop ® ND-1000 and ND-8000 8-Sample Spectrophotometers. BioTechniques 1997, 22, 474–481. [Google Scholar] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef] [PubMed]
- Paten, B.; Novak, A.; Haussler, D. Mapping to a Reference Genome Structure. arXiv, 2014; arXiv:1404.5010. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- SPSS Inc. SPSS Base 10.0 for Windows User’s Guide; SPSS Inc.: Chicago, IL, USA, 1999. [Google Scholar]
- Singh, U.M.; Yadav, S.; Dixit, S.; Ramayya, P.J.; Devi, M.N.; Raman, K.A.; Kumar, A. QTL Hotspots for Early Vigor and Related Traits under Dry Direct-Seeded System in Rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Wu, H.; Sen, S.; Churchill, G.A. R/QTL: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.J.R.; Ferreira, T.E.; Domiciano, S.; Paula, A.; Paiva, M.; Tecchio, M.A.; Leonel, S. Phenology, yield and fruit quality of four persimmon (Diospyros kaki L.) cultivars in So Paulos Midwest countryside, Brazil. Afr. J. Agric. Res. 2016, 11, 5171–5177. [Google Scholar]
- Podliesna, S.; Bezzina, C.R.; Lodder, E.M. Complex Genetics of Cardiovascular Traits in Mice: F2-Mapping of QTLs and Their Underlying Genes. Methods Mol. Biol. 2017, 1488, 431–454. [Google Scholar] [PubMed]
- Lander, E.; Kruglyak, L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat. Genet. 1995, 1, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Stuber, C.; Edwards, M.D.; Wendel, J.F. Molecular marker facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits. Crop Sci. 1987, 27, 639–648. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
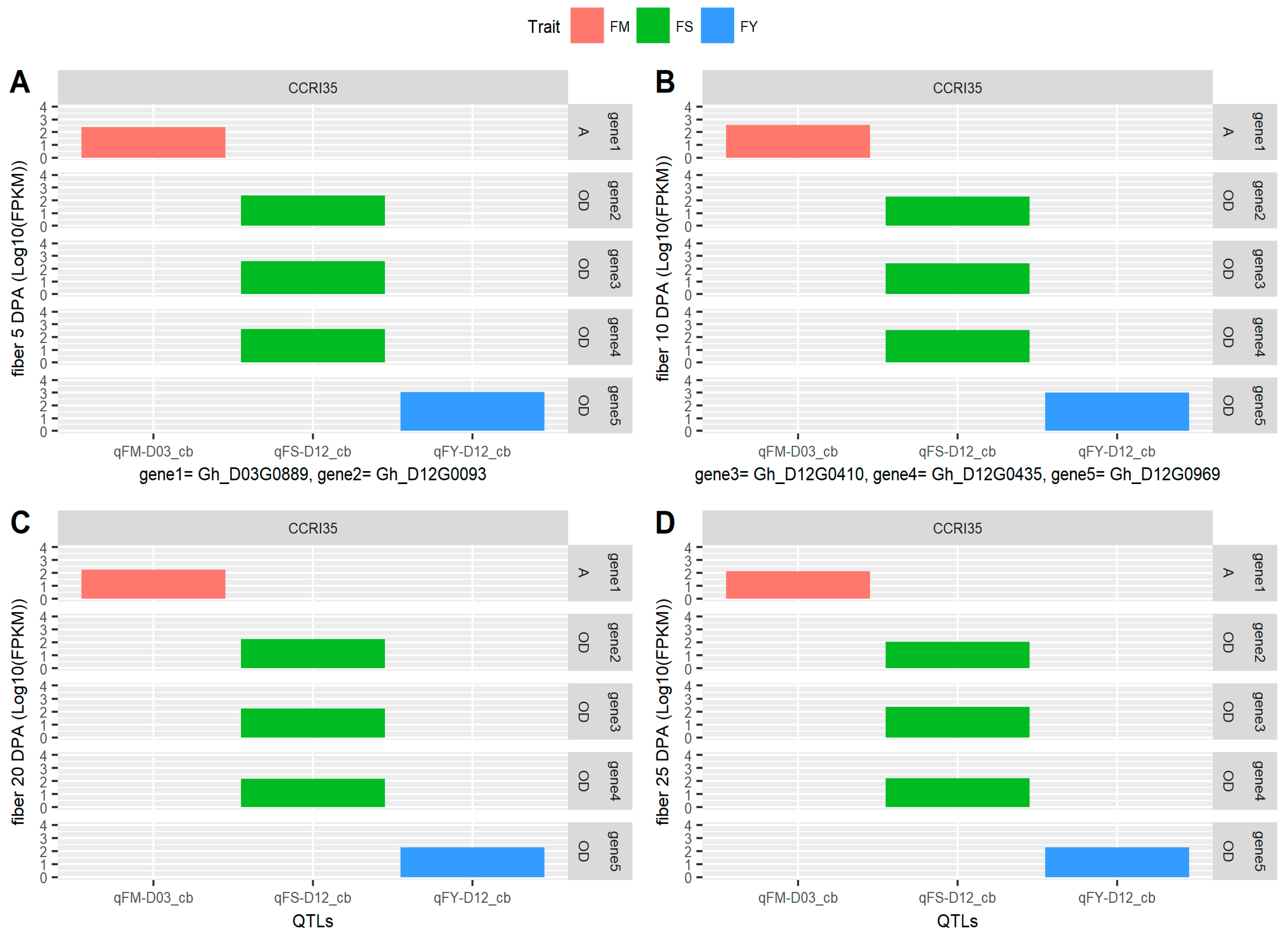{kind=link}
| Trait | Source | DF | SS | MS | F | Pr > F | Hb (%) | P1 | P2 | P1 − P2 | F2:3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Max | Min | Skew | Kurt | |||||||||||
| BW | e | 3 | 327.4 | 109.1 | 1.9 × 1010 | <0.0001 | 67.5 | - | - | 4.77 | 0.73 | 7.9 | 1.8 | −0.16 | 0.94 | |
| g | 276 | 728.6 | 2.6 | 4.59 × 1008 | <0.0001 | |||||||||||
| g*e | 828 | 710.5 | 0.9 | 1.49 × 1008 | <0.0001 | |||||||||||
| LP | e | 3 | 1856.7 | 619 | 9.5 × 1010 | <0.0001 | 82.1 | - | - | 35.97 | 3.71 | 54 | 16.08 | 0.09 | 1.2 | |
| g | 276 | 28,244 | 102.3 | 1.57 × 1010 | <0.0001 | |||||||||||
| g*e | 828 | 15,200.7 | 18.4 | 2.82 × 1009 | <0.0001 | |||||||||||
| FL | e | 3 | 1561.4 | 521 | 9.23 × 1024 | <0.0001 | 63.6 | 24.12 | 21.12 | 3 | 26.46 | 1.2 | 32.2 | 21.55 | 0.94 | 0.55 |
| g | 276 | 1530.1 | 5.5 | 9.83 × 1022 | <0.0001 | |||||||||||
| g*e | 828 | 1670.7 | 2 | 3.58 × 1022 | <0.0001 | |||||||||||
| FU | e | 3 | 1200.6 | 400 | 9 × 1023 | <0.0001 | 77.1 | 87.23 | 80.6 | 6.63 | 85.14 | 1.69 | 89.3 | 77.8 | −0.75 | 0.12 |
| g | 276 | 4844.2 | 17.6 | 3.95 × 1022 | <0.0001 | |||||||||||
| g*e | 828 | 3321.3 | 4 | 9.02 × 1021 | <0.0001 | |||||||||||
