Experimental Intrastriatal Applications of Botulinum Neurotoxin-A: A Review

Abstract
:1. Introduction
1.1. Idiopathic Parkinson Syndrome and Current Treatment Concepts
1.2. Botulinum Neurotoxins/Botulinum Neurotoxin-A
1.3. Experimental Intracerebral Applications of BoNTs in the Central Nervous System
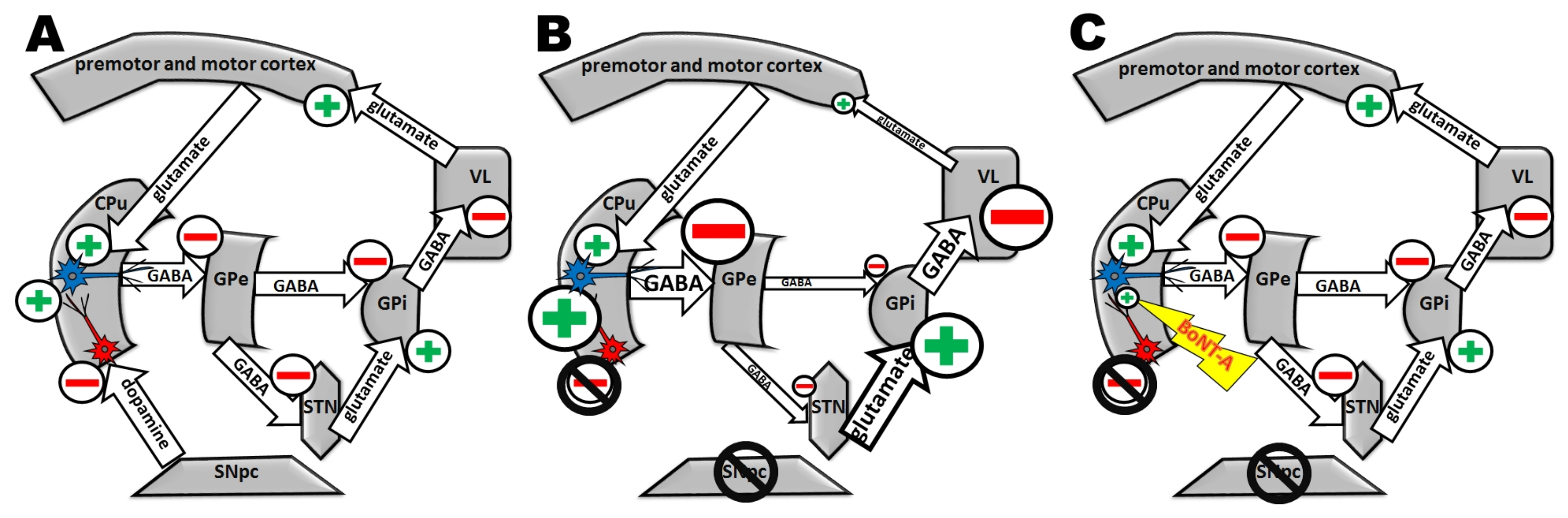
2. Intrastriatal BoNT-A Application
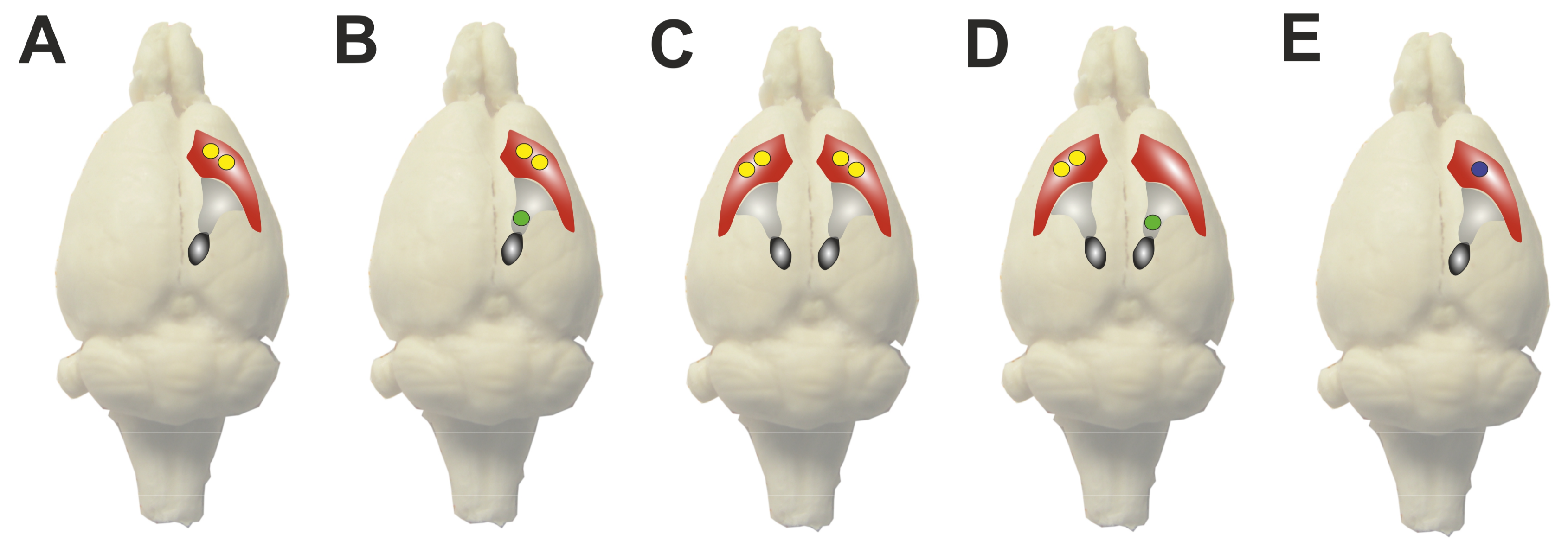
2.1. Dose Finding
2.2. Experimental Application in Rats
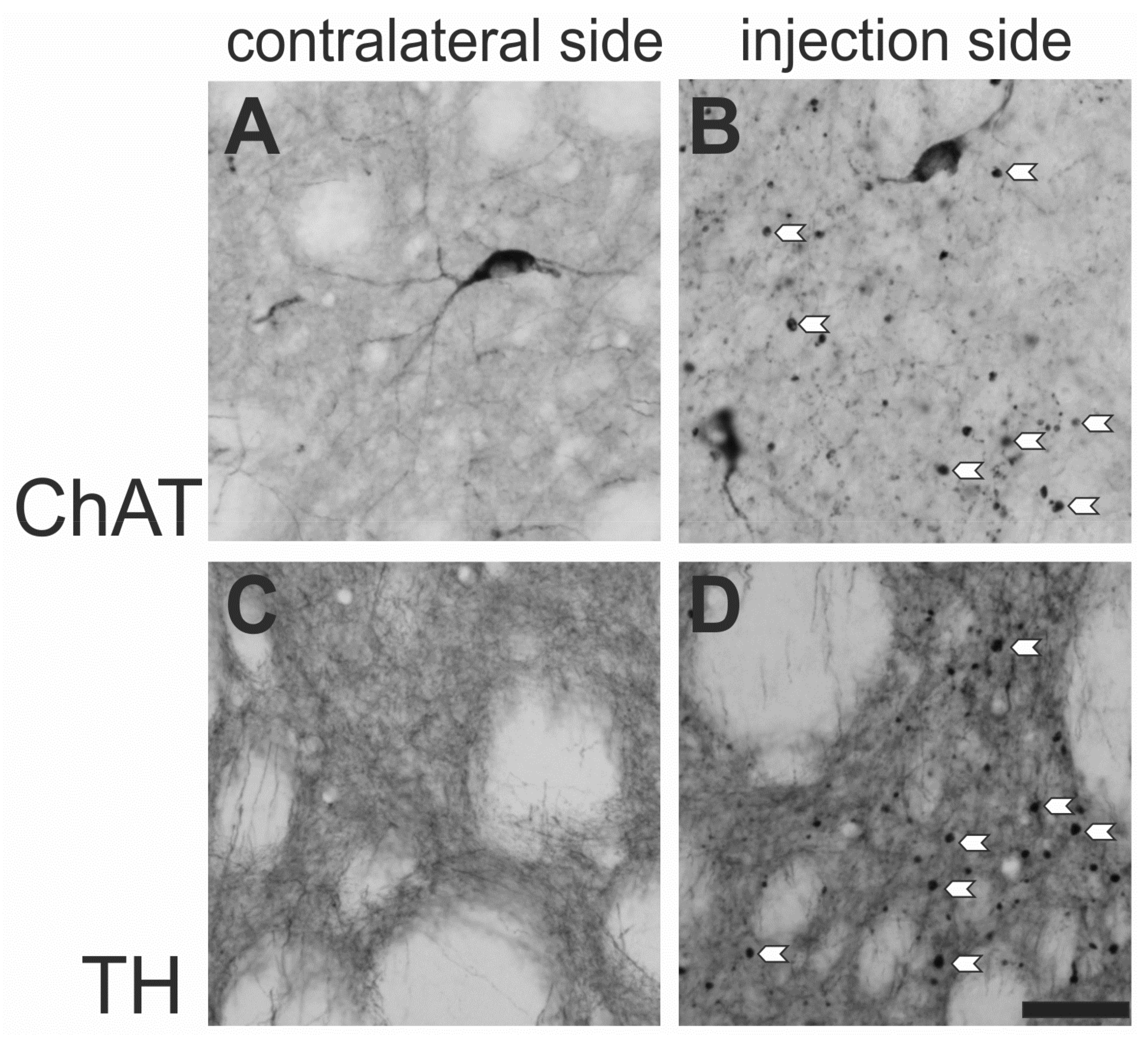
2.2.1. Unilateral Application in Naїve Rats
2.2.2. Unilateral Application of BoNT-A in the Hemiparkinsonian 6-OHDA Rat Model
BoNT-A Application Ipsilateral to the 6-OHDA-Lesioned Side
Application of BoNT-A Contralateral to the 6-OHDA-Lesioned Side
2.2.3. Bilateral BoNT-A Application in Rats and Consequences for Motor Behavior and Cognition
2.2.4. In Vitro Receptor Autoradiography and Positron Emission Tomography
2.3. Experimental Intrastriatal BoNT-A Application in Mice
3. Experimental Comparison of Intrastriatal Application BoNT-A Subtype 1 and BoNT-A Subtype 2
3.1. Main Findings
3.2. Discussion of Itakura’s Findings and Integration with Other Results
4. Discussion of the BoNT Doses
5. Future Prospects
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| BiVs | Botulinum neurotoxin induced varicosities |
| BoNT | Botulinum neurotoxin |
| ChAT | choline acetyltransferase |
| CNS | central nervous system |
| CPu | caudate-putamen/striatum |
| GPe | globus pallidus pars externa/external globus pallidus |
| GABA | gamma-aminobutyric acid |
| GPi | globus pallidus pars interna/internal globus pallidus |
| LD50 | median lethal dose |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| MSN | medium spiny neurons |
| PD | Parkinson’s disease |
| SNARE | soluble N-ethylmaleimide-sensitive-factor attachment receptor |
| SNpc | substantia nigra pars compacta |
| STN | subthalamic nucleus |
| SV2C | synaptic vesicle glycoprotein 2C |
| TH | tyrosine hydroxylase |
| VL | ventrolateral thalamic nucleus |
References
- Braak, H.; Bohl, J.R.; Müller, C.M.; Rüb, U.; de Vos, R.A.; Del Tredici, K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. 2006, 21, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Parkinsons Dis. 2017, 7, S73–S87. [Google Scholar] [CrossRef] [PubMed]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–545. [Google Scholar] [CrossRef]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Picconi, B.; Parnetti, L.; Di Filippo, M. A convergent model for cognitive dysfunctions in Parkinson’s disease: The critical dopamine-acetylcholine synaptic balance. Lancet Neurol. 2006, 5, 974–983. [Google Scholar] [CrossRef]
