The Effects of Proteasome Inhibitors on Telomerase Activity and Regulation in Multiple Myeloma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
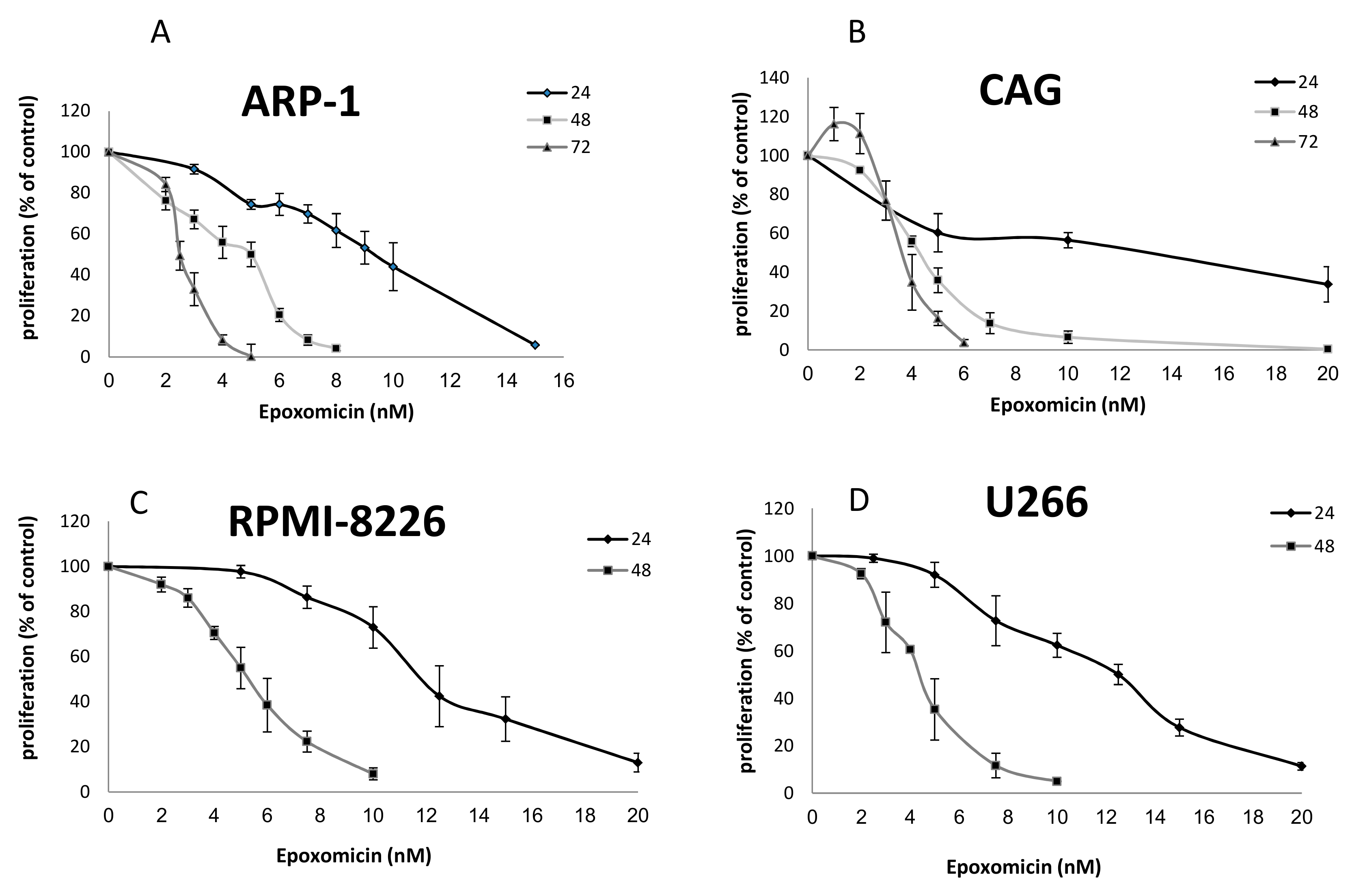
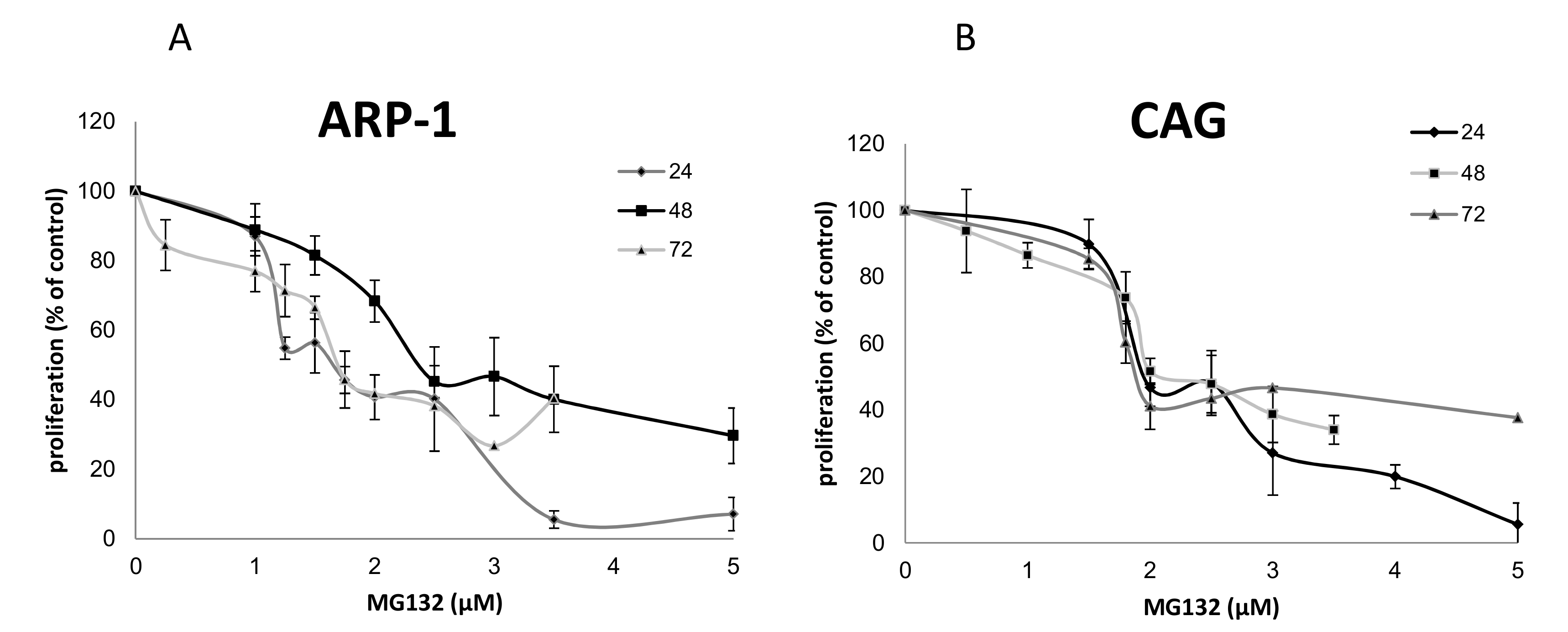
2.1. The Effects of Epoxomicin and MG-132 and on the Proliferation of MM Cells
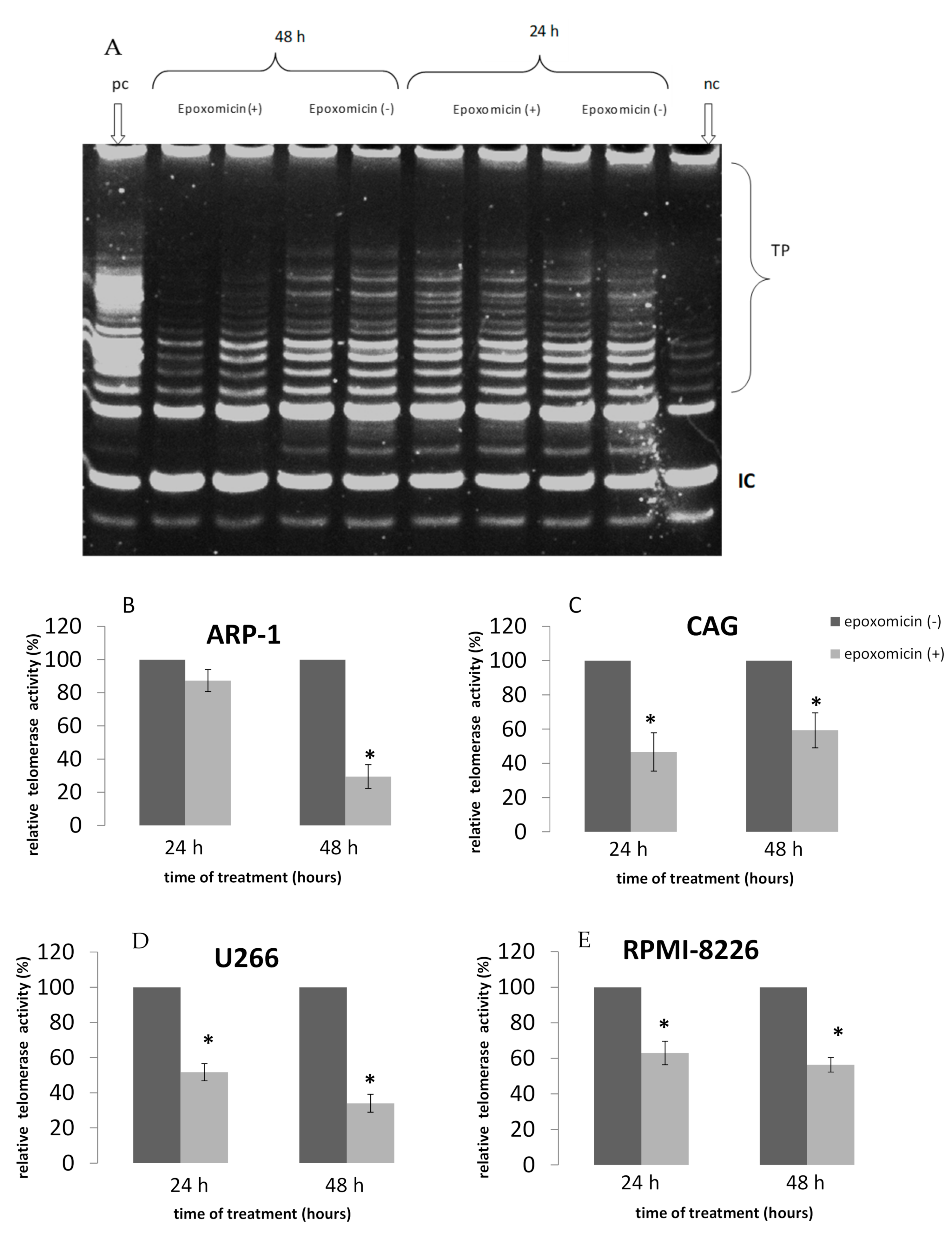
2.2. TA Following Treatment of MM Cell Lines by Epoxomicin
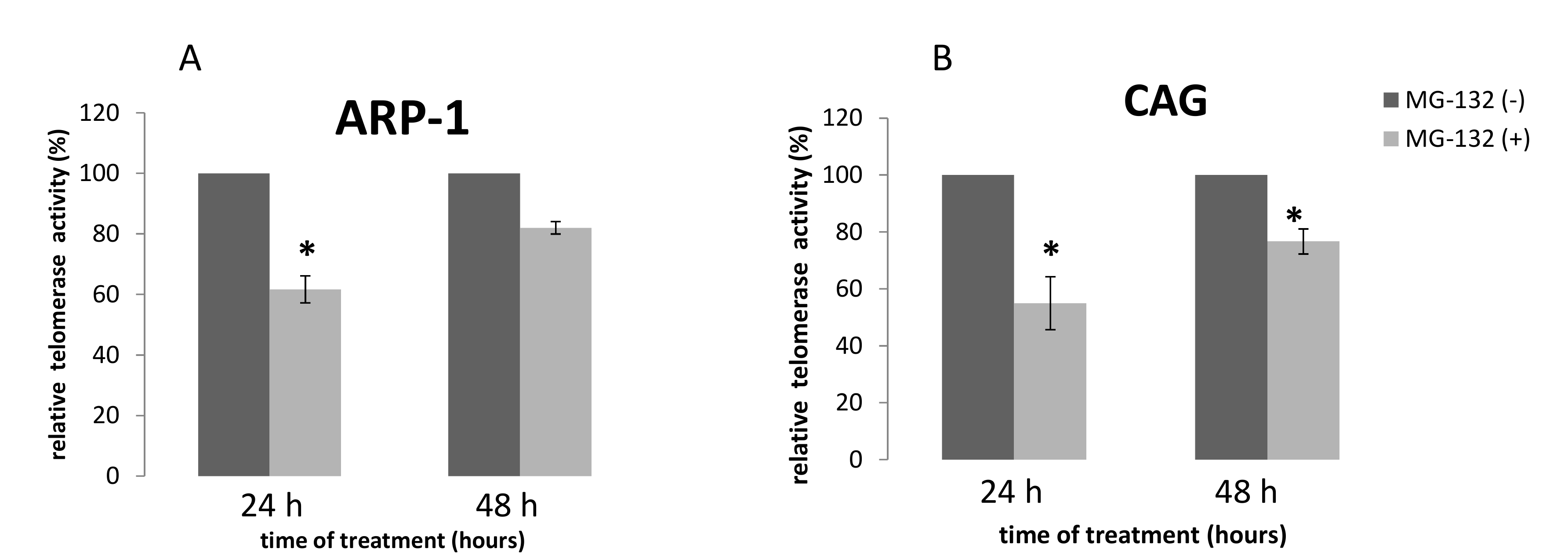
2.3. TA Following Treatment of MM Cell Lines by MG-132
2.4. The Effect of Epoxomicin on Telomerase Regulation
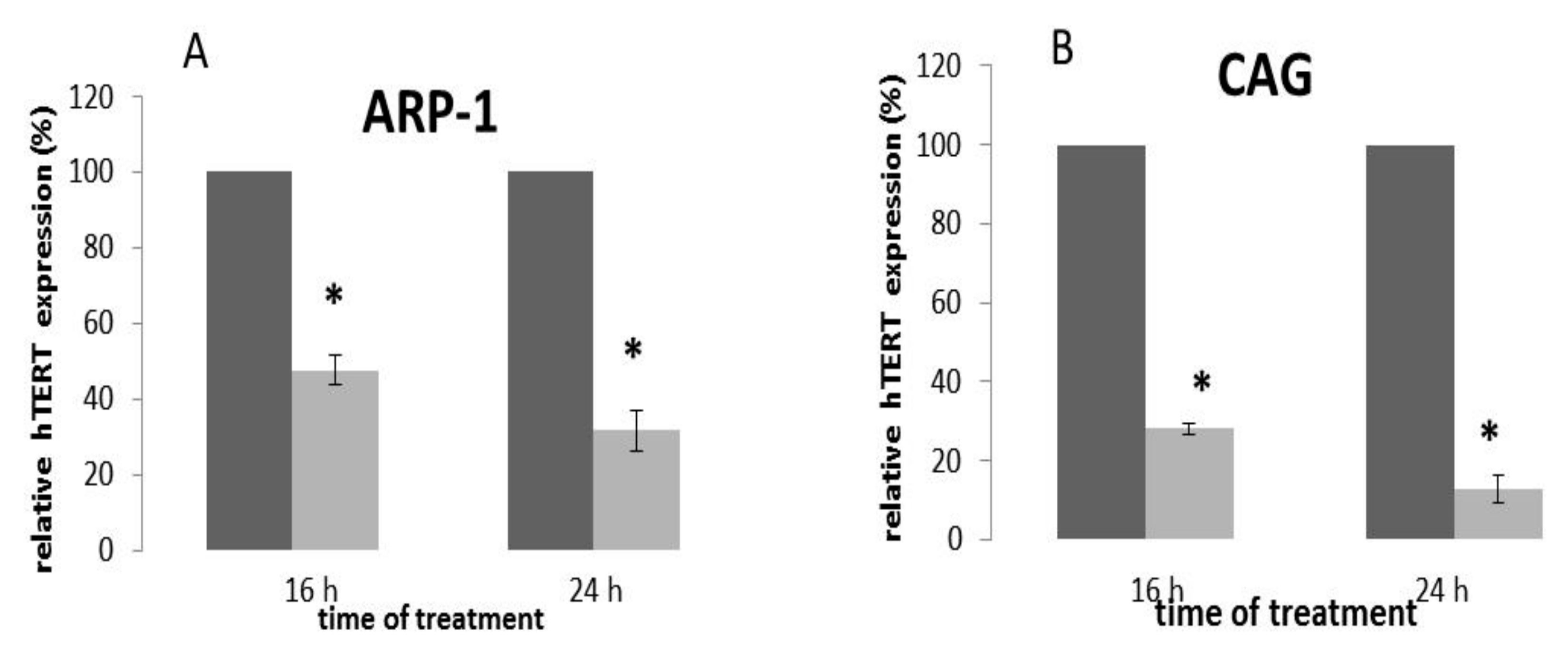
2.5. The Effect of Epoxomicin on the Transcription of the hTERT Gene
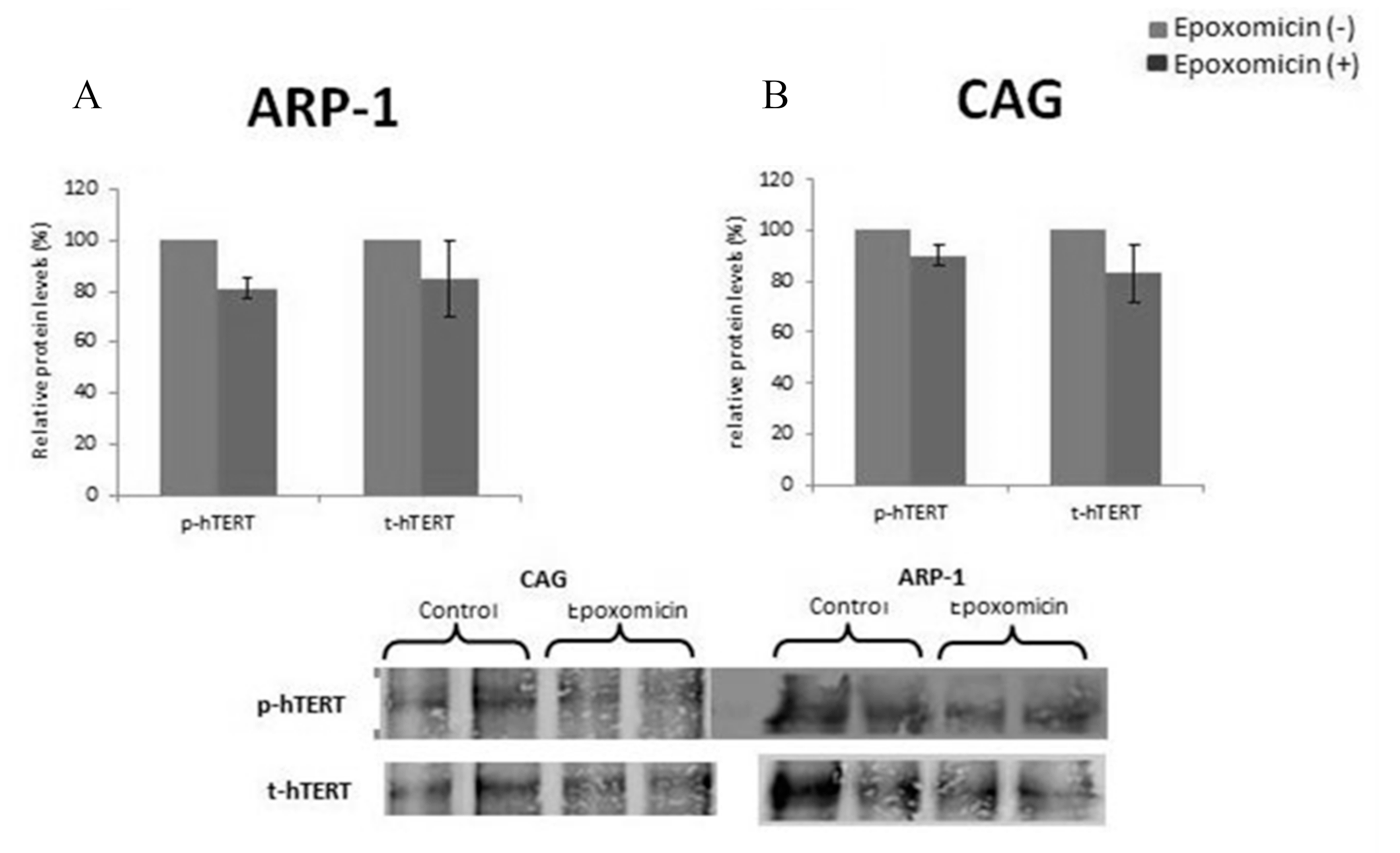
2.6. The Effect of Epoxomicin on the Post-Translational Modification of Telomerase
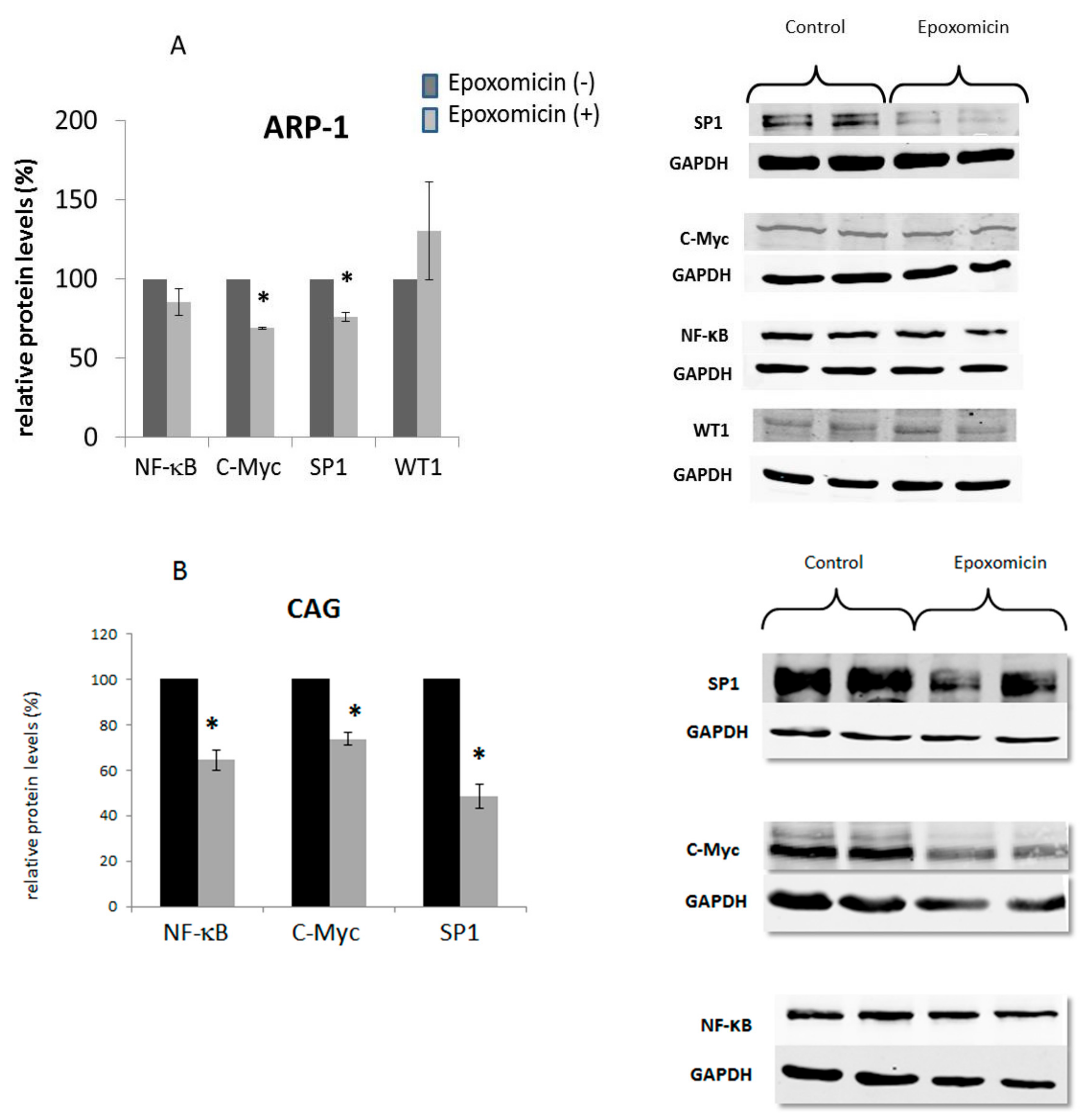
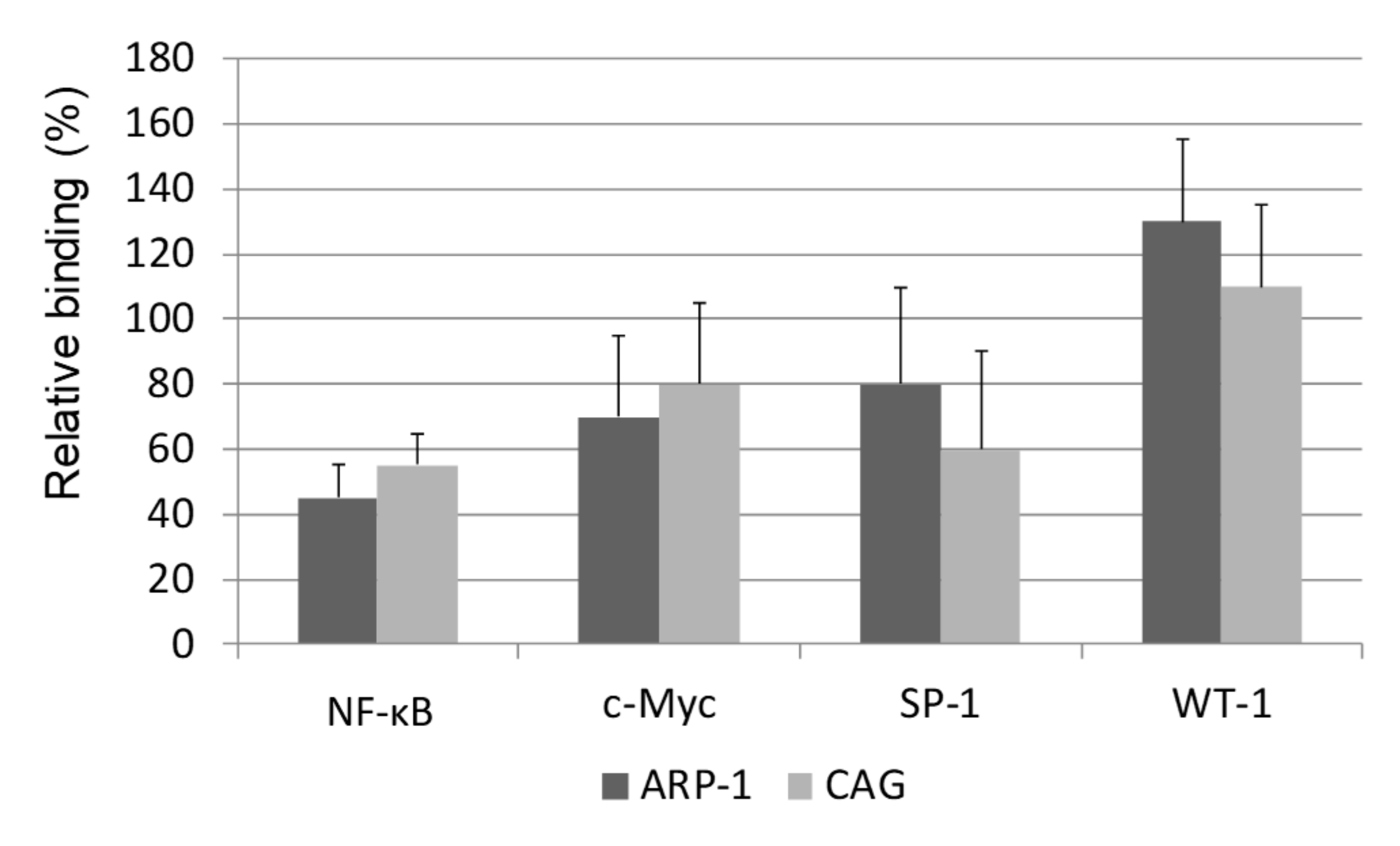
2.7. The Effect of Epoxomicin on the Cellular Levels and Promoter Binding of Several Transcription Factors of the Core Promoter of Telomerase
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability and Proliferation Assays