| FM | e | 3 | 12.700 | 4 | 2.19 × 1025 | <0.0001 | 92.4 | 5.61 | 5.32 | 0.29 | 4.57 | 0.72 | 6.75 | 2.2 | −0.19 | −0.15 |
| g | 276 | 1406.5 | 5.1 | 2.63 × 1025 | <0.0001 | |||||||||||
| g*e | 828 | 320.3 | 0.4 | 1.99 × 1024 | <0.0001 | |||||||||||
| FS | e | 3 | 2038.7 | 680 | 3.37 × 1025 | <0.0001 | 76.8 | 28.97 | 25.18 | 3.79 | 26.19 | 2.27 | 36.5 | 21 | 0.76 | 0.75 |
| g | 276 | 8851.8 | 32.1 | 1.59 × 1024 | <0.0001 | |||||||||||
| g*e | 828 | 6155.4 | 7.4 | 3.69 × 1023 | <0.0001 | |||||||||||
| FE | e | 3 | 10.800 | 4 | 2.15 × 1024 | <0.0001 | 61.8 | 6.83 | 6.87 | −0.04 | 6.51 | 0.3 | 8.1 | 4.5 | −1.02 | 0.02 |
| g | 276 | 137.1 | 0.5 | 2.96 × 1023 | <0.0001 | |||||||||||
| g*e | 828 | 157.5 | 0.2 | 1.13 × 1023 | <0.0001 | |||||||||||
| FR | e | 3 | 335.500 | 112 | 5.58 × 1022 | <0.0001 | 84.8 | - | - | 63.23 | 2.27 | 73.6 | 58.7 | 0.9 | 1.22 | |
| g | 276 | 11,577.9 | 41.9 | 2.09 × 1022 | <0.0001 | |||||||||||
| g*e | 828 | 5282.3 | 6.4 | 3.18 × 1021 | <0.0001 | |||||||||||
| FY | e | 3 | 38.2 | 12.7 | 8.03 × 1023 | <0.0001 | 86.3 | - | - | 7.83 | 0.61 | 9.6 | 5.5 | −0.32 | 1.19 | |
| g | 276 | 845.6 | 3.1 | 1.93 × 1023 | <0.0001 | |||||||||||
| g*e | 828 | 348 | 0.4 | 2.65 × 1022 | <0.0001 | |||||||||||
| SCI | e | 3 | 24,493.6 | 8164.5 | 1.39 × 1025 | <0.0001 | 86.6 | - | - | 122.26 | 11.57 | 167 | 82 | 0.09 | 0.97 | |
| g | 276 | 299,967 | 1086.8 | 1.85 × 1024 | <0.0001 | |||||||||||
| g*e | 828 | 120,302.4 | 145.3 | 2.48 × 1023 | <0.0001 | |||||||||||
| MI | e | 3 | 0.5 | 0.2 | 6.73 × 1025 | <0.0001 | 66.7 | - | - | 0.81 | 0.02 | 0.87 | 0.75 | −0.19 | −0.79 | |
| g | 276 | 0.7 | 0 | 1.18 × 1024 | <0.0001 | |||||||||||
| g*e | 828 | 0.5 | 0 | 2.77 × 1023 | <0.0001 | |||||||||||
| Group | Marker Number | Map Length (cM) | Av Distance (cM) | Max Gap (cM) | <10 cM | >10 cM | Ratio |
|---|---|---|---|---|---|---|---|
| A01(c1) | 448 | 146.704 | 0.33 | 8.505 | 447 | 0 | 1 |
| A02(c2) | 705 | 346.314 | 0.49 | 17.848 | 699 | 5 | 0.99 |
| A03(c3) | 323 | 213.937 | 0.66 | 17.145 | 319 | 3 | 0.99 |
| A04(c4) | 106 | 203.891 | 1.92 | 26.598 | 99 | 6 | 0.93 |
| A05(c5) | 378 | 385.092 | 1.02 | 21.198 | 365 | 12 | 0.97 |
| A06(c6) | 58 | 73.063 | 1.26 | 15.032 | 54 | 3 | 0.93 |
| A07(c7) | 279 | 205.892 | 0.74 | 11.622 | 276 | 2 | 0.99 |
| A08(c8) | 69 | 112.137 | 1.63 | 18.894 | 65 | 3 | 0.94 |
| A09(c9) | 98 | 138.501 | 1.41 | 19.234 | 95 | 2 | 0.97 |
| A10(c10) | 292 | 202.134 | 0.69 | 10.551 | 287 | 4 | 0.98 |
| A11(c11) | 51 | 70.548 | 1.38 | 23.241 | 49 | 1 | 0.96 |
| A12(c12) | 244 | 309.608 | 1.27 | 19.593 | 236 | 7 | 0.97 |
| A13(c13) | 262 | 203.61 | 0.78 | 17.425 | 256 | 5 | 0.98 |
| Subtotal At | 3313 | 2611.43 | 0.79 | 26.598 | 3247 | 53 | 0.98 |
| D01(c15) | 319 | 144.092 | 0.45 | 6.351 | 318 | 0 | 1 |
| D02(c14) | 454 | 313.268 | 0.69 | 14.541 | 450 | 3 | 0.99 |
| D03(c17) | 133 | 170.555 | 1.28 | 14.993 | 131 | 1 | 0.98 |
