Transcriptomic Signature of Right Ventricular Failure in Experimental Pulmonary Arterial Hypertension: Deep Sequencing Demonstrates Mitochondrial, Fibrotic, Inflammatory and Angiogenic Abnormalities

 ,
,
Abstract
:
1. Introduction
2. Results
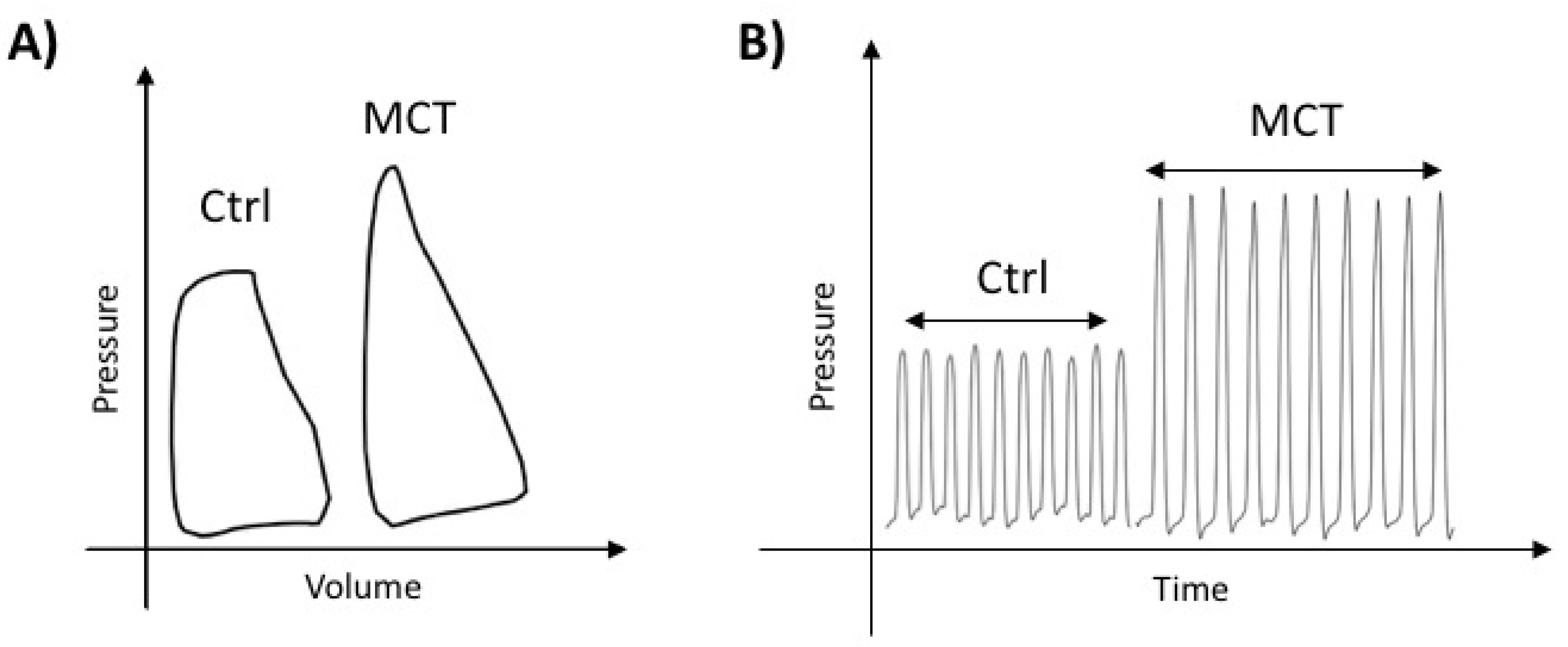
2.1. Characterization of RVF in MCT-PAH
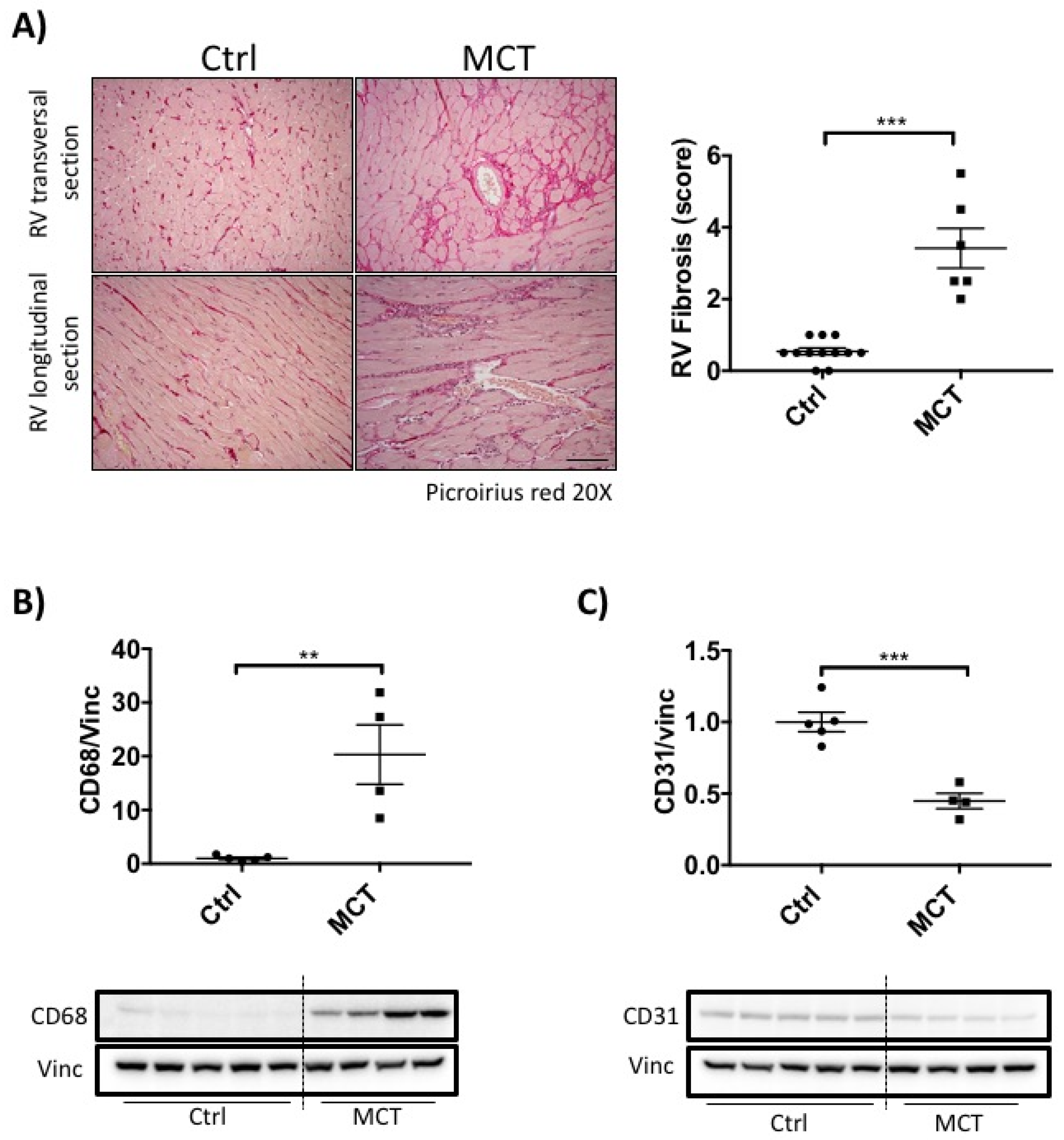
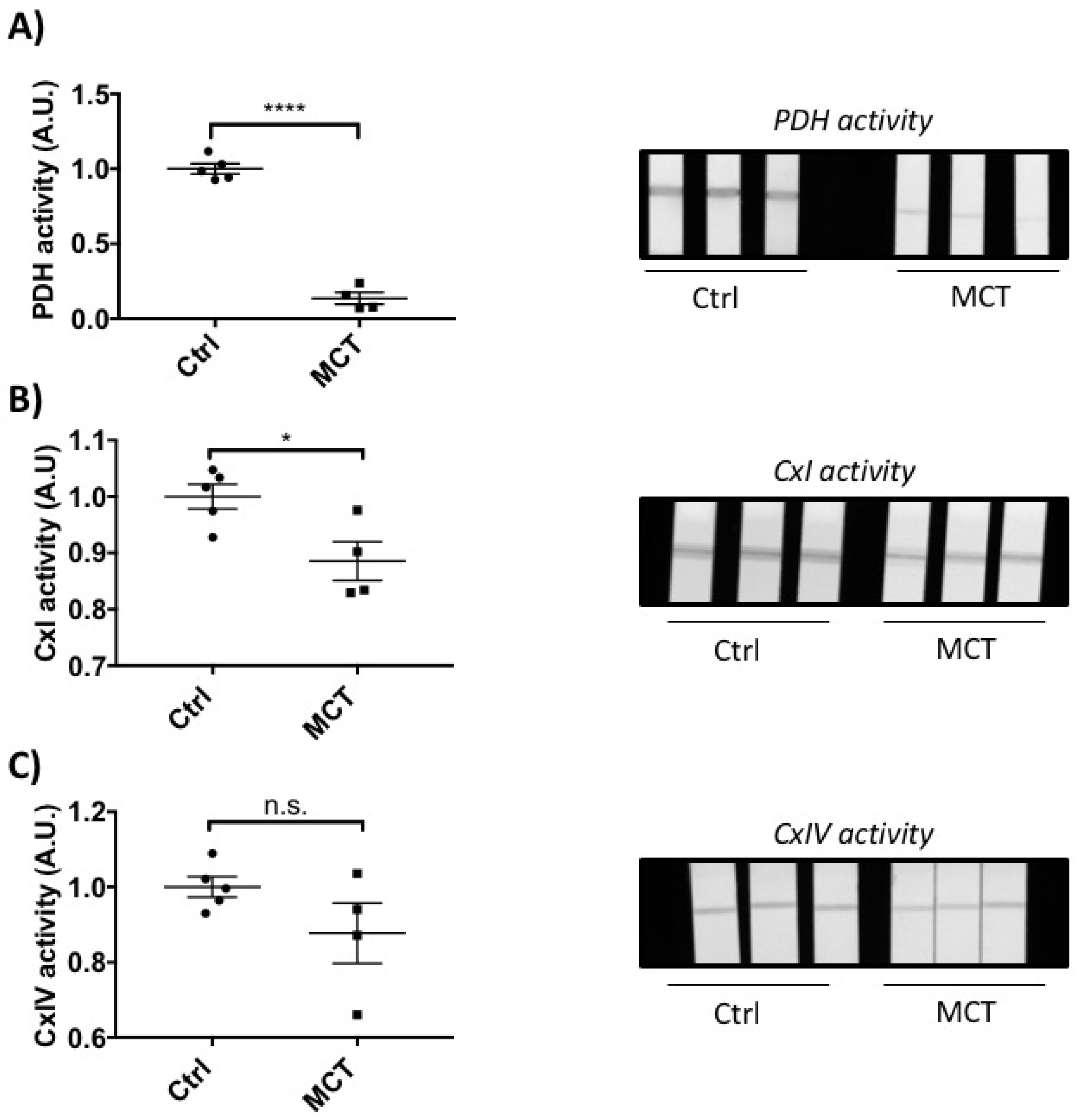
2.2. RVF Is Associated with Increased Fibrosis, Inflammation Decreased Angiogenesis and Mitochondrial/Metabolic Dysfunction
2.3. RNA Sequencing
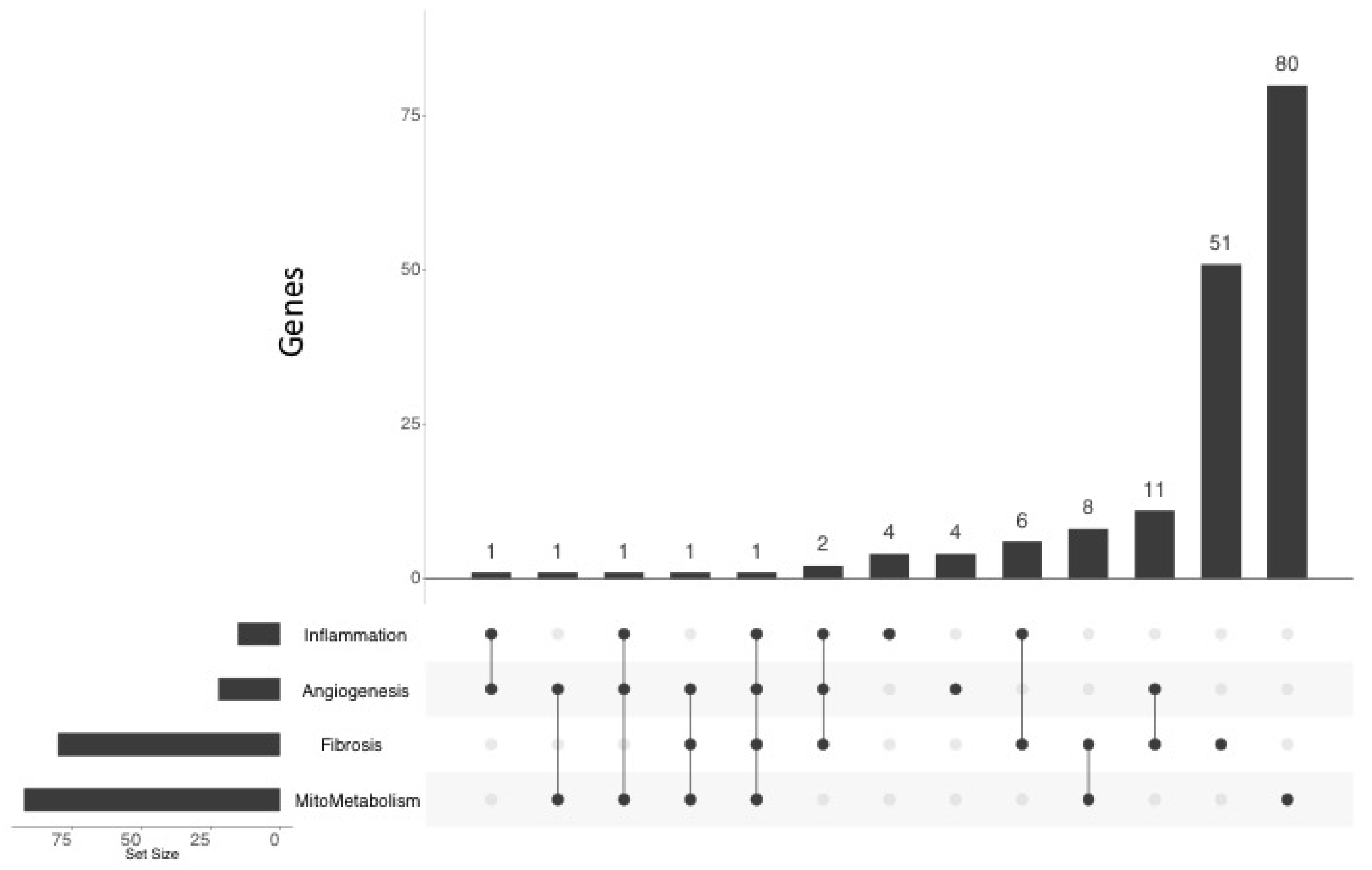
2.4. Functional Annotation of the MCT RV Transcriptome
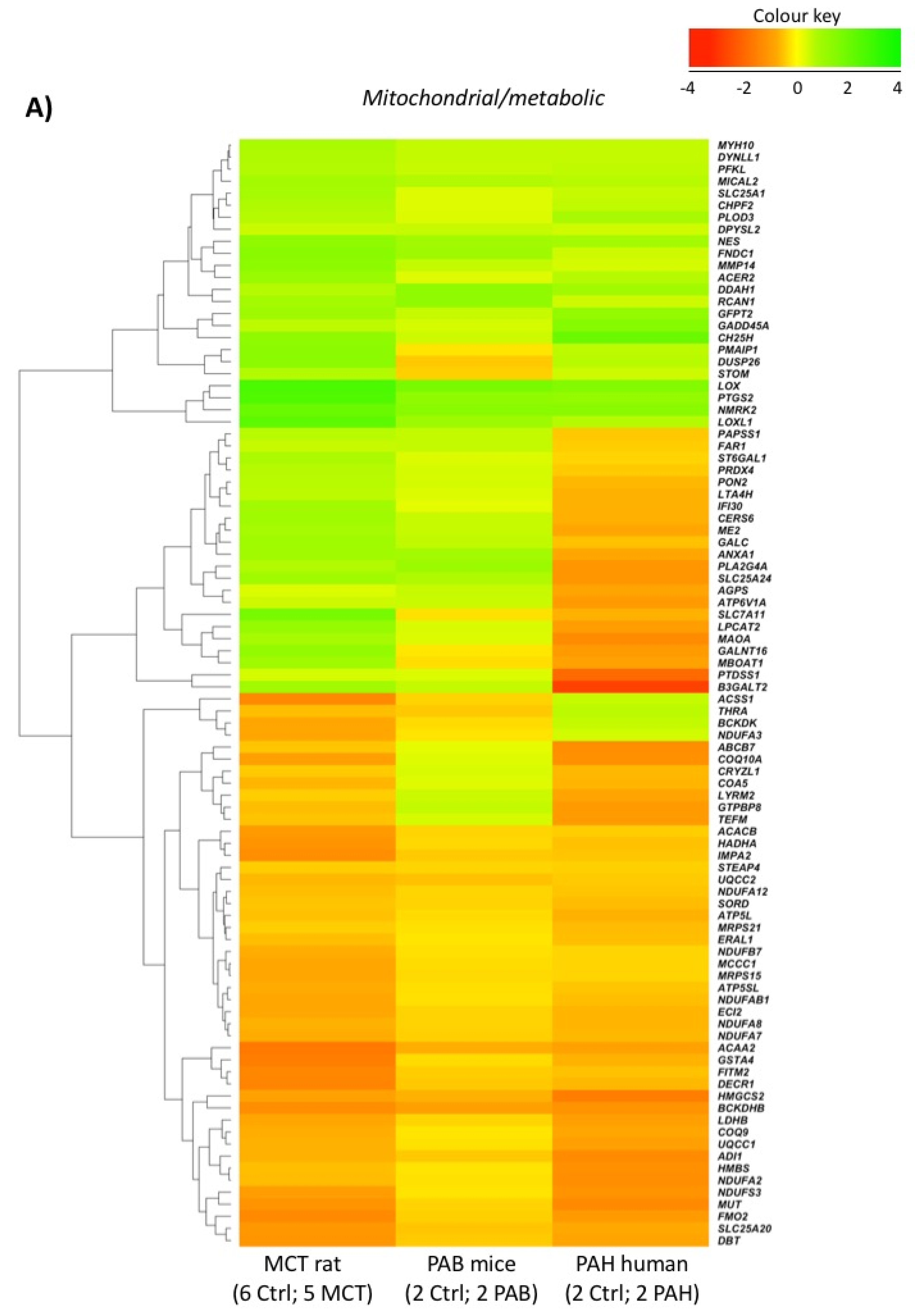
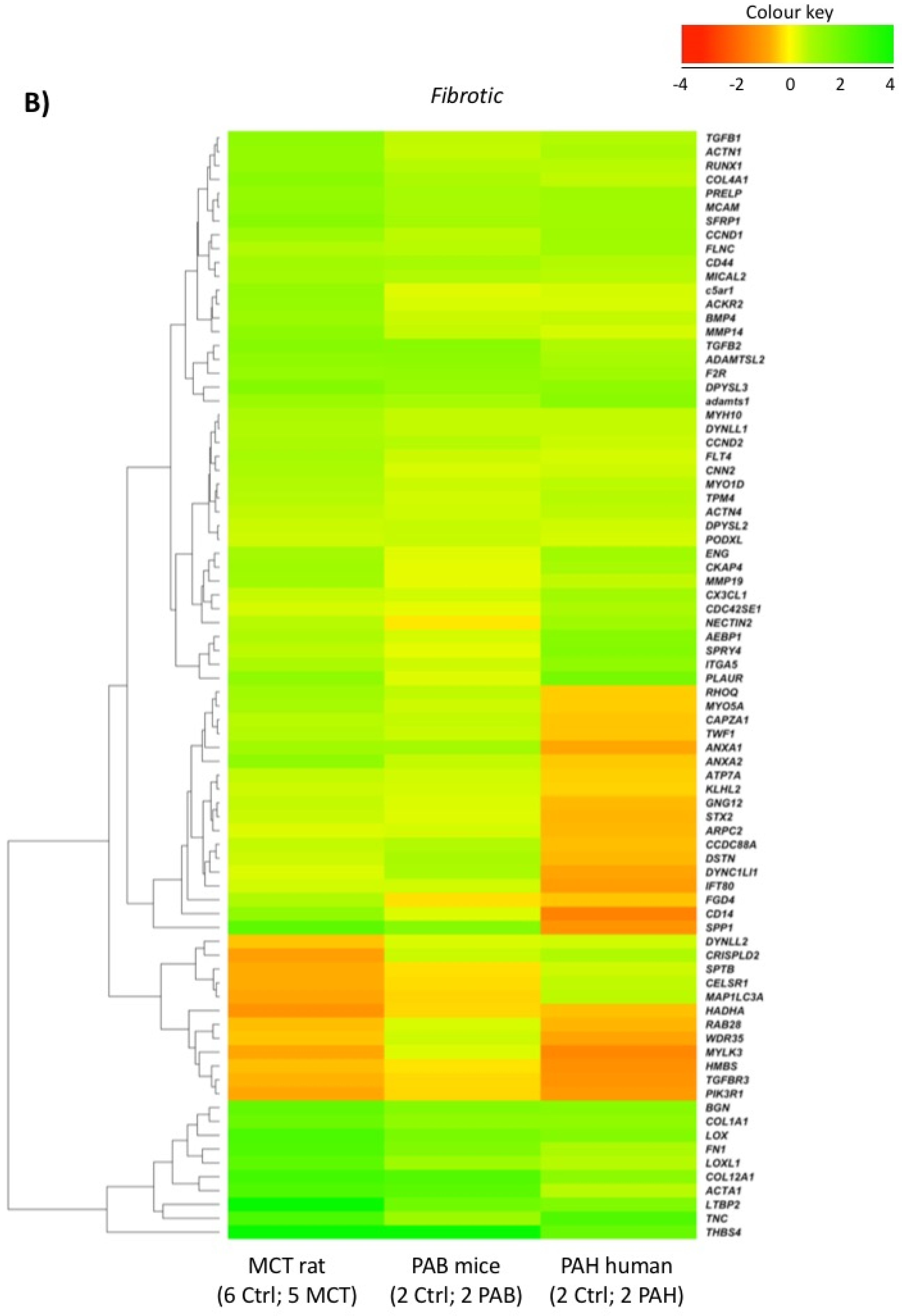
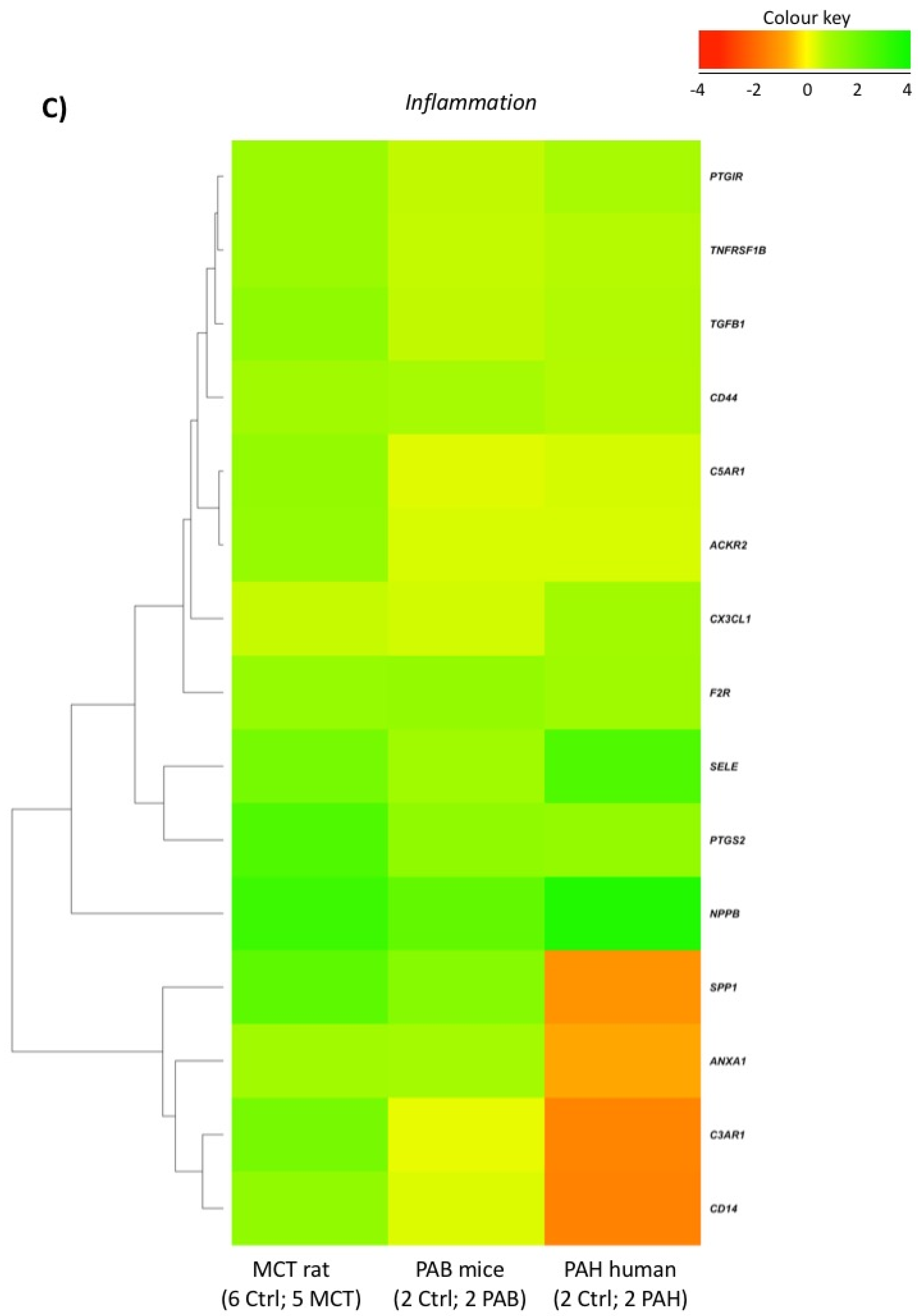
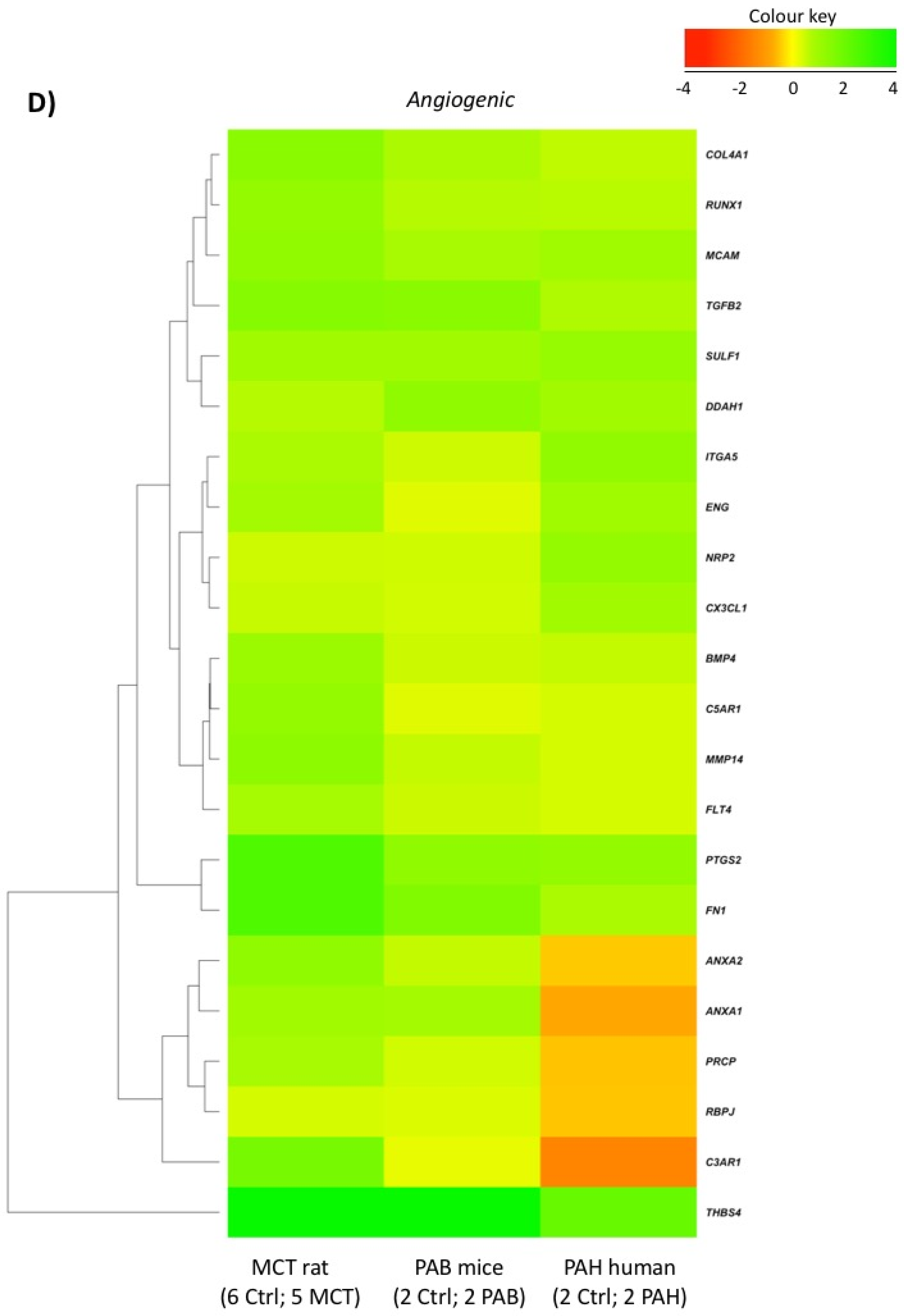
2.5. Comparison with Mouse and Human RV Transcriptomic Data
3. Discussion
4. Materials and Methods
4.1. Monocrotaline-Induced PAH Animal Model
4.2. Hemodynamic Parameters
4.3. Right Heart Catheterization (RHC)
4.4. Immunoblotting and Histology
4.5. Mitochondrial Enzyme Activity
4.6. RNA Sequencing
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vonk Noordegraaf, A.; Galiè, N. The role of the right ventricle in pulmonary arterial hypertension. Eur. Respir. Rev. 2011, 20, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Veerdonk, M.C.; Kind, T.; Marcus, J.T.; Mauritz, G.J.; Heymans, M.W.; Bogaard, H.J.; Boonstra, A.; Marques, K.M.; Westerhof, N.; Vonk-Noordegraaf, A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J. Am. Coll. Cardiol. 2011, 58, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Wijeratne, D.T.; Lajkosz, K.; Brogly, S.B.; Lougheed, M.D.; Jiang, L.; Housin, A.; Barber, D.; Johnson, A.; Doliszny, K.M.; Archer, S.L. Increasing Incidence and Prevalence of World Health Organization Groups 1 to 4 Pulmonary Hypertension. Circ. Cardiovasc. Qual. Outcomes 2018, 11, e003973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef] [PubMed]
- Sydykov, A.; Mamazhakypov, A.; Petrovic, A.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Ghofrani, H.A.; Schermuly, R.T. Inflammatory Mediators Drive Adverse Right Ventricular Remodeling and Dysfunction and Serve as Potential Biomarkers. Front. Physiol. 2018, 9, 609. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Archer, S.L. The Right Ventricle in Pulmonary Arterial Hypertension: Disorders of Metabolism, Angiogenesis and Adrenergic Signaling in Right Ventricular Failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Rain, S.; Andersen, S.; Najafi, A.; Gammelgaard, S.J.; da Silva Gonçalves, B.D.; Handoko, M.L.; Bogaard, H.J.; Vonk-Noordegraaf, A.; Andersen, A.; van der Velden, J.; et al. Right Ventricular Myocardial Stiffness in Experimental Pulmonary Arterial HypertensionClinical Perspective. Circ. Hear Fail. 2016, 9, e002636. [Google Scholar] [CrossRef] [PubMed]
- Freed, B.H.; Gomberg-Maitland, M.; Chandra, S.; Mor-Avi, V.; Rich, S.; Archer, S.L.; Jamison, E.B., Jr.; Lang, R.M.; Patel, A.R. Late gadolinium enhancement cardiovascular magnetic resonance predicts clinical worsening in patients with pulmonary hypertension. J. Cardiovasc. Magn. Reson. 