4.3. Telomerase Activity Assay
4.4. Real-Time PCR
4.5. Chromatin Immunoprecipitation Assay
| For the region of c-Myc and SP-1 binding site: |
| Forward primer: AGTGGATTCGCGGGCACAGA; |
| Reverse primer: TTCCCACGTGCGCAGCAGGA |
| For the region of NF-κB binding site: |
| Forward primer: GCCTCCTAGCTCTGCAGT |
| Reverse primer: ACCCGAGGACGCATTGCT |
| For the binding site of WT1: |
| Forward primer: TTTGCCCTAGTGGCAGAGAC |
| Reverse primer: GCCGGAGGAAATTG |
4.6. Immunoprecipitation and Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef]
- Raab, M.S.; Podar, K.; Breitkreutz, I.; Richardson, P.G.; Anderson, K.C. Multiple myeloma. Lancet 2009, 374, 324–339. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Adams, J.; Palombella, V.J.; Elliott, P.J. Proteasome inhibition: A new strategy in cancer treatment. Invest New Drugs 2000, 18, 109–121. [Google Scholar] [CrossRef]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994, 78, 773–785. [Google Scholar]
- Read, M.A.; Neish, A.S.; Luscinskas, F.W.; Palombella, V.J.; Maniatis, T.; Collins, T. The proteasome pathway is required for cytokine-induced endothelial-leukocyte adhesion molecule expression. Immunity 1995, 2, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Delic, J.; Masdehors, P.; Omura, S.; Cosset, J.M.; Dumont, J.; Binet, J.L.; Magdelénat, H. The proteasome inhibitor lactacystin induces apoptosis and sensitizes chemo- and radioresistant human chronic lymphocytic leukaemia lymphocytes to TNF-alpha-initiated apoptosis. Br. J. Cancer. 1998, 77, 1103–1107. [Google Scholar] [CrossRef]
- Almond, J.B.; Cohen, G.M. The proteasome: A novel target for cancer chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar] [CrossRef]
- Adams, J.; Behnke, M.; Chen, S.; Cruickshank, A.A.; Dick, L.R.; Grenier, L.; Klunder, J.M.; Ma, Y.T.; Plamondon, L.; Stein, R.L. Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998, 8, 333–338. [Google Scholar] [CrossRef]
- Aghajanian, C.; Soignet, S.; Dizon, D.S.; Pien, C.S.; Adams, J.; Elliott, P.J.; Sabbatini, P.; Miller, V.; Hensley, M.L.; Pezzulli, S.; et al. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin. Cancer Res. 2002, 8, 2505–2511. [Google Scholar]
