Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management


Abstract
: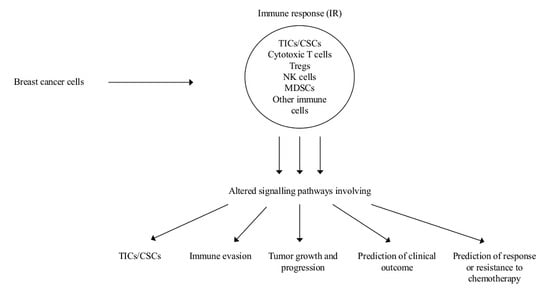
1. Introduction
2. Role of Myeloid Derived Suppressor Cells (MDSCs) on Tumor Initiating Cells (TICs) and of INFs on Cancer Stem Cells (CSCs)
2.1. MDSCs and TICs-Notch Signaling
2.2. INF, TIC Activities, and CSCs
2.3. Stabilization of PD-L1, Up-Regulation of CD47 in Cancer Cells, and ShcA Signaling as Mechanisms of Immune Evasion
2.3.1. PD-L1 Stabilization
2.3.2. CD47 Upregulation
2.3.3. Type III Chaperone Protein ShcA (ShcA) Signaling
2.4. Altered Intra-and Inter-Cellular Signaling in the Immune Microenvironment Affects Tumor Growth and Progression
2.4.1. C-C motif Chemokine Receptor (CCR)7 and Its Chemokine Ligands (CCL)19/(CCL)21
2.4.2. Annexin 1 (ANKA1) and Macrophages
2.4.3. Inflammatory Cells, NR4A1 TGF-β/SMAD Signaling
2.5. Prediction of Clinical Outcome
2.5.1. The NF-κB Pathway
2.5.2. Prognostic HTICS Signature Involving an IR
2.5.3. Long-Noncoding (Lnc) RNAs
2.6. Prediction of Response or Resistance to Chemotherapy
2.6.1. Two Immune-Based Gene Modules
2.6.2. Plasma Cells Inhibit Immunogenic Cell Death (ICD)
3. Perspectives and Conclusion
Conflicts of Interest
References
- Nicolini, A.; Carpi, A. Immune manipulation of advanced breast cancer: An interpretative model of the relationship between immune system and tumor cell biology. Med. Res. Rev. 2009, 29, 436–471. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Carpi, A.; Rossi, G. Cytokines in breast cancer. Cytokine Growth Factor Rev. 2006, 17, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Clarke, M.F. Self-renewal and solid tumor stem cells. Oncogene 2004, 23, 7274–7282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, A.; Ferrari, P.; Fini, M.; Borsari, V.; Fallahi, P.; Antonelli, A.; Berti, P.; Carpi, A.; Miccoli, P. Stem cells: Their role in breast cancer development and resistance to treatment. Curr. Pharm. Biotechnol. 2011, 12, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waight, J.D.; Hu, Q.; Miller, A.; Liu, S.; Abrams, S.I. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS ONE 2011, 6, e27690. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1β-secreting cells. J. Immunol. 2005, 175, 8200–8208. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007, 67, 10019–10026. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Gallina, G.; Dolcetti, L.; Serafini, P.; De Santo, C.; Marigo, I.; Colombo, M.P.; Basso, G.; Brombacher, F.; Borrello, I.; Zanovello, P.; et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Investig. 2006, 116, 2777–2790. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Serafini, P.; De Santo, C.; Marigo, I.; Tosello, V.; Mazzoni, A.; Segal, D.M.; Staib, C.; Lowel, M.; Sutter, G. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J. Immunol. 2003, 170, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celià-Terrassa, T.; Liu, D.D.; Choudhury, A.; Hang, X.; Wei, Y.; Zamalloa, J.; Alfaro-Aco, R.; Chakrabarti, R.; Jiang, Y.Z.; Koh, B.I.; et al. Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a-LCOR axis. Nat. Cell Biol. 2017, 19, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P.; Rossi, G.; Carpi, A. Tumour growth and immune evasion as targets for a new strategy in advanced cancer. Endocr. Relat. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betancur, P.A.; Abraham, B.J.; Yiu, Y.Y.; Willingham, S.B.; Khameneh, F.; Zarnegar, M.; Kuo, A.H.; McKenna, K.; Kojima, Y.; Leeper, N.J.; et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat. Commun. 