2012, 14, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blyth, K.G.; Groenning, B.A.; Martin, T.N.; Foster, J.E.; Mark, P.B.; Dargie, H.J.; Peacock, A.J. Contrast enhanced-cardiovascular magnetic resonance imaging in patients with pulmonary hypertension. Eur. Heart J. 2005, 26, 1993–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safdar, Z.; Tamez, E.; Chan, W.; Arya, B.; Ge, Y.; Deswal, A.; Bozkurt, B.; Frost, A.; Entman, M. Circulating Collagen Biomarkers as Indicators of Disease Severity in Pulmonary Arterial Hypertension. JACC Hear Fail. 2014, 2, 412–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.-Q.; Zhang, R.; Zhang, H.-D.; Yuan, P.; Wang, X.-J.; Zhao, Q.-H.; Wang, L.; Jiang, R.; Jan Bogaard, H.; Jing, Z.-C. Reversal of right ventricular remodeling by dichloroacetate is related to inhibition of mitochondria-dependent apoptosis. Hypertens. Res. 2016, 39, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Ferreira, P.; Maia-Rocha, C.; Adão, R.; Mendes, M.J.; Santos-Ribeiro, D.; Alves, B.S.; Cerqueira, R.J.; Castro-Chaves, P.; Lourenço, A.P.; De Keulenaer, G.W.; et al. Neuregulin-1 improves right ventricular function and attenuates experimental pulmonary arterial hypertension. Cardiovasc. Res. 2016, 109, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Harhay, M.O.; Tracy, R.P.; Bagiella, E.; Barr, R.G.; Pinder, D.; Hundley, W.G.; Bluemke, D.A.; Kronmal, R.A.; Lima, J.A.; Kawut, S.M. Relationship of CRP, IL-6, and fibrinogen with right ventricular structure and function: The MESA-Right Ventricle Study. Int. J. Cardiol. 2013, 168, 3818–3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, K.W.; Archer, S.L.; Pritzker, M.; Rose, L.; Weir, E.K.; Sharma, A.; Thenappan, T. Interleukin-6 is independently associated with right ventricular function in pulmonary arterial hypertension. J. Hear Lung Transplant. 2018, 37, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Mathai, S.C.; Hassoun, P.M. Pulmonary arterial hypertension in connective tissue diseases. Heart Fail. Clin. 2012, 8, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Landmesser, U.; Drexler, H. Molecular Mechanisms in Heart Failure: Focus on Cardiac Hypertrophy, Inflammation, Angiogenesis, and Apoptosis. J. Am. Coll. Cardiol. 2006, 48, A56–A66. [Google Scholar] [CrossRef]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis-Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Gomez-Arroyo, J.; Abbate, A.; Bogaard, H.J. Mechanisms of right heart failure—A work in progress and a plea for failure prevention. Pulm. Circ. 2013, 3, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Potus, F.; Ruffenach, G.; Dahou, A.; Thebault, C.; Breuils-Bonnet, S.; Tremblay, È.; Nadeau, V.; Paradis, R.; Graydon, C.; Wong, R.; et al. Downregulation of miR-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation 2015, 132, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Courtman, D.W.; Ng, D.S.; Robb, M.J.; Deng, Y.P.; Trogadis, J.; Han, R.N.; Stewart, D.J. Microvascular Regeneration in Established Pulmonary Hypertension by Angiogenic Gene Transfer. Am. J. Respir. Cell Mol. Biol. 2006, 35, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partovian, C.; Adnot, S.; Raffestin, B.; Louzier, V.; Levame, M.; Mavier, I.M.; Lemarchand, P.; Eddahibi, S. Adenovirus-Mediated Lung Vascular Endothelial Growth Factor Overexpression Protects against Hypoxic Pulmonary Hypertension in Rats. Am. J. Respir. Cell Mol. Biol. 2000, 23, 762–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, S.; Bernstein, D. Molecular Mechanisms of Right Ventricular Failure. Circulation 2015, 132, 1734–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini-Chohan, H.K.; Dakshinamurti, S.; Taylor, W.A.; Shen, G.X.; Murphy, R.; Sparagna, G.C.; Hatch, G.M. Persistent pulmonary hypertension results in reduced tetralinoleoyl-cardiolipin and mitochondrial complex II + III during the development of right ventricular hypertrophy in the neonatal pig heart. Am. J. Physiol. Circ. Physiol. 