Telomerase Inhibitor TMPyP4 Alters Adhesion and Migration of Breast-Cancer Cells MCF7 and MDA-MB-231

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
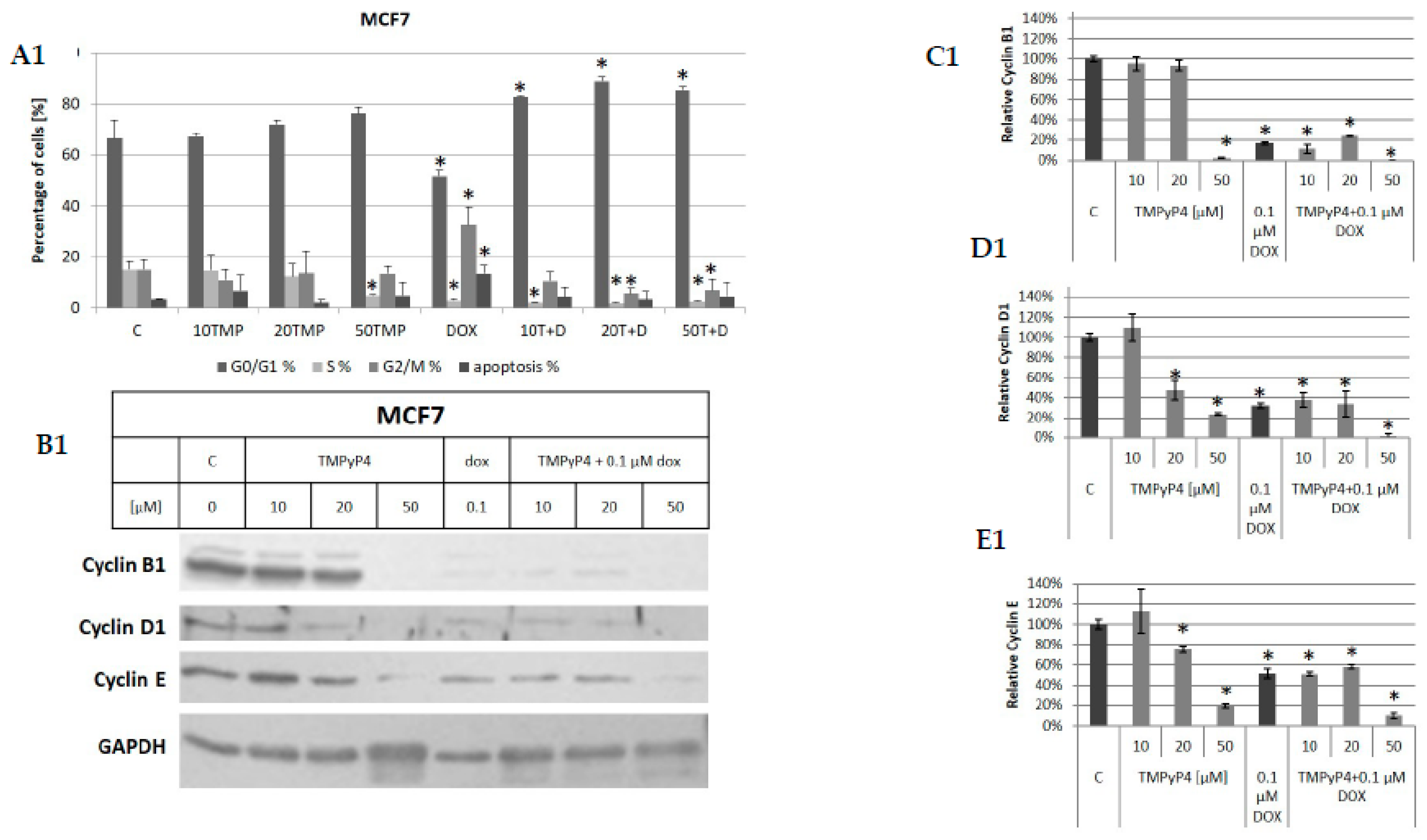