- Chu, H.Y.; McIver, E.L.; Kovaleski, R.F.; Atherton, J.F.; Bevan, M.D. Loss of Hyperdirect Pathway Cortico-Subthalamic Inputs Following Degeneration of Midbrain Dopamine Neurons. Neuron 2017, 95, 1306–1318. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Marin, C.; Rodriguez-Oroz, C.; Blesa, J.; Benitez-Temiño, B.; Mena-Segovia, J.; Rodríguez, M.; Olanow, C.W. The basal ganglia in Parkinson’s disease: Current concepts and unexplained observations. Ann. Neurol. 2008, 64 (Suppl. 2), 30–46. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodríguez-Oroz, M.C.; Benitez-Temino, B.; Blesa, F.J.; Guridi, J.; Marin, C.; Rodriguez, M. Functional organization of the basal ganglia: Therapeutic implications for Parkinson’s disease. Mov. Disord. 2008, 23 (Suppl. 3), 548–559. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Stamelou, M.; Bhatia, K.P.; Burn, D.J. The expanding universe of disorders of the basal ganglia. Lancet 2014, 384, 523–531. [Google Scholar] [CrossRef]
- Dautan, D.; Huerta-Ocampo, I.; Witten, I.B.; Deisseroth, K.; Bolam, J.P.; Gerdjikov, T.; Mena-Segovia, J. A major external source of cholinergic innervation of the striatum and nucleus accumbens originates in the brainstem. J. Neurosci. 2014, 34, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Albin, R.L. The cholinergic system and Parkinson disease. Behav. Brain Res. 2011, 221, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.A.; Reynolds, J.N. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience 2011, 198, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.A.; Kang, U.J.; McGehee, D.S. Striatal cholinergic interneuron regulation and circuit effects. Front. Synaptic Neurosci. 2014, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Ztaou, S.; Maurice, N.; Camon, J.; Guiraudie-Capraz, G.; Kerkerian-Le Goff, L.; Beurrier, C.; Liberge, M.; Amalric, M. Involvement of Striatal Cholinergic Interneurons and M1 and M4 Muscarinic Receptors in Motor Symptoms of Parkinson’s Disease. J. Neurosci. 2016, 36, 9161–9172. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.; Lindqvist, M.; Magnusson, T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 1957, 180, 1200. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Obeso, J.A. Levodopa toxicity and Parkinson disease: Still a need for equipoise. Neurology 2011, 77, 1416–1417. [Google Scholar] [CrossRef] [PubMed]
- Playfer, J.R. Parkinson’s disease. Postgrad. Med. J. 1997, 73, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Whitney, C.M. Medications for Parkinson’s disease. Neurologist 2007, 13, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Pahwa, R. Understanding Parkinson’s disease: An update on current diagnostic and treatment strategies. J. Am. Med. Dir. Assoc. 2006, 7 (Suppl. 2), 4–10. [Google Scholar] [PubMed]
- Fowler, J.S.; Logan, J.; Volkow, N.D.; Shumay, E.; McCall-Perez, F.; Jayne, M.; Wang, G.J.; Alexoff, D.L.; Apelskog-Torres, K.; Hubbard, B.; et al. Evidence that formulations of the selective MAO-B inhibitor, selegiline, which bypass first-pass metabolism, also inhibit MAO-A in the human brain. Neuropsychopharmacology 2015, 40, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Müller, T. Monoamine oxidase-B inhibitors in the treatment of Parkinson’s disease: Clinical-pharmacological aspects. J. Neural Transm. 2018. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S287–S296. [Google Scholar] [CrossRef] [PubMed]