| D04(c22) | 114 | 136.228 | 1.19 | 20.275 | 110 | 3 | 0.96 |
| D05(c19) | 153 | 218.788 | 1.43 | 27.062 | 148 | 4 | 0.97 |
| D06(c25) | 16 | 79.084 | 4.94 | 22.389 | 12 | 3 | 0.75 |
| D07(c16) | 169 | 235.366 | 1.39 | 26.041 | 161 | 7 | 0.95 |
| D08(c24) | 118 | 226.688 | 1.92 | 20.878 | 109 | 8 | 0.92 |
| D09(c23) | 40 | 136.744 | 3.42 | 14.48 | 33 | 6 | 0.83 |
| D10(c20) | 80 | 129.051 | 1.61 | 20.539 | 76 | 3 | 0.95 |
| D11(c21) | 98 | 89.782 | 0.92 | 27.564 | 95 | 2 | 0.97 |
| D12(c26) | 143 | 194.735 | 1.36 | 30.082 | 135 | 7 | 0.94 |
| D13(c18) | 28 | 82.286 | 2.94 | 20.917 | 25 | 2 | 0.89 |
| Subtotal Dt | 1865 | 2156.67 | 1.156 | 30.082 | 1803 | 49 | 0.97 |
| TOTAL (At + Dt) | 5178 | 4768.1 | 0.92 | 30.082 | 5050 | 102 | 0.98 |
| Trait | QTL | Chr | Start Marker | End Marker | Start Marker (bp) | End Marker (bp) | Start Marker (cM) | End Marker (cM) | Position (cM) | LOD | Ae | De | |d/a| | GA | R2 (%) | DPE |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FS | qFS-A02_15 | A02 | mk1761_A02 | mk1778_A02 | 80,488,799 | 81,766,125 | 0 | 17.848 | 5.01 | 3.761129 | −0.0052 | 1.5095 | 290.28846 | OD | 0.5295 | NH |
| qFS-A02_cb | A02 | mk1761_A02 | mk1778_A02 | 80,488,799 | 81,766,125 | 0 | 17.848 | 7.01 | 2.903366 | 0.0292 | 0.5368 | 18.383562 | OD | 0.0421 | CCRI35 | |
| qFS-A02_cb | A02 | mk1020_A02 | mk1022_A02 | 827,449 | 909,242 | 337.304 | 346.314 | 337.11 | 5.507058 | 0.2417 | 0.1944 | 0.8043029 | A | 5.6762 | CCRI35 | |
| SCI | qSCI-A02_15 | A02 | mk1761_A02 | mk1778_A02 | 80,488,799 | 8,176,6125 | 0 | 17.848 | 1.01 | 3.268187 | −0.7383 | 9.0221 | 12.2201 | OD | 1.2775 | NH |
| qSCI-A02_cb | A02 | mk1761_A02 | mk1778_A02 | 80,488,799 | 81,766,125 | 0 | 17.848 | 1.01 | 2.740499 | −0.4142 | 5.1274 | 12.379044 | OD | 0.9695 | NH | |
| qSCI-A02_cb | A02 | mk1018_A02 | mk1019_A02 | 822,030 | 827,340 | 334.259 | 337.053 | 336.31 | 3.27253 | 0.9943 | 4.7528 | 4.7800463 | OD | 0.277 | CCRI35 | |
| FL | qFL-A03_14 | A03 | mk1922_A03 | mk1927_A03 | 1,863,137 | 1,863,215 | 194.163 | 194.228 | 194.21 | 2.54506 | 0.1746 | 0.5337 | 3.056701 | OD | 0.4959 | CCRI35 |
| qFL-A03_15 | A03 | mk1989_A03 | mk2007_A03 | 2,881,061 | 2,936,448 | 168.853 | 169.141 | 168.91 | 2.599349 | 0.276 | 0.1484 | 0.5376812 | PD | 3.0929 | CCRI35 | |
| qFL-A03_cb | A03 | mk11099 | mk2084_A03 | 31,386 | 666,6259 | 94.16 | 102.466 | 102.21 | 3.565689 | 0.148 | 0.3166 | 2.1391892 | OD | 1.445 | CCRI35 | |
| qFL-A03_cb | A03 | mk2085_A03 | mk2087_A03 | 6,666,473 | 6,736,164 | 130.379 | 130.946 | 130.41 | 2.723127 | 0.1219 | 0.2515 | 2.0631665 | OD | 1.3067 | CCRI35 | |
| FM | qFM-A05_15 | A05 | mk2943_A05 | mk2952_A05 | 21,550,988 | 23,173,778 | 195.463 | 206.855 | 197.51 | 2.927253 | 0.1377 | −0.2204 | 1.600581 | OD | 4.6742 | CCRI35 |