2011, 301, H1415–H1424. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Fang, Y.-H.; Cadete, V.J.J.; Wietholt, C.; Urboniene, D.; Toth, P.T.; Marsboom, G.; Zhang, H.J.; Haber, I.; Rehman, J.; et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J. Mol. Med. 2010, 88, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Neuber-Hess, M.; Mewburn, J.; Dasgupta, A.; Dunham-Snary, K.; Wu, D.; Chen, K.H.; Hong, Z.; Sharp, W.W.; Kutty, S.; et al. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J. Mol. Med. 2017, 95, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Philip, J.; Vinnakota, K.C.; Van den Bergh, F.; Tabima, D.M.; Hacker, T.; Beard, D.A.; Chesler, N.C. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol. Rep. 2017, 5, e13157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Potus, F.; Wu, D.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Lima, P.; Archer, S.L. Increased Drp1-Mediated Mitochondrial Fission Promotes Proliferation and Collagen Production by Right Ventricular Fibroblasts in Experimental Pulmonary Arterial Hypertension. Front. Physiol. 2018, 9, 828. [Google Scholar] [CrossRef] [PubMed]
- Meyrick, B.; Gamble, W.; Reid, L. Development of Crotalaria pulmonary hypertension: Hemodynamic and structural study. Am. J. Physiol. Circ. Physiol. 1980, 239, H692–H702. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Grosse Kreymborg, K.; Uchida, S.; Gellert, P.; Schneider, A.; Boettger, T.; Voswinckel, R.; Wietelmann, A.; Szibor, M.; Weissmann, N.; Ghofrani, A.H.; et al. Identification of right heart-enriched genes in a murine model of chronic outflow tract obstruction. J. Mol. Cell. Cardiol. 2010, 49, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for Right Ventricular Lipotoxicity in Heritable Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naeije, R.; Manes, A. The right ventricle in pulmonary arterial hypertension. Eur. Respir. Rev. 2014, 23, 476–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, R.; Sasayama, S.; Matsumori, A. Left ventricular pressure-volume relationship in a murine model of congestive heart failure due to acute viral myocarditis. J. Am. Coll. Cardiol. 2002, 40, 1506–1514. [Google Scholar] [CrossRef]
- Yu, Q.; Chan, S.Y. Mitochondrial and Metabolic Drivers of Pulmonary Vascular Endothelial Dysfunction in Pulmonary Hypertension. Adv. Exp. Med. Biol. 2017, 967, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafikov, R.; Sun, X.; Rafikova, O.; Meadows, M.L.; Desai, A.A.; Khalpey, Z.; Yuan, J.X.; Fineman, J.R.; Black, S.M. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox. Biol. 2015, 6, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redout, E.; Wagner, M.; Zuidwijk, M.; Boer, C.; Musters, R.J.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res. 2007, 75, 770–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norris, R.A.; Damon, B.; Mironov, V.; Kasyanov, V.; Ramamurthi, A.; Moreno-Rodriguez, R.; Trusk, T.; Potts, J.D.; Goodwin, R.L.; Davis, J.; et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J. Cell. Biochem. 