- Weaver, F.M.; Follett, K.; Stern, M.; Hur, K.; Harris, C.; Marks, W.J., Jr.; Rothlind, J.; Sagher, O.; Reda, D.; Moy, C.S.; et al. Bilateral deep brain stimulation vs. best medical therapy for patients with advanced Parkinson disease: A randomized controlled trial. JAMA 2009, 301, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; Jaggi, J.L.; Hurtig, H.I.; Siderowf, A.D.; Colcher, A.; Stern, M.B.; Baltuch, G.H. Deep brain stimulation for movement disorders: Morbidity and mortality in 109 patients. J. Neurosurg. 2003, 98, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Angot, E.; Steiner, J.A.; Hansen, C.; Li, J.Y.; Brundin, P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010, 9, 1128–1138. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Dubow, J.; Singer, C. Is levodopa toxic? Insights from a brain bank. Neurology 2011, 77, 1414–1415. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Diamond, M.I. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 2010, 11, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Prusiner, S.B. Is Parkinson’s disease a prion disorder? Proc. Natl. Acad. Sci. USA 2009, 106, 12571–12572. [Google Scholar] [CrossRef] [PubMed]
- Wijeyekoon, R.; Barker, R.A. Cell replacement therapy for Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Duvoisin, R.C. Cholinergic-anticholinergic antagonism in Parkinsonism. Arch. Neurol. 1967, 17, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H.A.; Machover, S.; Rabiner, A. A study of the effectiveness of drug therapy in Parkinsonism. J. Nerv. Ment. Dis. 1954, 119, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.E. Medical management of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2002, 72 (Suppl. 1), I22–I27. [Google Scholar] [CrossRef] [PubMed]
- Katzenschlager, R.; Sampaio, C.; Costa, J.; Lees, A. Anticholinergics for symptomatic management of Parkinson’s disease. Cochrane Database Syst. Rev. 2003, CD003735. [Google Scholar] [CrossRef]
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. SV2 is the protein receptor for botulinum neurotoxin A. Science 2006, 312, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Pirazzini, M.; Montecucco, C. Botulinum neurotoxins: Genetic, structural and mechanistic insights. Nat. Rev. Microbiol. 2014, 12, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Rossetto, O.; Grumelli, C.; Frassoni, C.; Montecucco, C.; Matteoli, M. Entering neurons: Botulinum toxins and synaptic vesicle recycling. EMBO Rep. 2006, 7, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Hagenah, R.; Benecke, R.; Wiegand, H. Effects of type A botulinum toxin on the cholinergic transmission at spinal Renshaw cells and on the inhibitory action at ia inhibitory interneurones. Naunyn Schmiedebergs Arch. Pharmacol. 1977, 299, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Luvisetto, S.; Rossetto, O.; Montecucco, C.; Pavone, F. Toxicity of botulinum neurotoxins in central nervous system of mice. Toxicon 2003, 41, 475–481. [Google Scholar] [CrossRef]