- Orlowski, R.Z.; Stinchcombe, T.E.; Mitchell, B.S.; Shea, T.C.; Baldwin, A.S.; Stahl, S.; Adams, J.; Esseltine, D.L.; Elliott, P.J.; Pien, C.S.; et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin. Oncol. 2002, 20, 4420–4427. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Mohan, R.; Kwok, B.H.; Elofsson, M.; Sin, N.; Crews, C.M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA 1999, 96, 10403–10408. [Google Scholar] [CrossRef] [PubMed]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trentin, L.; Ballon, G.; Ometto, L.; Perin, A.; Basso, U.; Chieco-Bianchi, L.; Semenzato, G.; De Rossi, A. Telomerase activity in chronic lymphoproliferative disorders of B-cell lineage. Br. J. Haematol. 1999, 106, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. Tracking telomerase. Cell 2004, 116, 83–86. [Google Scholar] [CrossRef]
- Shay, J.W.; Zou, Y.; Hiyama, E.; Wright, W.E. Telomerase and cancer. Hum. Mol. Genet. 2001, 10, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Mor-Tzuntz, R.; Uziel, O.; Shpilberg, O.; Lahav, J.; Raanani, P.; Bakhanashvili, M.; Rabizadeh, E.; Zimra, Y.; Lahav, M.; Granot, G. Effect of imatinib on the signal transduction cascade regulating telomerase activity in K562 (BCR-ABL-positive) cells sensitive and resistant to imatinib. Exp. Hematol. 2010, 38, 27–37. [Google Scholar] [CrossRef]
- Uziel, O.; Fenig, E.; Nordenberg, J.; Beery, E.; Reshef, H.; Sandbank, J.; Birenbaum, M.; Bakhanashvili, M.; Yerushalmi, R.; Luria, D.; et al. Imatinib mesylate (Gleevec) downregulates telomerase activity and inhibits proliferation in telomerase-expressing cell lines. Br. J. Cancer 2005, 92, 10. [Google Scholar] [CrossRef]
- Dong, X.; Liu, A.; Zer, C.; Feng, J.; Zhen, Z.; Yang, M.; Zhong, L. siRNA inhibition of telomerase enhances the anti-cancer effect of doxorubicin in breast cancer cells. BMC Cancer 2009, 9, 133. [Google Scholar] [CrossRef]