2007, 101, 695–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.Y.; Zheng, H.; Ouyang, G. Periostin, a multifunctional matricellular protein in inflammatory and tumor microenvironments. Matrix Biol. 2014, 37, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of Tenascin-C into the Extracellular Matrix by Periostin Underlies an Extracellular Meshwork Architecture. J. Biol. Chem. 2010, 285, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wu, H.; Xia, W.; Chen, X.; Zhu, S.; Zhang, S.; Shao, Y.; Ma, W.; Yang, D.; Zhang, J. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J. Cardiol. 2014, 63, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Gavins, F.N.E.; Hickey, M.J. Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 2012, 3, 354. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Yi, B.; Chen, Y.; Huang, Q.; Wang, H.; Lu, K.; Fu, W. The ET-1-mediated carbonylation and degradation of ANXA1 induce inflammatory phenotype and proliferation of pulmonary artery smooth muscle cells in HPS. PLoS ONE 2017, 12, e0175443. [Google Scholar] [CrossRef] [PubMed]
- De Coupade, C.; Ajuebor, M.N.; Russo-Marie, F.; Perretti, M.; Solito, E. Cytokine modulation of liver annexin 1 expression during experimental endotoxemia. Am. J. Pathol. 2001, 159, 1435–1443. [Google Scholar] [CrossRef]
- Fredman, G.; Kamaly, N.; Spolitu, S.; Milton, J.; Ghorpade, D.; Chiasson, R.; Kuriakose, G.; Perretti, M.; Farokzhad, O.; Tabas, I. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 2015, 7, 275ra20. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, M.; de Jong, R.; Rossaint, J.; Viola, J.R.; Leoni, G.; Wang, J.M.; Grommes, J.; Hinkel, R.; Kupatt, C.; Weber, C.; et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ. Res. 2015, 116, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Kusters, D.H.M.; Chatrou, M.L.; Willems, B.A.G.; De Saint-Hubert, M.; Bauwens, M.; van der Vorst, E.; Bena, S.; Biessen, E.A.; Perretti, M.; Schurgers, L.J.; et al. Pharmacological Treatment with Annexin A1 Reduces Atherosclerotic Plaque Burden in LDLR-/- Mice on Western Type Diet. PLoS ONE 2015, 10, e0130484. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; Di Filippo, C.; La, M.; Solito, E.; McLean, P.G.; Flower, R.J.; Oliani, S.M.; Perretti, M. Lipocortin 1 reduces myocardial ischemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. 2000, 14, 1867–1869. [Google Scholar] [CrossRef] [PubMed]
- Gavins, F.N.E.; Kamal, A.M.; D’Amico, M.; Oliani, S.M.; Perretti, M. Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct. FASEB J. 2005, 19, 100–102. [Google Scholar] [CrossRef] [PubMed]
- La, M.; D’Amico, M.; Bandiera, S.; Di Filippo, C.; Oliani, S.M.; Gavins, F.N.; Flower, R.J.; Perretti, M. Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: Analysis of their mechanism of action. FASEB J. 2001, 15, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Consalvo, A.P.; Ray, V.; Di Filippo, C.; D’Amico, M.; Mehta, N.; Perretti, M. Proresolving and tissue-protective actions of annexin A1-based cleavage-resistant peptides are mediated by formyl peptide receptor 2/lipoxin A4 receptor. J. Immunol. 2013, 190, 6478–6487. [Google Scholar] [CrossRef] [PubMed]