- Luvisetto, S.; Marinelli, S.; Rossetto, O.; Montecucco, C.; Pavone, F. Central injection of botulinum neurotoxins: Behavioural effects in mice. Behav. Pharmacol. 2004, 15, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Luvisetto, S.; Marinelli, S.; Lucchetti, F.; Marchi, F.; Cobianchi, S.; Rossetto, O.; Montecucco, C.; Pavone, F. Botulinum neurotoxins and formalin-induced pain: Central vs. peripheral effects in mice. Brain Res. 2006, 1082, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Chaddock, J.A.; Purkiss, J.R.; Alexander, F.C.; Doward, S.; Fooks, S.J.; Friis, L.M.; Hall, Y.H.; Kirby, E.R.; Leeds, N.; Moulsdale, H.J.; et al. Retargeted clostridial endopeptidases: Inhibition of nociceptive neurotransmitter release in vitro, and antinociceptive activity in in vivo models of pain. Mov. Disord. 2004, 19 (Suppl. 8), 42–47. [Google Scholar] [CrossRef] [PubMed]
- Caleo, M.; Restani, L.; Gianfranceschi, L.; Costantin, L.; Rossi, C.; Rossetto, O.; Montecucco, C.; Maffei, L. Transient synaptic silencing of developing striate cortex has persistent effects on visual function and plasticity. J. Neurosci. 2007, 27, 4530–4540. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Rossi, C.; Gianfranceschi, L.; Rossetto, O.; Caleo, M. Long-distance retrograde effects of botulinum neurotoxin A. J. Neurosci. 2008, 28, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Antonucci, F.; Gianfranceschi, L.; Rossi, C.; Rossetto, O.; Caleo, M. Evidence for anterograde transport and transcytosis of botulinum neurotoxin A (BoNT/A). J. Neurosci. 2011, 31, 15650–15659. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Kobayashi, S.; Waki, H.; Kon, K.; Fukui, F.; Tadenuma, T.; Iwamoto, M.; Takeda, Y.; Izumiyama, N. Animal model of dementia induced by entorhinal synaptic damage and partial restoration of cognitive deficits by BDNF and carnitine. J. Neurosci. Res. 2002, 70, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lackovic, Z.; Rebic, V.; Riederer, P.F. Single intracerebroventricular injection of botulinum toxin type A produces slow onset and long-term memory impairment in rats. J. Neural Transm. 2009, 16, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- De Leonibus, E.; Costantini, V.J.; Massaro, A.; Mandolesi, G.; Vanni, V.; Luvisetto, S.; Pavone, F.; Oliverio, A. Cognitive and neural determinants of response strategy in the dual-solution plus-maze task. Learn. Mem. 2011, 18, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Costantin, L.; Bozzi, Y.; Richichi, C.; Viegi, A.; Antonucci, F.; Funicello, M.; Gobbi, M.; Mennini, T.; Rossetto, O.; Montecucco, C.; et al. Antiepileptic effects of botulinum neurotoxin E. J. Neurosci. 2005, 25, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Bozzi, Y.; Caleo, M. Intrahippocampal infusion of botulinum neurotoxin E (BoNT/E) reduces spontaneous recurrent seizures in a mouse model of mesial temporal lobe epilepsy. Epilepsia 2009, 50, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Gasior, M.; Tang, R.; Rogawski, M.A. Long-lasting attenuation of amygdala-kindled seizures after convection-enhanced delivery of botulinum neurotoxins A and B into the amygdala in rats. J. Pharmacol. Exp. Ther. 2013, 346, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Cerri, C.; Vetencourt, J.F.; Caleo, M. Acute