2017, 8, 14802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPα) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.H. Emerging properties of animal gene regulatory networks. Nature 2010, 468, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Ahn, R.; Sabourin, V.; Bolt, A.M.; Hébert, S.; Totten, S.; De Jay, N.; Festa, M.C.; Young, Y.K.; Im, Y.K.; Pawson, T. The Shc1 adaptor simultaneously balances Stat1 and Stat3 activity to promote breast cancer immune suppression. Nat. Commun. 2017, 8, 14638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Eyob, H.; Ekiz, H.A.; Derose, Y.S.; Waltz, S.E.; Williams, M.A.; Welm, A.L. Inhibition of ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 2013, 3, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Ursini-Siegel, J.; Cory, S.; Zuo, D.; Hardy, W.R.; Rexhepaj, E.; Lam, S.; Schade, B.; Jirstrom, K.; Bjur, E.; Piccirillo, C.A. Receptor tyrosine kinase signaling favors a protumorigenic state in breast cancer cells by inhibiting the adaptive immune response. Cancer Res. 2010, 70, 7776–7787. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.R.; Siegel, P.M.; Ursini-Siegel, J. The Tyrosine Kinome Dictates Breast Cancer Heterogeneity and Therapeutic Responsiveness. J. Cell. Biochem. 2016, 117, 1971–1990. [Google Scholar] [CrossRef] [PubMed]
- Sonbul, S.N.; Gorringe, K.L.; Aleskandarany, M.A.; Mukherjee, A.; Green, A.R.; Ellis, I.O.; Rakha, E.A. Chemokine (C-C motif) receptor 7 (CCR7) associates with the tumour immune microenvironment but not progression in invasive breast carcinoma. J. Pathol. Clin. Res. 2017, 3, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutunea-Fatan, E.; Majumder, M.; Xin, X.; Lala, P.K. The role of CCL21/CCR7 chemokine axis in breast cancer-induced lymphangiogenesis. Mol. Cancer. 2015, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.J.; Du, C.L.; Fu, Y.F.; Zhang, Y.N.; Wang, R.W. Silencing of CCR7 inhibits the growth, invasion and migration of prostate cancer cells induced by VEGFC. Int. J. Clin. Exp. Pathol. 2015, 8, 12533–12540. [Google Scholar] [PubMed]
- Cunningham, H.D.; Shannon, L.A.; Calloway, P.A.; Fassold, B.C.; Dunwiddie, I.; Vielhauer, G.; Zhang, M.; Vines, C.M. Expression of the C-C chemokine receptor 7 mediates metastasis of breast cancer to the lymph nodes in mice. Transl. Oncol. 2010, 3, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Cabioglu, N.; Assi, H.; Sabourin, J.C.; Delaloge, S.; Sahin, A.; Broglio, K.; Spano, J.P.; Combadiere, C.; Bucana, C.; et al. Expression of chemokine receptors predicts the site of metastatic relapse in patients with axillary node positive primary breast cancer. Ann. Oncol. 2006, 17, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ji, R.; Li, J.; Gu, Q.; Zhao, X.; Sun, T.; Wang, J.; Li, J.; Du, Q.; Sun, B. Correlation effect of EGFR and CXCR4 and CCR7 chemokine receptors in predicting breast cancer metastasis and prognosis. J. Exp. Clin. Cancer Res. 2010, 29, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.L.; Burchell, J.; Grimshaw, M.J. Endothelins induce CCR7 expression by breast tumor cells via endothelin receptor A and hypoxia-inducible factor-1. Cancer Res. 2006, 66, 11802–11807. [Google Scholar] [CrossRef] [PubMed]