- Gavins, F.N.E.; Dalli, J.; Flower, R.J.; Granger, D.N.; Perretti, M. Activation of the annexin 1 counter-regulatory circuit affords protection in the mouse brain microcirculation. FASEB J. 2007, 21, 1751–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relton, J.K.; Strijbos, P.J.; O’Shaughnessy, C.T.; Carey, F.; Forder, R.A.; Tilders, F.J.; Rothwell, N.J. Lipocortin-1 is an endogenous inhibitor of ischemic damage in the rat brain. J. Exp. Med. 1991, 174, 305–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.K.; Gil, C.D.; Oliani, S.M.; Gavins, F.N.E. Targeting formyl peptide receptor 2 reduces leukocyte-endothelial interactions in a murine model of stroke. FASEB J. 2015, 29, 2161–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N. Formyl-Peptide Receptor 2/3/Lipoxin A4 Receptor Regulates Neutrophil-Platelet Aggregation and Attenuates Cerebral Inflammation: Impact for Therapy in Cardiovascular Disease. Circulation 2016, 133, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.X.; Finlayson, S.B.; Al-Sharea, A.; Tate, M.; De Blasio, M.J.; Deo, M.; Rosli, S.; Prakoso, D.; Thomas, C.J.; Kiriazis, H.; et al. Endogenous Annexin-A1 Regulates Haematopoietic Stem Cell Mobilisation and Inflammatory Response Post Myocardial Infarction in Mice In Vivo. Sci. Rep. 2017, 7, 16615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babiychuk, E.B.; Atanassoff, A.P.; Monastyrskaya, K.; Brandenberger, C.; Studer, D.; Allemann, C.; Draeger, A. The Targeting of Plasmalemmal Ceramide to Mitochondria during Apoptosis. PLoS ONE 2011, 6, e23706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohnishi, M.; Tokuda, M.; Masaki, T.; Fujimura, T.; Tai, Y.; Itano, T.; Matsui, H.; Ishida, T.; Konishi, R.; Takahara, J.; et al. Involvement of annexin-I in glucose-induced insulin secretion in rat pancreatic islets. Endocrinology 1995, 136, 2421–2426. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Bindel, F.; Grimm, C.; Lin, S.J.; Wappler, J.; Klinger, B.; Blüthgen, N.; Du Bois, I.; Schmeck, B.; Lehrach, H.; et al. Annexin A1 sustains tumor metabolism and cellular proliferation upon stable loss of HIF1A. Oncotarget 2016, 7, 6693–6710. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Schnitzer, J.E. Impaired tumor growth, metastasis, angiogenesis and wound healing in annexin A1-null mice. Proc. Natl. Acad. Sci. USA 2009, 106, 17886–17891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamanlidis, G.; Bautista-Hernandez, V.; Fynn-Thompson, F.; Del Nido, P.; Tian, R. Impaired Mitochondrial Biogenesis Precedes Heart Failure in Right Ventricular Hypertrophy in Congenital Heart Disease. Circ. Hear Fail. 2011, 4, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Lu, D.; Dong, W.; Zhang, L.; Zhang, X.; Quan, X.; Ma, C.; Lian, H.; Zhang, L. Expression of CYP2E1 increases oxidative stress and induces apoptosis of cardiomyocytes in transgenic mice. FEBS J. 2011, 278, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; Tigchelaar, W.; Ruifrok, W.P.T.; van Gilst, W.H.; de Boer, R.A.; Silljé, H.H. DHRS7c, a novel cardiomyocyte-expressed gene that is down-regulated by adrenergic stimulation and in heart failure. Eur. J. Heart Fail. 2012, 14, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Machado, R.F.; Shimoda, L. Pulmonary Vascular and Ventricular Dysfunction in the Susceptible Patient (2015 Grover Conference Series). Pulm. Circ. 2016, 6, 426–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topol, E.J.; McCarthy, J.; Gabriel, S.; Moliterno, D.J.; Rogers, W.J.; Newby, L.K.; Freedman, M.; Metivier, J.; Cannata, R.; O’Donnell, C.J.; et al. Single nucleotide polymorphisms in multiple novel thrombospondin genes may be associated with familial premature myocardial infarction. Circulation 2001, 104, 2641–2644. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.G.; Sopko, N.; Blech, L.; Popovic, Z.B.; Li, J.; Vasanji, A.; Drumm, C.; Krukovets, I.; Jain, M.K.; Penn, M.S.; et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 2012, 26, 2363–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oklü, R.; Hesketh, R. The latent transforming growth factor beta binding protein (LTBP) family. Biochem. J. 2000, 352 Pt 3, 601–610. [Google Scholar] [CrossRef]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, P.; Zhang, X.; Huang, J.; Hu, S.; Wei, Y. LTBP-2 acts as a novel marker in human heart failure—A preliminary study. Biomarkers 2012, 17, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.R.; Iacobazzi, D.; Abdul-Ghani, S.; Ghorbel, M.; Heesom, K.; Wilson, M.; Gillett, C.; George, S.J.; Caputo, M.; Suleiman, S.; et al. Changes in contractile protein expression are linked to ventricular stiffness in infants with pulmonary hypertension or right ventricular hypertrophy due to congenital heart disease. Open Hear 2018, 5, e000716. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ctrl (n = 15) | MCT (n = 8) | |
|---|---|---|
| Parameters | ||
| HR (BPM) | 308.8 (7.33) | 248.9 (18.85) |
| RVSP (mmHg) | 23.51 (0.9742) | 51.33 * (6.589) |
| mPAP (mmHg) | 16.34 (0.5943) | 33.31 * (4.019) |
| EDV (µL) | 241.4 (21.12) | 424.8 * (43.04) |
| SV (µL) | 296.3 (8.3) | 278.3 (23.97) |
| CO (µL/min) | 91,650 (2672) | 70,209 * (4425) |
| Ea (mmHg/µL) | 0.1871 (0.01736) | 0.26 * (0.01962) |
| Emax (mmHg/µL) | 0.2828 (0.04747) | 0.199 (0.05393) |
| PAAT (ms) | 32.72 (0.8737) | 21.17 * (0.7032) |
| RVFW (%) | 106.7 (3.926) | 28.33 * (8.476) |
| TPR (mmHg/mL/min) | 0.1911 (0.007972) | 0.4552 * (0.04644) |
| Fulton index | 0.2725 (0.0054) | 0.6354 * (0.0302) |
| Systolic Indices | ||
| TAPSE (mm) | 3.088 (0.05361) | 2.16 * (0.1136) |
| EF (%) | 66.36 (1.769) | 52.69 * (2.343) |
| dP/dtmax (mmHg/s) | 651.1 (22.4) | 1048 * (117.8) |
| SW (mJoule) | 0.4639 (0.03557) | 0.9711 * (0.188) |
| Diastolic indices | ||
| dP/dtmin (−mmHg/s) | 577.4 (20.42) | 1063 * (151.9) |
| Tau Mirnsky (mS) | 29.11 (0.3685) | 38.46 * (2.082) |
| Contractibility indices | ||
| PRSW (mmHg) | 14.12 (0.6412) | 23.38 * (2.75) |
| PVA/EDV (mmHg) | 0.001571 (0.0001373) | 0.002625 * (0.0003239) |
| Ventricular-Arterial Coupling | ||
| Emax/Ea | 1.516 (0.1886) | 0.7298 * (0.1884) |
| Term | Category | Function | Gene Count | p Value |
|---|---|---|---|---|
| GO:0005739~mitochondrion | GOTERM_CC_DIRECT | MitoMetabolism | 414 | 1.85 × 10−56 |
| Mitochondrion | UP_KEYWORDS | MitoMetabolism | 196 | 4.16 × 10−37 |
| GO:0005743~mitochondrial inner membrane | GOTERM_CC_DIRECT | MitoMetabolism | 104 | 1.37 × 10−22 |
| Mitochondrion inner membrane | UP_KEYWORDS | MitoMetabolism | 65 | 7.55 × 10−18 |
| transit peptide:Mitochondrion | UP_SEQ_FEATURE | MitoMetabolism | 100 | 2.10 × 10−17 |
| rno01100:Metabolic pathways | KEGG_PATHWAY | MitoMetabolism | 279 | 3.38 × 10−15 |
| rno00190:Oxidative phosphorylation | KEGG_PATHWAY | MitoMetabolism | 56 | 5.89 × 10−13 |
| GO:0005747~mitochondrial respiratory chain complex I | GOTERM_CC_DIRECT | MitoMetabolism | 29 | 4.58 × 10−12 |