neuroprotection by the synaptic blocker botulinum neurotoxin E in a rat model of focal cerebral ischaemia. Neuroscience 2010, 169, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Mix, E.; Hawlitschka, A.; Antipova, V.; Witt, M.; Schmitt, O.; Benecke, R. Intrastriatal botulinum toxin abolishes pathologic rotational behaviour and induces axonal varicosities in the 6-OHDA rat model of Parkinson’s disease. Neurobiol. Dis. 2011, 41, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Hawlitschka, A.; Holzmann, C.; Witt, S.; Spiewok, J.; Neumann, A.M.; Schmitt, O.; Wree, A.; Antipova, V. Intrastriatally injected botulinum neurotoxin-A differently effects cholinergic and dopaminergic fibers in C57BL/6 mice. Brain Res. 2017, 1676, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Montecucco, C. Tetanus and botulism neurotoxins: Isolation and assay. Methods Enzymol. 1995, 248, 643–652. [Google Scholar] [PubMed]
- Montecucco, C.; Schiavo, G. Structure and function of tetanus and botulinum neurotoxins. Q. Rev. Biophys. 1995, 28, 423–472. [Google Scholar] [CrossRef] [PubMed]
- Mann, T.; Zilles, K.; Dikow, H.; Hellfritsch, A.; Cremer, M.; Piel, M.; Rösch, F.; Hawlitschka, A.; Schmitt, O.; Wree, A. Dopamine, Noradrenaline and Serotonin Receptor Densities in the Striatum of Hemiparkinsonian Rats following Botulinum Neurotoxin-A Injection. Neuroscience 2018, 374, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Antipova, V.; Hawlitschka, A.; Mix, E.; Schmitt, O.; Dräger, D.; Benecke, R.; Wree, A. Behavioral and structural effects of unilateral intrastriatal injections of botulinum neurotoxin a in the rat model of Parkinson’s disease. J. Neurosci. Res. 2013, 91, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Mehlan, J.; Brosig, H.; Schmitt, O.; Mix, E.; Wree, A.; Hawlitschka, A. Intrastriatal injection of botulinum neurotoxin-A is not cytotoxic in rat brain—A histological and stereological analysis. Brain Res. 2016, 1630, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, C.; Dräger, D.; Mix, E.; Hawlitschka, A.; Antipova, V.; Benecke, R.; Wree, A. Effects of intrastriatal botulinum neurotoxin A on the behavior of Wistar rats. Behav. Brain Res. 2012, 234, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Antipova, V.A.; Holzmann, C.; Schmitt, O.; Wree, A.; Hawlitschka, A. Botulinum Neurotoxin A Injected Ipsilaterally or Contralaterally into the Striatum in the Rat 6-OHDA Model of Unilateral Parkinson’s Disease Differently Affects Behavior. Front. Behav. Neurosci. 2017, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Mann, T.; Kurth, J.; Hawlitschka, A.; Stenzel, J.; Lindner, T.; Polei, S.; Hohn, A.; Krause, B.J.; Wree, A. [18F] fallypride-PET/CT Analysis of the Dopamine D2/D3 Receptor in the Hemiparkinsonian Rat Brain Following Intrastriatal Botulinum Neurotoxin A Injection. Molecules 2018, 23, 587. [Google Scholar] [CrossRef] [PubMed]