| qFM-A05_cb | A05 | mk2943_A05 | mk2952_A05 | 21,550,988 | 23,173,778 | 195.463 | 206.855 | 197.51 | 3.040174 | 0.0684 | −0.0932 | 1.3625731 | OD | 5.0485 | CCRI35 | |
| LP | qLP-A05_14 | A05 | mk2943_A05 | mk2952_A05 | 21,550,988 | 23,173,778 | 195.463 | 206.855 | 195.51 | 4.76873 | 0.9516 | −0.8514 | 0.8947037 | D | 11.2688 | CCRI35 |
| qLP-A05_cb | A05 | mk2943_A05 | mk2952_A05 | 21,550,988 | 23,173,778 | 195.463 | 206.855 | 195.51 | 2.673181 | 0.4032 | −1.1877 | 2.9456845 | OD | 3.4428 | CCRI35 | |
| BW | qBW-A09_15 | A09 | mk6774_A09 | mk6775_A09 | 60,948,395 | 62,054,979 | 7.252 | 15.619 | 15.61 | 2.599349 | 0.1034 | 0.335 | 3.2398453 | OD | 0.2668 | CCRI35 |
| qBW-A09_15 | A09 | mk6764_A09 | mk6772_A09 | 59,295,756 | 59,503,467 | 25.198 | 26.005 | 25.21 | 2.744843 | 0.0794 | 0.3894 | 4.9042821 | OD | 0.0173 | CCRI35 | |
| qBW-A09_cb | A09 | mk6764_A09 | mk6772_A09 | 59,295,756 | 59,503,467 | 25.198 | 26.005 | 25.21 | 2.831705 | 0.004 | 0.2838 | 70.95 | OD | 0.6913 | CCRI35 | |
| FU | qFU-A09_16 | A09 | mk6410_A09 | mk6462_A09 | 4,242,475 | 7,339,105 | 115.638 | 134.872 | 117.71 | 2.62975 | −0.0221 | 0.6762 | 30.597285 | OD | 0.1419 | NH |
| qFU-A09_cb | A09 | mk8762 | mk6732_A09 | 13,222 | 55,126,525 | 35.486 | 44.202 | 40.51 | 2.586319 | −0.0343 | 0.6945 | 20.247813 | OD | 1.3251 | NH | |
| SCI | qSCI-A09_15 | A09 | mk18838 | mk6517_A09 | 64,093 | 33,530,295 | 79.726 | 79.934 | 79.91 | 4.60152 | 2.2915 | 9.0716 | 3.9588043 | OD | 0.5063 | CCRI35 |
| qSCI-A09_15 | A09 | mk6528_A09 | mk6531_A09 | 37,420,628 | 37,694,390 | 87.803 | 91.571 | 88.81 | 3.400651 | 2.6194 | 7.2082 | 2.7518516 | OD | 1.1267 | CCRI35 | |
| qSCI-A09_cb | A09 | mk6491_A09 | mk6493_A09 | 15,408,937 | 17,834,574 | 73.597 | 73.647 | 73.61 | 2.875136 | 3.6441 | 4.9294 | 1.3527071 | OD | 0.804 | CCRI35 | |
| qSCI-A09_cb | A09 | mk18838 | mk6517_A09 | 64,093 | 33,530,295 | 79.726 | 79.934 | 79.91 | 3.413681 | 0.7693 | 5.3779 | 6.9906408 | OD | 0.0048 | CCRI35 | |
| qSCI-A09_cb | A09 | mk6528_A09 | mk6531_A09 | 37,420,628 | 37,694,390 | 87.803 | 91.571 | 88.81 | 2.686211 | 1.0069 | 4.6458 | 4.6139637 | OD | 0.1298 | CCRI35 | |
| FM | qFM-A10_15 | A10 | mk7018_A10 | mk7020_A10 | 15,617,318 | 15,617,342 | 165.83 | 165.941 | 165.91 | 2.421281 | 0.0767 | −0.322 | 4.1981747 | OD | 2.1861 | CCRI35 |
| qFM-A10_15 | A10 | mk11965 | mk6991_A10 | 8852 | 12,815,805 | 171.292 | 171.66 | 171.31 | 3.339848 | 0.0919 | −0.3583 | 3.898803 | OD | 3.1176 | CCRI35 | |
| qFM-A10_cb | A10 | mk18875 | mk18876 | 355 | 389 | 58.679 | 59.519 | 58.71 | 2.912052 | 0.0306 | −0.1769 | 5.7810458 | OD | 2.1098 | CCRI35 | |
| qFM-A10_cb | A10 | mk11965 | mk6991_A10 | 8852 | 12,815,805 | 171.292 | 171.66 | 171.31 | 2.779587 | 0.0402 | −0.1538 | 3.8258706 | OD | 2.621 | CCRI35 | |
| FS | qFS-A10_16 | A10 | mk19550 | mk7479_A10 | 62,129 | 69,679,150 | 93.736 | 94.208 | 93.81 | 2.877307 | −0.4206 | 0.0644 | 0.1531146 | A | 7.1603 | NH |