| Oxidoreductase | UP_KEYWORDS | MitoMetabolism | 109 | 3.00 × 10−11 |
| Electron transport | UP_KEYWORDS | MitoMetabolism | 31 | 7.73 × 10−11 |
| GO:0055114~oxidation-reduction process | GOTERM_BP_DIRECT | MitoMetabolism | 129 | 1.09 × 10−8 |
| GO:0006979~response to oxidative stress | GOTERM_BP_DIRECT | MitoMetabolism | 44 | 1.40 × 10−8 |
| Ubiquinone | UP_KEYWORDS | MitoMetabolism | 18 | 2.05 × 10−8 |
| Respiratory chain | UP_KEYWORDS | MitoMetabolism | 19 | 4.37 × 10−7 |
| GO:0008137~NADH dehydrogenase (ubiquinone) activity | GOTERM_MF_DIRECT | MitoMetabolism | 18 | 4.64 × 10−7 |
| GO:0005759~mitochondrial matrix | GOTERM_CC_DIRECT | MitoMetabolism | 43 | 6.38 × 10−7 |
| Tricarboxylic acid cycle | UP_KEYWORDS | MitoMetabolism | 12 | 9.93 × 10−6 |
| GO:0000086~G2/M transition of mitotic cell cycle | GOTERM_BP_DIRECT | MitoMetabolism | 16 | 1.11 × 10−5 |
| GO:0006099~tricarboxylic acid cycle | GOTERM_BP_DIRECT | MitoMetabolism | 14 | 1.16 × 10−5 |
| GO:0006635~fatty acid beta-oxidation | GOTERM_BP_DIRECT | MitoMetabolism | 18 | 1.27 × 10−5 |
| GO:0050660~flavin adenine dinucleotide binding | GOTERM_MF_DIRECT | MitoMetabolism | 22 | 5.42 × 10−5 |
| FAD | UP_KEYWORDS | MitoMetabolism | 26 | 5.49 × 10−5 |
| GO:0031966~mitochondrial membrane | GOTERM_CC_DIRECT | MitoMetabolism | 27 | 6.32 × 10−5 |
| NAD | UP_KEYWORDS | MitoMetabolism | 34 | 1.43 × 10−4 |
| Flavoprotein | UP_KEYWORDS | MitoMetabolism | 26 | 1.89 × 10−4 |
| rno00020:Citrate cycle (TCA cycle) | KEGG_PATHWAY | MitoMetabolism | 14 | 2.28 × 10−4 |
| ATP synthesis | UP_KEYWORDS | MitoMetabolism | 8 | 2.47 × 10−4 |
| Mitochondrion outer membrane | UP_KEYWORDS | MitoMetabolism | 20 | 8.23 × 10−4 |
| TOTAL GENES (excluding duplicates) | 647 | |||
| GO:0031012~extracellular matrix | GOTERM_CC_DIRECT | Fibrosis | 72 | 5.83 × 10−12 |
| GO:0030027~lamellipodium | GOTERM_CC_DIRECT | Fibrosis | 47 | 3.85 × 10−9 |
| GO:0005925~focal adhesion | GOTERM_CC_DIRECT | Fibrosis | 90 | 4.02 × 10−9 |
| Actin-binding | UP_KEYWORDS | Fibrosis | 48 | 4.60 × 10−9 |
| Cytoskeleton | UP_KEYWORDS | Fibrosis | 109 | 8.70 × 10−9 |
| GO:0051015~actin filament binding | GOTERM_MF_DIRECT | Fibrosis | 39 | 5.91 × 10−7 |
| Extracellular matrix | UP_KEYWORDS | Fibrosis | 35 | 5.50 × 10−6 |
| GO:0005604~basement membrane | GOTERM_CC_DIRECT | Fibrosis | 28 | 9.53 × 10−6 |
| GO:0005884~actin filament | GOTERM_CC_DIRECT | Fibrosis | 24 | 9.77 × 10−6 |
| GO:0042060~wound healing | GOTERM_BP_DIRECT | Fibrosis | 34 | 1.09 × 10−5 |
| rno04510:Focal adhesion | KEGG_PATHWAY | Fibrosis | 54 | 1.34 × 10−5 |
| GO:0031100~organ regeneration | GOTERM_BP_DIRECT | Fibrosis | 27 | 1.77 × 10−5 |
| Microtubule | UP_KEYWORDS | Fibrosis | 39 | 2.53 × 10−5 |
| GO:0005578~proteinaceous extracellular matrix | GOTERM_CC_DIRECT | Fibrosis | 54 | 4.28 × 10−5 |
| GO:0005874~microtubule | GOTERM_CC_DIRECT | Fibrosis | 51 | 5.13 × 10−5 |
| GO:0002102~podosome | GOTERM_CC_DIRECT | Fibrosis | 13 | 1.49 × 10−4 |
| rno04512:ECM-receptor interaction | KEGG_PATHWAY | Fibrosis | 27 | 1.83 × 10−4 |
| GO:0031252~cell leading edge | GOTERM_CC_DIRECT | Fibrosis | 18 | 4.54 × 10−4 |
| rno04810:Regulation of actin cytoskeleton | KEGG_PATHWAY | Fibrosis | 50 | 5.84 × 10−4 |
| Collagen | UP_KEYWORDS | Fibrosis | 18 | 7.28 × 10−4 |
| TOTAL GENES (excluding duplicates) | 412 | |||
| Inflammatory response | UP_KEYWORDS | Inflammation | 24 | 2.35 × 10−5 |