- Wedekind, F.; Oskamp, A.; Lang, M.; Hawlitschka, A.; Zilles, K.; Wree, A.; Bauer, A. Intrastriatal administration of botulinum neurotoxin A normalizes striatal D2 R binding and reduces striatal D1 R binding in male hemiparkinsonian rats. J. Neurosci. Res. 2018, 96, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Itakura, M.; Kohda, T.; Kubo, T.; Semi, Y.; Azuma, Y.T.; Nakajima, H.; Kozaki, S.; Takeuchi, T. Botulinum neurotoxin A subtype 2 reduces pathological behaviors more effectively than subtype 1 in a rat Parkinson’s disease model. Biochem. Biophys. Res. Commun. 2014, 447, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Itakura, M.; Kohda, T.; Kubo, T.; Semi, Y.; Nishiyama, K.; Azuma, Y.T.; Nakajima, H.; Kozaki, S.; Takeuchi, T. Botulinum neurotoxin type A subtype 2 confers greater safety than subtype 1 in a rat Parkinson’s disease model. J. Vet. Med. Sci. 2014, 76, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Armentero, M.T. Animal models of Parkinson’s disease. FEBS J. 2012, 279, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Deumens, R.; Blokland, A.; Prickaerts, J. Modeling Parkinson’s disease in rats: An evaluation of 6-OHDA lesions of the nigrostriatal pathway. Exp. Neurol. 2002, 175, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Meissner, W.G.; Frasier, M.; Gasser, T.; Goetz, C.G.; Lozano, A.; Piccini, P.; Obeso, J.A.; Rascol, O.; Schapira, A.; Voon, V.; et al. Priorities in Parkinson’s disease research. Nat. Rev. Drug Discov. 2011, 10, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Sonsalla, P.K.; Chesselet, M.F. Animal models of Parkinson’s disease progression. Acta Neuropathol. 2008, 115, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Schwarting, R.K.; Huston, J.P. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog. Neurobiol. 1996, 50, 275–331. [Google Scholar] [CrossRef]
- Ungerstedt, U.; Ljungberg, T.; Steg, G. Behavioral, physiological, and neurochemical changes after 6-hydroxydopamine-induced degeneration of the nigro-striatal dopamine neurons. Adv. Neurol. 1974, 5, 421–426. [Google Scholar] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Schallert, T.; Tillerson, J.L. Intervention strategies for degeneration of dopamine neurons in Parkinsonism. Optimizing behavioral assessment of outcome. In Central Nervous System Diseases: Innovative Animal Models from Lab to Clinic; Emerich, D.F., Dean, R.L., Sandberg, P.R., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 131–151. [Google Scholar]
- Bohnen, N.I.; Müller, M.L.; Koeppe, R.A.; Studenski, S.A.; Kilbourn, M.A.; Frey, K.A.; Albin, R.L. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology 2009, 73, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Karachi, C.; Grabli, D.; Bernard, F.A.; Tandé, D.; Wattiez, N.; Belaid, H.; Bardinet, E.; Prigent, A.; Nothacker, H.P.; Hunot, S.; et al. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J. Clin. Investig. 2010, 120, 2745–2754. [Google Scholar] [CrossRef] [PubMed]
- Rabin, L.A.; Tanapat, P.; Relkin, N. Cholinergic components of frontal lobe function and dysfunction. Handb. Clin. Neurol. 2008, 88, 1–30. [Google Scholar] [PubMed]