| qFS-A10_16 | A10 | MulMa189-m_A10 | mk7438_A10 | 65,789,277 | 67,378,405 | 106.543 | 109.182 | 106.81 | 3.170467 | 0.0048 | 0.3517 | 73.270833 | OD | 0.4704 | CCRI35 | |
| qFS-A10_cb | A10 | mk6982_A10 | mk6986_A10 | 1,111,5569 | 12,645,781 | 177.095 | 185.191 | 177.11 | 3.676439 | −0.1987 | 0.1727 | 0.8691495 | D | 6.206 | NH | |
| FE | qFE-A12_14 | A12 | mk9173_A12 | mk9187_A12 | 79,355,806 | 81,262,301 | 23.53 | 38.305 | 31.51 | 2.54506 | −0.0177 | 0.1178 | 6.6553672 | OD | 3.0701 | NH |
| qFE-A12_16 | A12 | mk8958_A12 | mk8961_A12 | 59,776,633 | 6,102,5182 | 161.739 | 173.999 | 163.81 | 2.838219 | 0.0006 | 0.0458 | 76.333333 | OD | 0.0468 | CCRI35 | |
| FE | qFE-D01_15 | D01 | mk10708_D01 | mk10809_D01 | 42,734,090 | 44,178,280 | 105.061 | 106.117 | 106.11 | 2.551574 | −0.0001 | 0.1905 | 1905 | OD | 0.4005 | NH |
| qFE-D01_cb | D01 | mk10832_D01 | MulMa266-m_D01 | 46,916,080 | 51,199,148 | 77.706 | 82.287 | 79.71 | 4.141151 | 0.0444 | 0.0955 | 2.1509009 | OD | 0.7434 | CCRI35 | |
| qFE-D01_cb | D01 | mk10708_D01 | mk10709_D01 | 42,734,090 | 42,734,155 | 106.1 | 106.117 | 106.11 | 2.998914 | 0.0009 | 0.0961 | 106.77778 | OD | 0.4425 | CCRI35 | |
| SCI | qSCI-D02_15 | D02 | mk11587_D02 | mk11605_D02 | 51,060,053 | 5,122,5737 | 120.178 | 120.621 | 120.61 | 2.690554 | 1.4485 | −6.7494 | 4.6595789 | OD | 2.84 | CCRI35 |
| qSCI-D02_cb | D02 | mk11595_D02 | mk11603_D02 | 51,118,850 | 51,193,929 | 115.939 | 116.818 | 116.01 | 2.521173 | 0.2862 | −4.485 | 15.67086 | OD | 1.8379 | CCRI35 | |
| BW | qBW-D03_15 | D03 | mk12041_D03 | mk12042_D03 | 2,290,601 | 2,894,288 | 22.573 | 37.566 | 30.61 | 2.644951 | −0.0899 | 0.4957 | 5.5139043 | OD | 2.9343 | NH |
| qBW-D03_16 | D03 | mk12031_D03 | mk12032_D03 | 1,002,704 | 1,037,917 | 3.775 | 9.172 | 3.81 | 2.870793 | 0.0006 | 0.4782 | 797 | OD | 0.7748 | CCRI35 | |
| FM | qFM-D03_15 | D03 | mk12142_D03 | mk12159_D03 | 36,697,656 | 3,861,6587 | 125.137 | 134.584 | 130.21 | 7.52443 | 0.2667 | 0.1477 | 0.5538058 | PD | 8.7707 | CCRI35 |
| qFM-D03_15 | D03 | mk12152_D03 | mk12159_D03 | 37,665,167 | 38,616,587 | 134.584 | 141.665 | 136.01 | 7.685125 | 0.2661 | 0.0664 | 0.2495303 | PD | 10.0325 | CCRI35 | |
| qFM-D03_15 | D03 | mk12153_D03 | mk12158_D03 | 37,668,262 | 37,938,158 | 143.823 | 145.337 | 144.81 | 6.304017 | 0.2329 | 0.0922 | 0.3958781 | PD | 7.4939 | CCRI35 | |
| qFM-D03_cb | D03 | mk12085_D03 | mk12086_D03 | 25,573,334 | 25,700,132 | 87.285 | 87.516 | 87.31 | 4.95114 | 0.0975 | 0.0212 | 0.2174359 | PD | 6.3845 | CCRI35 | |
| qFM-D03_cb | D03 | mk12119_D03 | mk12123_D03 | 30,535,745 | 30,566,883 | 95.393 | 95.724 | 95.41 | 6.644951 | 0.1133 | 0.0204 | 0.180053 | A | 8.5806 | CCRI35 | |
| qFM-D03_cb | D03 | mk12108_D03 | mk12115_D03 | 2,763,8133 | 29,511,299 | 101.991 | 103.218 | 101.31 | 4.827362 | 0.095 | 0.061 | 0.6421053 | PD | 5.0024 | CCRI35 | |