| TOTAL GENES (excluding duplicates) | 24 | |||
| Gene | Functional Pathway | Sequencing (Ctrl/MCT) | PCR Validation (Ctrl/MCT) | ||
|---|---|---|---|---|---|
| (log) Mean Diff. | (log) Mean Diff. | SE of Diff. | Adjusted p Value | ||
| POSTN | Cell Adhesion | 4.564263838 | 4.326 | 1.213 | 0.0065 |
| THBS4 | Angiogenesis Fibrosis | 4.401212936 | 6.798 | 1.248 | <0.0001 |
| LTBP2 | Fibrosis | 4.16038822 | 3.545 | 1.112 | 0.0163 |
| RND1 | Cell Adhesion | 2.639368653 | 2.687 | 0.9778 | 0.0451 |
| SCN3B | Ion Transport | 2.587455526 | 3.327 | 1.06 | 0.0183 |
| CNTFR | Signal Transduction | −2.883345535 | −5.146 | 1.352 | 0.0036 |
| Gene | Functional Pathway | Sequencing (Ctrl/MCT) | PCR Validation (Ctrl/MCT) | ||
|---|---|---|---|---|---|
| (log) Mean Diff. | (log) Mean Diff. | SE of Diff. | Adjusted p Value | ||
| PRSS35 | mito/metabolism | 4.23 | 4.31 | 0.8278 | 0.0001 |
| CDK1 | fibrosis mito/metabolism | 2.11 | 0.5525 | 0.1917 | 0.0342 |
| ADAM12 | angiogenesis mito/metabolism | 2.03 | 0.8509 | 0.2286 | 0.005 |
| BCAT1 | mito/metabolism | 1.68 | 0.451 | 0.1891 | 0.1005 |
| LDHD | mito/metabolism | −1.51 | −0.8941 | 0.26 | 0.0089 |
| ADH1 | fibrosis mito/metabolism | −1.55 | −0.8194 | 0.2765 | 0.0294 |
| EHHADH | mito/metabolism | −1.56 | −0.9721 | 0.2564 | 0.0038 |
| GSTA4 | mito/metabolism | −1.57 | −0.7991 | 0.2441 | 0.0133 |
| ECI1 | mito/metabolism | −1.62 | −0.8955 | 0.25 | 0.0063 |
| ACAA2 | mito/metabolism | −1.63 | −0.8883 | 0.2471 | 0.0061 |
| MACROD1 | mito/metabolism | −1.8 | −0.9501 | 0.2723 | 0.0079 |
| UCP3 | mito/metabolism | −1.86 | −0.9768 | 0.3092 | 0.0057 |
| HADH | mito/metabolism | −2.12 | −0.9813 | 0.2654 | 0.0048 |
| CYP2E1 | mito/metabolism | −3.9 | −6.394 | 1.469 | 0.0009 |
| DECR1 | mito/metabolism | −1.42 | −0.8718 | 0.2272 | 0.0034 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potus, F.; Hindmarch, C.C.T.; Dunham-Snary, K.J.; Stafford, J.; Archer, S.L. Transcriptomic Signature of Right Ventricular Failure in Experimental Pulmonary Arterial Hypertension: Deep Sequencing Demonstrates Mitochondrial, Fibrotic, Inflammatory and Angiogenic Abnormalities. Int. J. Mol. Sci. 2018, 19, 2730. https://doi.org/10.3390/ijms19092730
Potus F, Hindmarch CCT, Dunham-Snary KJ, Stafford J, Archer SL. Transcriptomic Signature of Right Ventricular Failure in Experimental Pulmonary Arterial Hypertension: Deep Sequencing Demonstrates Mitochondrial, Fibrotic, Inflammatory and Angiogenic Abnormalities. International Journal of Molecular Sciences. 2018; 19(9):2730. https://doi.org/10.3390/ijms19092730
Chicago/Turabian StylePotus, Francois, Charles Colin Thomas Hindmarch, Kimberly J. Dunham-Snary, Jeff Stafford, and Stephen L. Archer. 2018. "Transcriptomic Signature of Right Ventricular Failure in Experimental Pulmonary Arterial Hypertension: Deep Sequencing Demonstrates Mitochondrial, Fibrotic, Inflammatory and Angiogenic Abnormalities" International Journal of Molecular Sciences 19, no. 9: 2730. https://doi.org/10.3390/ijms19092730
APA StylePotus, F., Hindmarch, C. C. T., Dunham-Snary, K. J., Stafford, J., & Archer, S. L. (2018). Transcriptomic Signature of Right Ventricular Failure in Experimental Pulmonary Arterial Hypertension: Deep Sequencing Demonstrates Mitochondrial, Fibrotic, Inflammatory and Angiogenic Abnormalities. International Journal of Molecular Sciences, 19(9), 2730. https://doi.org/10.3390/ijms19092730