- Konieczny, J.; Czarnecka, A.; Lenda, T.; Kamińska, K.; Antkiewicz-Michaluk, L. The significance of rotational behavior and sensitivity of striatal dopamine receptors in hemiparkinsonian rats: A comparativestudy of lactacystin and 6-OHDA. Neuroscience 2017, 340, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Kroken, A.R.; Blum, F.C.; Zuverink, M.; Barbieri, J.T. Entry of Botulinum Neurotoxin Subtypes A1 and A2 into Neurons. Infect. Immun. 2016, 85, e00795-16. [Google Scholar] [CrossRef] [PubMed]
- Seiden, L.S.; Sabol, K.E.; Ricaurte, G.A. Amphetamine: Effects on catecholamine systems and behavior. Annu. Rev. Pharmacol. Toxicol. 1993, 33, 639–677. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef] [PubMed]
- Hawlitschka, A.; Antipova, V.; Schmitt, O.; Witt, M.; Benecke, R.; Mix, E.; Wree, A. Intracerebrally applied botulinum neurotoxin in experimental neuroscience. Curr. Pharm. Biotechnol. 2013, 14, 124–130. [Google Scholar] [PubMed]
- Day, M.; Wang, Z.; Ding, J.; An, X.; Ingham, C.A.; Shering, A.F.; Wokosin, D.; Ilijic, E.; Sun, Z.; Sampson, A.R.; et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 2006, 9, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Bernardi, G.; Ding, J.; Surmeier, D.J. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007, 30, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Horstink, M.; Tolosa, E.; Bonuccelli, U.; Deuschl, G.; Friedman, A.; Kanovsky, P.; Larsen, J.P.; Lees, A.; Oertel, W.; Poewe, W.; et al. Review of the therapeutic management of Parkinson’s disease. Report of a joint task force of the European Federation of Neurological Societies and the Movement Disorder Society-European Section. Part I: Early (uncomplicated) Parkinson’s disease. Eur. J. Neurol. 2006, 13, 1170–1185. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, T. Medikamentöse behandlung der idiopathischen Parkinson-krankheit. Nervenarzt 2003, 74, S12–S21. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, H.H. Updates in the medical management of Parkinson disease. Clevel. Clin. J. Med. 2012, 79, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, S. Clinical uses of botulinum neurotoxins: Current indications, limitations and future developments. Toxins 2012, 4, 913–939. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. An update on new and unique uses of botulinum toxin in movement disorders. Toxicon 2017, 147, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Botulinum toxin: State of the art. Mov. Disord. 2017, 32, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Orsini, M.; Leite, M.A.A.; Chung, T.M.; Bocca, W.; de Souza, J.A.; de Souza, O.G.; Moreira, R.P.; Bastos, V.H.; Teixeira, S.; Oliveira, A.B.; et al. Botulinum neurotoxin type A in neurology: Update. Neurol. Int. 2015, 7, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Yue, Z.; Kirik, D.; Spillantini, M.G. Animal models of Parkinson’s disease: Limits and relevance to neuroprotection studies. Mov. Disord. 