- Weiss, C.; Uziel, O.; Wolach, O.; Nordenberg, J.; Beery, E.; Bulvick, S.; Kanfer, G.; Cohen, O.; Ram, R.; Bakhanashvili, M.; et al. Differential downregulation of telomerase activity by bortezomib in multiple myeloma cells-multiple regulatory pathways in vitro and ex vivo. Br. J. Cancer 2012, 107, 1844–1852. [Google Scholar] [CrossRef]
- Uziel, O.; Cohen, O.; Beery, E.; Nordenberg, J.; Lahav, M. The effect of Bortezomib and Rapamycin on Telomerase Activity in Mantle Cell Lymphoma. Transl Oncol. 2014, 7, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Shalem-Cohavi, S. The effect of Proteosome Inhibitors on Telomerase Regulation in Multiple Myeloma Cell Lines. Master’s Thesis, Tel Aviv University, Tel Aviv, Israel, 31 July 2012. [Google Scholar]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar]
- Cong, Y.S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 3, 407–425. [Google Scholar] [CrossRef]
- Suske, G. The Sp-family of transcription factors. Gene 1999, 238, 291–300. [Google Scholar] [CrossRef]
- Kyo, S.; Takakura, M.; Taira, T.; Kanaya, T.; Itoh, H.; Yutsudo, M.; Ariga, H.; Inoue, M. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000, 28, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, P.; Hastie, N.D. The many facets of the Wilms’ tumour gene, WT1. Hum. Mol. Genet. 2006, 15, R196–R201. [Google Scholar] [CrossRef]
- Ueda, T.; Oji, Y.; Naka, N.; Nakano, Y.; Takahashi, E.; Koga, S.; Asada, M.; Ikeba, A.; Nakatsuka, S.; Abeno, S.; et al. Overexpression of the Wilms’tumor gene WT1 in human bone and soft-tissue sarcomas. Cancer Sci. 2003, 94, 271–276. [Google Scholar] [CrossRef]
- Sitaram, R.T.; Degerman, S.; Ljungberg, B.; Andersson, E.; Oji, Y.; Sugiyama, H.; Roos, G.; Li, A. Wilms’ tumour 1 can suppress hTERT gene expression and telomerase activity in clear cell renal cell carcinoma via multiple pathways. Br. J. Cancer 2010, 103, 1255–1262. [Google Scholar] [CrossRef]
- Das, P.M.; Ramachandran, K.; van Wert, J.; Singal, R. Chromatin immunoprecipitation assay. Biotechniques 2004, 37, 961–969. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalem-Cohavi, N.; Beery, E.; Nordenberg, J.; Rozovski, U.; Raanani, P.; Lahav, M.; Uziel, O. The Effects of Proteasome Inhibitors on Telomerase Activity and Regulation in Multiple Myeloma Cells. Int. J. Mol. Sci. 2019, 20, 2509. https://doi.org/10.3390/ijms20102509
Shalem-Cohavi N, Beery E, Nordenberg J, Rozovski U, Raanani P, Lahav M, Uziel O. The Effects of Proteasome Inhibitors on Telomerase Activity and Regulation in Multiple Myeloma Cells. International Journal of Molecular Sciences. 2019; 20(10):2509. https://doi.org/10.3390/ijms20102509
Chicago/Turabian StyleShalem-Cohavi, Naama, Einat Beery, Jardena Nordenberg, Uri Rozovski, Pia Raanani, Meir Lahav, and Orit Uziel. 2019. "The Effects of Proteasome Inhibitors on Telomerase Activity and Regulation in Multiple Myeloma Cells" International Journal of Molecular Sciences 20, no. 10: 2509. https://doi.org/10.3390/ijms20102509
APA StyleShalem-Cohavi, N., Beery, E., Nordenberg, J., Rozovski, U., Raanani, P., Lahav, M., & Uziel, O. (2019). The Effects of Proteasome Inhibitors on Telomerase Activity and Regulation in Multiple Myeloma Cells. International Journal of Molecular Sciences, 20(10), 2509. https://doi.org/10.3390/ijms20102509