| FY | qFY-D03_15 | D03 | mk12142_D03 | mk12159_D03 | 36,697,656 | 38,616,587 | 125.137 | 134.584 | 130.21 | 2.610206 | 0.1828 | −0.0298 | 0.1630197 | A | 4.7288 | CCRI35 |
| qFY-D03_15 | D03 | mk12154_D03 | mk12155_D03 | 37,676,414 | 37,682,981 | 141.665 | 142.221 | 141.71 | 3.806732 | 0.184 | −0.1844 | 1.0021739 | D | 6.9294 | CCRI35 | |
| qFY-D03_15 | D03 | mk12158_D03 | mk12161_D03 | 37,938,158 | 39,407,242 | 145.337 | 150.198 | 148.31 | 4.210641 | 0.2038 | −0.2276 | 1.1167812 | D | 8.0817 | CCRI35 | |
| qFY-D03_cb | D03 | mk12109_D03 | mk12111_D03 | 27,707,667 | 29,136,194 | 105.804 | 106.491 | 105.81 | 2.571118 | 0.0879 | −0.0931 | 1.0591581 | D | 4.7115 | CCRI35 | |
| LP | qLP-D03_15 | D03 | mk12142_D03 | mk12159_D03 | 36,697,656 | 38,616,587 | 125.137 | 134.584 | 129.21 | 9.233442 | 1.7674 | −0.2922 | 0.1653276 | A | 15.4584 | CCRI35 |
| qLP-D03_15 | D03 | mk12152_D03 | mk12160_D03 | 37,665,167 | 38,832,736 | 134.986 | 141.665 | 138.01 | 8.214984 | 1.6425 | −0.399 | 0.2429224 | PD | 13.9321 | CCRI35 | |
| qLP-D03_16 | D03 | mk12152_D03 | mk12160_D03 | 37,665,167 | 38,832,736 | 134.986 | 141.665 | 138.01 | 5.439739 | 1.4767 | −0.9737 | 0.6593756 | PD | 9.8343 | CCRI35 | |
| qLP-D03_16 | D03 | mk12153_D03 | mk12158_D03 | 37,668,262 | 37,938,158 | 143.823 | 145.337 | 144.81 | 4.621064 | 1.2806 | −1.0766 | 0.8406997 | D | 7.9495 | CCRI35 | |
| qLP-D03_cb | D03 | mk12152_D03 | mk12160_D03 | 37,665,167 | 38,832,736 | 134.986 | 141.665 | 139.01 | 8.169381 | 1.2356 | −0.1607 | 0.1300583 | A | 13.3867 | CCRI35 | |
| qLP-D03_cb | D03 | mk12158_D03 | mk12161_D03 | 37,938,158 | 39,407,242 | 145.337 | 150.198 | 147.31 | 7.090119 | 1.1317 | −0.3152 | 0.278519 | PD | 12.0557 | CCRI35 | |
| FR | qFR-D04_15 | D04 | MulMa448_D04 | MulMa451_D04 | 49,867,798 | 50,187,192 | 4.736 | 8.337 | 5.01 | 2.660152 | −0.1841 | −1.5832 | 8.5996741 | OD | 0.0079 | NH |
| qFR-D04_cb | D04 | MulMa448_D04 | MulMa451_D04 | 49,867,798 | 50,187,192 | 4.736 | 8.337 | 5.01 | 2.781759 | −0.227 | −0.8557 | 3.7696035 | OD | 0.6158 | NH | |
| FM | qFM-D05_15 | D05 | mk12822_D05 | mk12824_D05 | 30,214,244 | 30,216,527 | 152.95 | 153.434 | 153.01 | 5.061889 | 0.2063 | 0.008 | 0.0387785 | A | 6.8075 | CCRI35 |
| qFM-D05_cb | D05 | MulMa463-m_D05 | mk12861_D05 | 30,373,354 | 31,354,896 | 144.668 | 148.809 | 146.71 | 4.021716 | 0.08 | −0.1214 | 1.5175 | OD | 6.6096 | CCRI35 | |
| qFM-D05_cb | D05 | mk12822_D05 | mk12824_D05 | 30,214,244 | 30,216,527 | 152.95 | 153.434 | 153.41 | 4.627579 | 0.0886 | −0.0879 | 0.9920993 | D | 7.3098 | CCRI35 | |
| SCI | qSCI-D05_15 | D05 | MulMa463-m_D05 | mk12861_D05 | 30,373,354 | 31,354,896 | 144.668 | 148.809 | 144.71 | 2.566775 | −2.7495 | 3.5799 | 1.3020185 | OD | 4.5444 | NH |