- Weitzenfeld, P.; Kossover, O.; Körner, C.; Meshel, T.; Wiemann, S.; Seliktar, D.; Legler, D.F.; Ben-Baruch, A. Chemokine axes in breast cancer: Factors of the tumor microenvironment reshape the CCR7-driven metastatic spread of luminal-A breast tumors. J. Leukoc. Biol. 2016, 99, 1009–1025. [Google Scholar] [CrossRef] [PubMed]
- Xuan, W.; Qu, Q.; Zheng, B.; Xiong, S.; Fan, G.H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2015, 97, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Na, I.K.; Busse, A.; Scheibenbogen, C.; Ghadjar, P.; Coupland, S.E.; Letsch, A.; Loddenkemper, C.; Stroux, A.; Bauer, S.; Thiel, E.; et al. Identification of truncated chemokine receptor 7 in human colorectal cancer unable to localize to the cell surface and unreactive to external ligands. Int. J. Cancer 2008, 123, 1565–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci. Rep. 2017, 7, 17925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussunoor, S.; Murray, G.I. The role of annexins in tumour development and progression. J. Pathol. 2008, 216, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Bist, P.; Leow, S.C.; Phua, Q.H.; Shu, S.; Zhuang, Q.; Loh, W.T.; Nguyen, T.H.; Zhou, J.B.; Hooi, S.C.; Lim, L.H. Annexin-1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF-κB: Implication in breast cancer metastasis. Oncogene 2011, 30, 3174–3185. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Anbalagan, D.; Lee, L.H.; Samy, R.P.; Shanmugam, M.K.; Kumar, A.P.; Sethi, G.; Lobie, P.E.; Lim, L.H. ANXA1 inhibits miRNA-196a in a negative feedback loop through NF-κB and c-Myc to reduce breast cancer proliferation. Oncotarget 2016, 7, 27007–27020. [Google Scholar] [CrossRef] [PubMed]
- Khau, T.; Langenbach, S.Y.; Schuliga, M.; Harris, T.; Johnstone, C.N.; Anderson, R.L.; Stewart, A.G. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J. 2011, 25, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investg. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, B.L.; Francis, P.A.; Parker, B.S.; Anderson, R.L. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat. Rev. Drug Discov. 2012, 11, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Drabsch, Y.; Dekker, T.J.; de Vinuesa, A.G.; Li, Y.; Hawinkels, L.J.; Sheppard, K.A.; Goumans, M.J.; Luwor, R.B.; de Vries, C.J. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat. Commun. 2014, 5, 3388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmierer, B.; Hill, C.S. TGFβ-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell. Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Liu, C.; Derynck, R. New regulatory mechanisms of TGF-β receptor function. Trends Cell Biol. 2009, 19, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Allavena, P.; Mantovani, A. Cancer related inflammation: The macrophage connection. Cancer Lett. 2008, 267, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Kioi, M.; Nakashima, H.; Sugiura, K.; Mitsudo, K.; Aoki, I.; Taniguchi, H.; Tohnai, I. M2-polarized macrophages contribute to neovasculogenesis, leading to relapse of oral cancer following radiation. Sci. Rep. 2016, 6, 27548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschendorff, A.E.; Miremadi, A.; Pinder, S.E.; Ellis, I.O.; Caldas, C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007, 8, R157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhani, S.R.; Jacquemier, J.; Sloane, J.P.; Gusterson, B.A.; Anderson, T.J.; van de Vijver, M.J.; Farid, L.M.; Venter, D.; Antoniou, A.; Storfer-Isser, A.; et al. Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J. Natl. Cancer Inst. 1998, 90, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Kreike, B.; van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; van de Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, R65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jézéquel, P.; Loussouarn, D.; Guérin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verrièle, V.; et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: Importance of immune response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Haddock, P.; De Matos Simoes, R.; Parkes, E.; Irwin, G.; Emmert-Streib, F.; McQuaid, S.; Kennedy, R.; Mullan, P. A BRCA1 deficient, NFκB driven immune signal predicts good outcome in triple negative breast cancer. Oncotarget 2016, 7, 19884–19896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.C.; Zacksenhouse, M.; Eisen, A.; Nofech-Mozes, S.; Zacksenhaus, E. Identification of cell proliferation, immune response and cell migration as critical pathways in a prognostic signature for HER2+:ERα- breast cancer. PLoS ONE 2017, 12, e0179223. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Kong, D.; Chen, Q.; Ping, Y.; Pang, D. Oncogenic long noncoding RNA landscape in breast cancer. Mol. Cancer 2017, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Foukakis, T.; Lövrot, J.; Matikas, A.; Zerdes, I.; Lorent, J.; Tobin, N.; Suzuki, C.; Brage, S.E.; Carlsson, L.; Einbeigi, Z. Immune gene expression and response to chemotherapy in advanced breast cancer. Br. J. Cancer 2018, 118, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Orthmann, A.; Peiker, L.; Fichtner, I.; Hoffmann, A.; Hilger, R.A.; Zeisig, R. Improved Treatment of MT-3 Breast Cancer and Brain Metastases in a Mouse Xenograft by LRP-Targeted Oxaliplatin Liposomes. J. Biomed. Nanotechnol. 2016, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, A.; Aroldi, F.; Bertocchi, P.; Prochilo, T.; Mutti, S.; Savelli, G.; Fraccon, A.P.; Zaniboni, A. GEMOX: An Active Regimen for the Treatment of Luminal and Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. Chemotherapy 2017, 62, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.; Wang, Z.; Hu, X.; Wang, B.; Cao, J.; Lv, F.; Zhen, C.; Zhang, S.; Shao, Z. A phase II trial of biweekly vinorelbine and oxaliplatin in second- or third-line metastatic triple-negative breast cancer. Cancer Biol. Ther. 2015, 16, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xiao, Y.; Wei, W.; Guo, J.X.; Liu, Y.C.; Huang, X.H.; Zhang, R.X.; Wu, Y.J.; Zhou, J. Clinical efficacy of administering oxaliplatin combined with S-1 in the treatment of advanced triple-negative breast cancer. Exp. Ther. Med. 2015, 10, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| IR Factor or Mediator | Mechanism | Result | Perspective | Ref. |
|---|---|---|---|---|
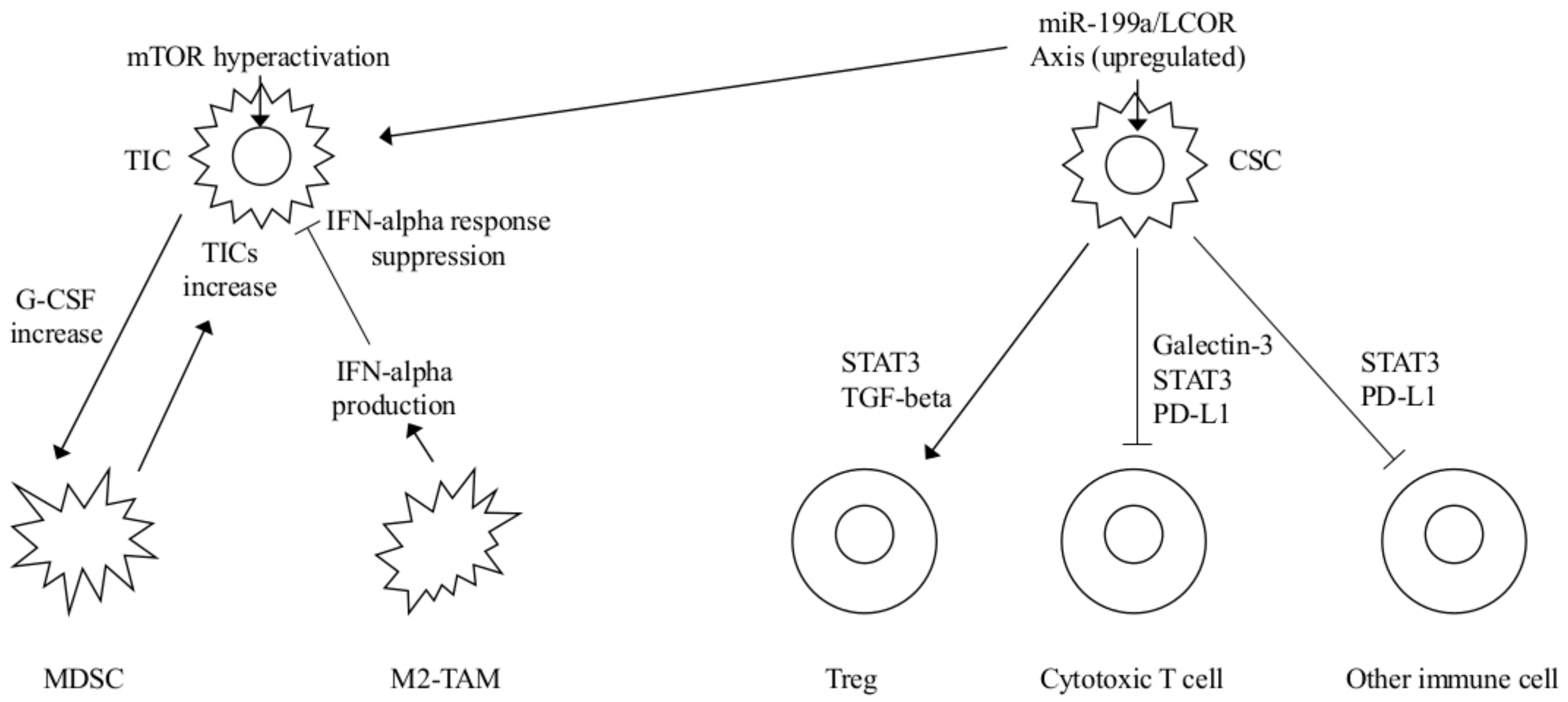
| MDSCs | Hyperactivated Akt-mTOR pathway G-CSF increased expression MDSC mediated Notch stemness-related genes upregulation | TICs mediated MDSCs accumulation Increased TICs frequency | mTOR plus checkpoint inhibitors FGFR or G-CSF inhibitors | [5] |
| INF-α | miR-199a overexpression LCOR repression and modulation of the INF-α mediated suppressive effects | CSCs protected by INF-mediated effects MaSC-enriched basal vs luminal population | INF-α plus miR-199-LCOR targeting as adjuvant therapy | [15] |