2013, 28, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Cremer, J.N.; Amunts, K.; Graw, J.; Piel, M.; Rösch, F.; Zilles, K. Neurotransmitter receptor density changes in Pitx3ak mice—A model relevant to Parkinson’s disease. Neuroscience 2015, 285, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Cremer, J.N.; Amunts, K.; Schleicher, A.; Palomero-Gallagher, N.; Piel, M.; Rösch, F.; Zilles, K. Changes in the expression of neurotransmitter receptors in Parkin and DJ-1 knockout mice—A quantitative multireceptor study. Neuroscience 2015, 311, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Yuan, L. Genetic variants and animal models in SNCA and Parkinson disease. Ageing Res. Rev. 2014, 15, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Pinto, M.; Moraes, C.T. Mouse models of Parkinson’s disease associated with mitochondrial dysfunction. Mol. Cell. Neurosci. 2013, 55, 87–94. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Publication | Species | 6-OHDA Lesion | BoNT-A | BoNT-A Influence on Drug-Induced Turning Rate | Readjustment of Forelimb Use after Unilateral 6-OHDA Lesion | Improvement of Forelimb Akinesia (Stepping Test) | Readjustment of Lateralized Sensorimotor Integration | Numeric Density BiVs | D2 Receptor Binding Potential after BoNT-A | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ipsi-Lateral | Contra-Lateral | Bi-Lateral | Apo-Morphine | Amphetamine/Meth-Amphetamine | Cylinder Test | Steps of Forelimb Ipsilateral to 6-OHDA | Steps of Forelimb Contralateral to 6-OHDA | Corridor Task | ChAT Positive | TH Positive | Receptor Autoradiography or PET/CT | |||
| Wree et al. (2010) [54] | Wistar rat | right MFB | 1 ng | ↓ | − | + | − | |||||||
| 2 ng | ↓ | + | ++ | − | ||||||||||
| 0.1 ng | ↓ | + | + | |||||||||||
| 1 ng | ↓ | + | + | |||||||||||
| 2 ng | ↓ | ++ | ++ | |||||||||||
| Holzmann et al. (2012) [61] | Wistar rat | 1 ng | ||||||||||||
| Antipova et al. (2013) [59] | Wistar rat | right MFB | 1 ng | ↓ | − | − | ||||||||
| 2 ng | ↓ | ↑ | + | |||||||||||
| Antipova et al. (2017) [62] | Wistar rat | right MFB | 1 ng | ↓ | ↑ | − | − | − | − | |||||
| 1 ng | ↑ | −↓ | + | + | + | + | ||||||||
| Mehlan et al. (2016) [60] | Wistar rat | 1 ng | + | + | ||||||||||
| Hawlitschka et al. (2017) [55] | C57BL/6 mouse | 0.025–0.2 ng | + | − | ||||||||||
| Wedekind et al. (2017) [64] | Wistar rat | right MFB | 1 ng | ↓ | − | ↓ | ||||||||
| Mann et al. (2018a) [58] | Wistar rat | right MFB | 1 ng | ↓ | ↓ | |||||||||
| 1 ng | ↓ | ↓ | ||||||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawlitschka, A.; Wree, A. Experimental Intrastriatal Applications of Botulinum Neurotoxin-A: A Review. Int. J. Mol. Sci. 2018, 19, 1392. https://doi.org/10.3390/ijms19051392
Hawlitschka A, Wree A. Experimental Intrastriatal Applications of Botulinum Neurotoxin-A: A Review. International Journal of Molecular Sciences. 2018; 19(5):1392. https://doi.org/10.3390/ijms19051392
Chicago/Turabian StyleHawlitschka, Alexander, and Andreas Wree. 2018. "Experimental Intrastriatal Applications of Botulinum Neurotoxin-A: A Review" International Journal of Molecular Sciences 19, no. 5: 1392. https://doi.org/10.3390/ijms19051392
APA StyleHawlitschka, A., & Wree, A. (2018). Experimental Intrastriatal Applications of Botulinum Neurotoxin-A: A Review. International Journal of Molecular Sciences, 19(5), 1392. https://doi.org/10.3390/ijms19051392