| qSCI-D05_cb | D05 | MulMa463-m_D05 | mk12861_D05 | 30,373,354 | 31,354,896 | 144.668 | 148.809 | 144.71 | 2.579805 | −1.778 | 1.3725 | 0.7719348 | PD | 4.7561 | NH | |
| FR | qFR-D08_15 | D08 | mk15992_D08 | mk15995_D08 | 56,628,640 | 56,628,844 | 181.945 | 182.082 | 182.01 | 2.655809 | 0.587 | −0.2932 | 0.4994889 | PD | 4.6002 | CCRI35 |
| qFR-D08_cb | D08 | mk15992_D08 | mk15995_D08 | 56,628,640 | 56,628,844 | 181.945 | 182.082 | 182.01 | 3.441911 | 0.408 | −0.2977 | 0.7296569 | PD | 6.2705 | CCRI35 | |
| FL | qFL-D08_14 | D08 | MulMa514_D08 | mk16004_D08 | 54,937,781 | 58,533,805 | 187.587 | 196.342 | 196.31 | 3.583062 | 0.3322 | −0.0152 | 0.0457556 | A | 5.8804 | CCRI35 |
| qFL-D08_cb | D08 | MulMa514_D08 | mk16004_D08 | 54,937,781 | 5,853,3805 | 187.587 | 196.342 | 196.31 | 3.639522 | 0.1799 | 0.0892 | 0.495831 | PD | 4.9405 | CCRI35 | |
| qFL-D08_cb | D08 | mk16017_D08 | mk16020_D08 | 59,691,087 | 59,698,388 | 208.553 | 208.76 | 208.61 | 3.255157 | 0.1595 | 0.1876 | 1.1761755 | D | 3.2928 | CCRI35 | |
| FE | qFE-D10_14 | D10 | mk17141_D10 | MulMa593-m_D10 | 56,817,432 | 56,887,380 | 102.161 | 106.55 | 106.21 | 2.523344 | −0.0328 | 0.082 | 2.5 | OD | 3.7527 | NH |
| qFE-D10_15 | D10 | MulMa575-m_D10 | MulMa581_D10 | 24,793,863 | 24,918,141 | 1.604 | 2.047 | 1.91 | 2.959826 | 0.0751 | 0.0306 | 0.4074567 | PD | 3.9966 | CCRI35 | |
| FL | qFL-D10_14 | D10 | mk17141_D10 | MulMa593-m_D10 | 56,817,432 | 56,887,380 | 102.161 | 106.55 | 105.21 | 3.411509 | −0.1394 | 0.7384 | 5.2969871 | OD | 3.6287 | NH |
| qFL-D10_cb | D10 | mk2492 | MulMa366-m | 519 | 33,037 | 78.155 | 98.694 | 83.21 | 2.614549 | −0.045 | 0.464 | 10.311111 | OD | 2.0017 | NH | |
| FL | qFL-D11_16 | D11 | mk17462_D11 | mk17463_D11 | 15,695,804 | 15,711,598 | 88.706 | 88.935 | 88.71 | 2.831705 | 0.0011 | 0.1765 | 160.45455 | OD | 0.8561 | CCRI35 |
| qFL-D11_cb | D11 | mk17464_D11 | mk17514_D11 | 15,711,711 | 21,298,890 | 61.142 | 88.706 | 82.21 | 3.079262 | −0.0461 | 0.5185 | 11.247289 | OD | 2.2603 | NH | |
| FS | qFS-D12_14 | D12 | mk18221_D12 | MulMa605_D12 | 50,554,371 | 5,129,3378 | 148.99 | 160.984 | 153.01 | 3.270358 | 0.0077 | 0.8261 | 107.28571 | OD | 0.6079 | CCRI35 |
| qFS-D12_15 | D12 | mk17994_D12 | mk17997_D12 | 3,798,8313 | 3,8143,957 | 65.608 | 66.056 | 65.61 | 3.743757 | 0.3487 | −0.7277 | 2.0868942 | OD | 5.7325 | CCRI35 | |
| qFS-D12_cb | D12 | mk19853 | mk17913_D12 | 101,319 | 13,479,261 | 16.392 | 16.946 | 16.41 | 4.049946 | 0.1807 | −0.3037 | 1.6806862 | OD | 6.3446 | CCRI35 | |
| FY | qFY-D12_15 | D12 | mk17995_D12 | mk18057_D12 | 38,058,755 | 41,722,495 | 66.861 | 96.943 | 70.91 | 2.677524 | 0.047 | −0.4721 | 10.044681 | OD | 2.0175 | CCRI35 |
| qFY-D12_cb | D12 | mk1009 | mk17992_D12 | 18,989 | 37,732,030 | 66.348 | 66.861 | 66.41 | 3.252986 | 0.0211 | −0.2739 | 12.981043 | OD | 1.9523 | CCRI35 | |