| PD-L1 | N192, N200, N219 glycosylation induces PD-L1 stability and antagonizes PD-L1 GSK3-β interactions as well as EGF and other EGFR ligands | Immunesuppression | Targeting PD-L1 stabilization | [18] |
| CD47 | TNF-NF-κB mediated CD47 upregulation by SEs CD47 SIRP α binding on macrophages | Cancer cells protection from phagocytosis | Increased macrophage phagocytosis by TNF-NF-κB inhibition | [19] |
| Y239/Y240-Shc-A phosphory-lation | Antitumor STAT-1 activity decrease STAT-3 mediated immune suppression increase | Immunesuppression | Constitutive binding or specific Y239/Y240-Shc-A inhibitors to sensitize to immunotherapies | [25] |
| IR Factor or Mediator | Mechanism | Result | Perspective | Ref. |
|---|---|---|---|---|
| CCR7 | Membrane CCR7-CCL19/CCL21 interaction; No cytoplasmic CCR7-CCL19/CCL21 interaction | Treg and macrophage attraction to the microenvironment; inversely associated with CD3+ cells in the stroma | Better evaluation of CCR7 role in membrane and cytoplasm | [31] |
| Annexin-1 | FPR2-ERK-NF-κB pathway activation, M2 phenotype macrophages polarization | Angiogenesis, tumor progression, immune suppression | Targeting FPR2-ERK signaling | [41] |
| NR4A1 | NR4A1 hyperexpression T-βRI activation, SMAD 2/3 phosphorylation, intense SMAD signaling | EMT and cell migration, poor prognosis | Targeting TGF-β and NR4A1 | [49] |
| Immune Genes/s or IR Mediator | Mechanism | Result | Perspective | Ref. |
|---|---|---|---|---|
| TNBC with BRCA1 dysfunction | “NF-κB on” signal, M1-type macrophages microenvironment, and CD8+ infiltration | Better outcome | Checkpoint inhibitors in addition to conventional FEC CT | [56] |
| 17-gene HTICs signature | IR, proliferation and migration as critical biological pathways | Worse prognosis and benefit from trastuzumab in HER2+ ER- BC | More appropriate adjuvant therapy in HER2+ ER-BC | [57] |
| LncRNAs | Regulation of the immune system activation by 30 hyper- and 25 hypo- expressed Lnc RNAs | Tumor progression and worse survival | Prognosis and complementation of conventional parameters in specific subtypes | [58] |
| Immune module SCORE | Activated immune microenvironment | Prediction of response to CT in ER+ and Luminal BCs | Better patient selection and design of combined chemo-immunotherapies | [59] |
| Immune suppressive plasma cells expressing IGA, IL-10 and PDL-1 | Inhibition of oxaliplatin tumor directed CTL activation and ICD | Poor response to oxaliplatin | Inhibition of IGA+ plasmocytes in oxaliplatin treated patients | [65] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolini, A.; Ferrari, P.; Diodati, L.; Carpi, A. Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management. Int. J. Mol. Sci. 2018, 19, 2733. https://doi.org/10.3390/ijms19092733
Nicolini A, Ferrari P, Diodati L, Carpi A. Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management. International Journal of Molecular Sciences. 2018; 19(9):2733. https://doi.org/10.3390/ijms19092733
Chicago/Turabian StyleNicolini, Andrea, Paola Ferrari, Lucrezia Diodati, and Angelo Carpi. 2018. "Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management" International Journal of Molecular Sciences 19, no. 9: 2733. https://doi.org/10.3390/ijms19092733
APA StyleNicolini, A., Ferrari, P., Diodati, L., & Carpi, A. (2018). Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management. International Journal of Molecular Sciences, 19(9), 2733. https://doi.org/10.3390/ijms19092733