| SCI | qSCI-D12_14 | D12 | mk18202_D12 | mk18207_D12 | 48,411,387 | 48,718,111 | 132.168 | 133.627 | 133.61 | 3.14658 | 1.3176 | −0.8128 | 0.6168792 | PD | 6.2241 | CCRI35 |
| qSCI-D12_15 | D12 | mk19857 | mk17916_D12 | 117,142 | 15,801,265 | 23.692 | 24.245 | 23.71 | 3.072747 | 2.636 | 4.6094 | 1.7486343 | OD | 1.4248 | CCRI35 | |
| FL | qFL-D12_14 | D12 | mk18210_D12 | mk18214_D12 | 48,923,084 | 49133419 | 135.788 | 138.113 | 135.81 | 2.851249 | 0.281 | −0.216 | 0.7686833 | PD | 5.2886 | CCRI35 |
| qFL-D12_cb | D12 | mk18221_D12 | MulMa605_D12 | 50,554,371 | 51,293,378 | 148.99 | 160.984 | 158.01 | 2.896851 | 0.1755 | 0.1739 | 0.9908832 | D | 3.2967 | CCRI35 | |
| qFL-D12_cb | D12 | MulMa604-m_D12 | mk18232_D12 | 51,286,859 | 52,905,207 | 161.79 | 172.395 | 165.81 | 2.773073 | 0.1653 | 0.1375 | 0.8318209 | D | 3.0451 | CCRI35 | |
| FL | qFL-D13_14 | D13 | mk18377_D13 | MulMa619_D13 | 4,171,037 | 34,392,983 | 36.052 | 56.969 | 46.11 | 3.237785 | 0.1336 | −0.7846 | 5.8727545 | OD | 3.0376 | CCRI35 |
| qFL-D13_15 | D13 | mk18378_D13 | mk18379_D13 | 4,310,490 | 4,329,364 | 64.756 | 65.409 | 64.81 | 2.529859 | 0.2624 | 0.2724 | 1.0381098 | D | 2.1912 | CCRI35 | |
| qFL-D13_cb | D13 | mk18516_D13 | mk18533_D13 | 41,759,681 | 47,859,575 | 2.023 | 10.429 | 9.01 | 3.051031 | 0.17 | −0.0572 | 0.3364706 | PD | 5.3187 | CCRI35 | |
| qFL-D13_cb | D13 | mk20382 | mk18378_D13 | 141 | 4,310,490 | 65.409 | 72.523 | 65.41 | 3.072747 | 0.1705 | −0.0658 | 0.3859238 | PD | 5.4653 | CCRI35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diouf, L.; Magwanga, R.O.; Gong, W.; He, S.; Pan, Z.; Jia, Y.H.; Kirungu, J.N.; Du, X. QTL Mapping of Fiber Quality and Yield-Related Traits in an Intra-Specific Upland Cotton Using Genotype by Sequencing (GBS). Int. J. Mol. Sci. 2018, 19, 441. https://doi.org/10.3390/ijms19020441
Diouf L, Magwanga RO, Gong W, He S, Pan Z, Jia YH, Kirungu JN, Du X. QTL Mapping of Fiber Quality and Yield-Related Traits in an Intra-Specific Upland Cotton Using Genotype by Sequencing (GBS). International Journal of Molecular Sciences. 2018; 19(2):441. https://doi.org/10.3390/ijms19020441
Chicago/Turabian StyleDiouf, Latyr, Richard Odongo Magwanga, Wenfang Gong, Shoupu He, Zhaoe Pan, Yin Hua Jia, Joy Nyangasi Kirungu, and Xiongming Du. 2018. "QTL Mapping of Fiber Quality and Yield-Related Traits in an Intra-Specific Upland Cotton Using Genotype by Sequencing (GBS)" International Journal of Molecular Sciences 19, no. 2: 441. https://doi.org/10.3390/ijms19020441
APA StyleDiouf, L., Magwanga, R. O., Gong, W., He, S., Pan, Z., Jia, Y. H., Kirungu, J. N., & Du, X. (2018). QTL Mapping of Fiber Quality and Yield-Related Traits in an Intra-Specific Upland Cotton Using Genotype by Sequencing (GBS). International Journal of Molecular Sciences, 19(2), 441. https://doi.org/10.3390/ijms19020441