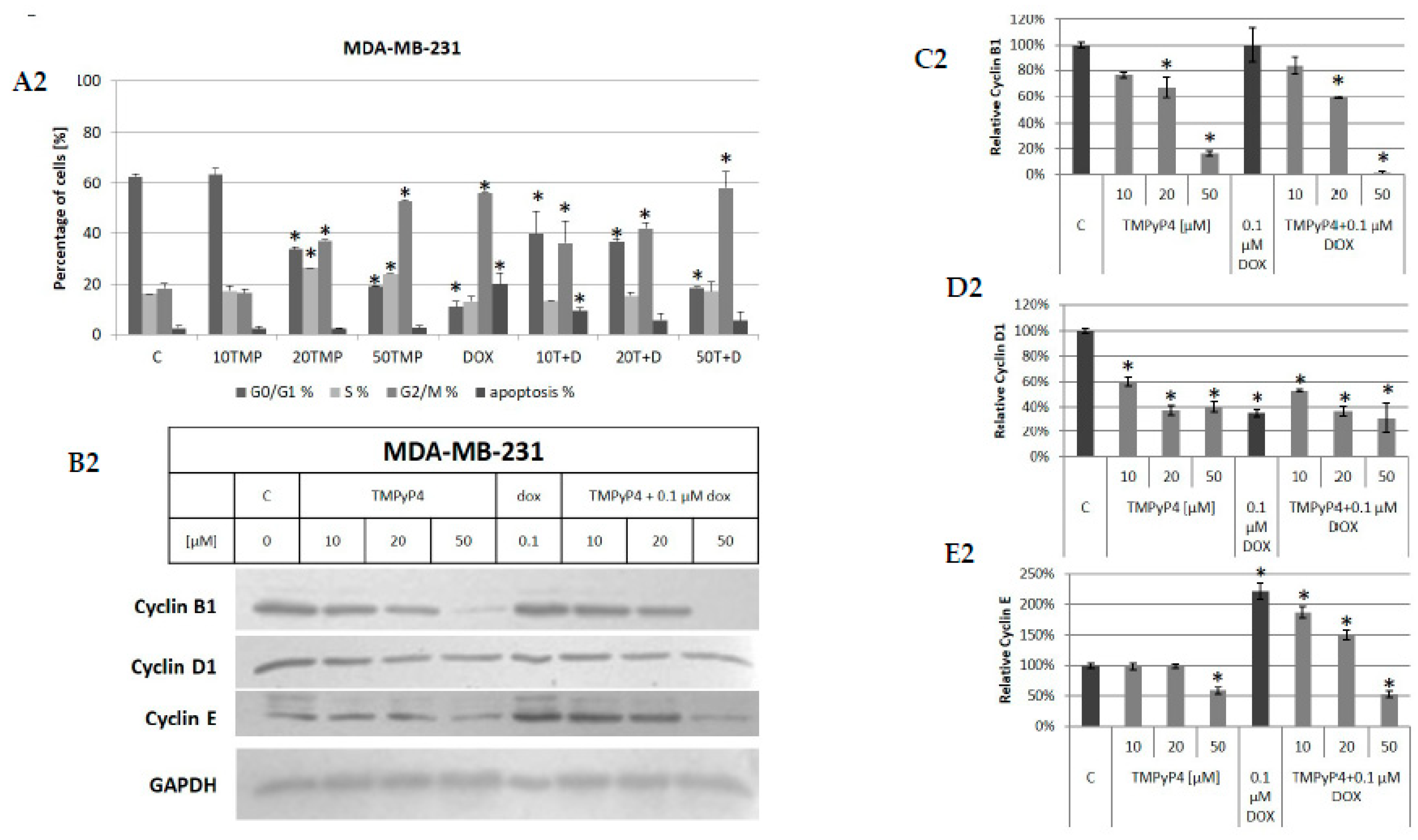
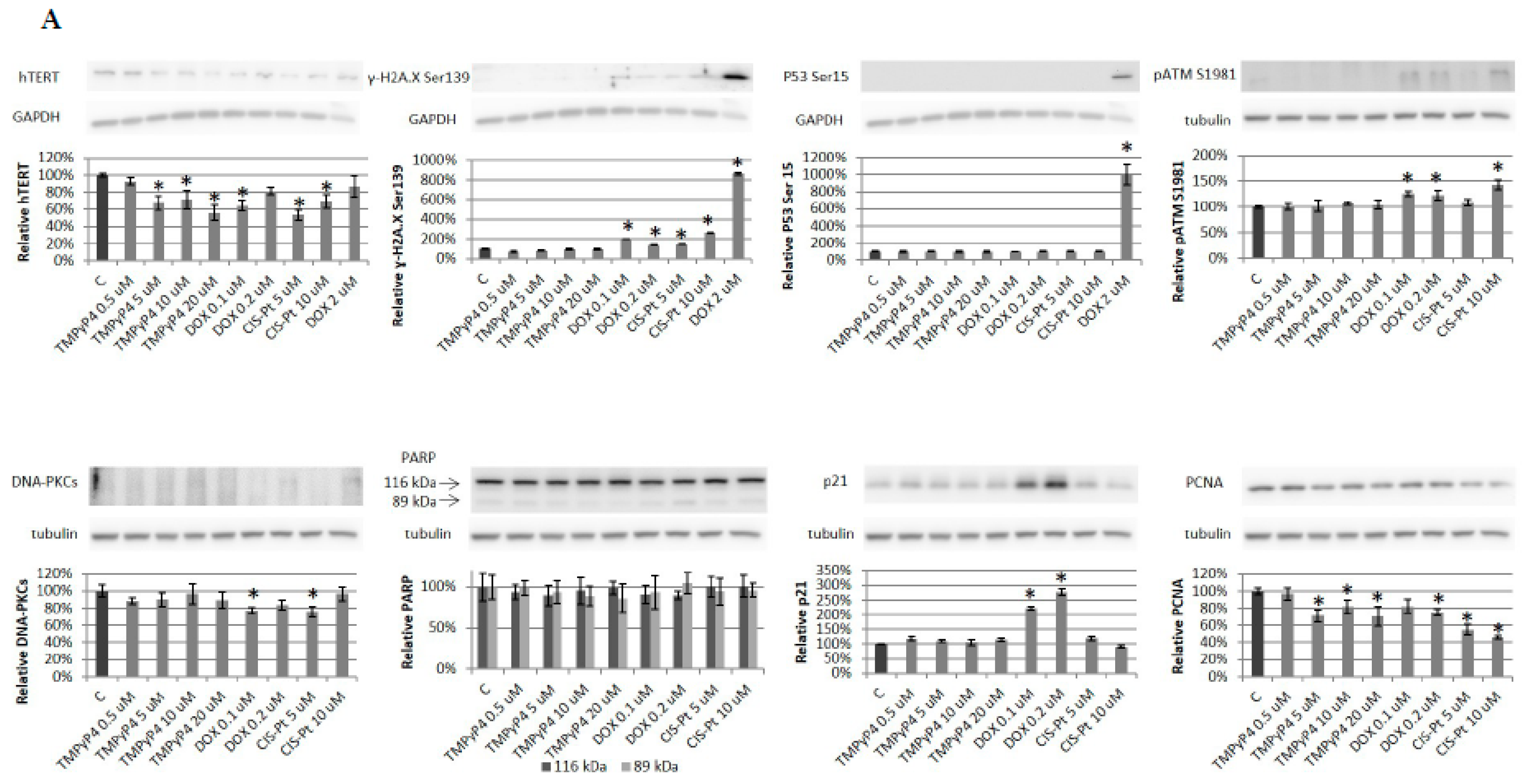
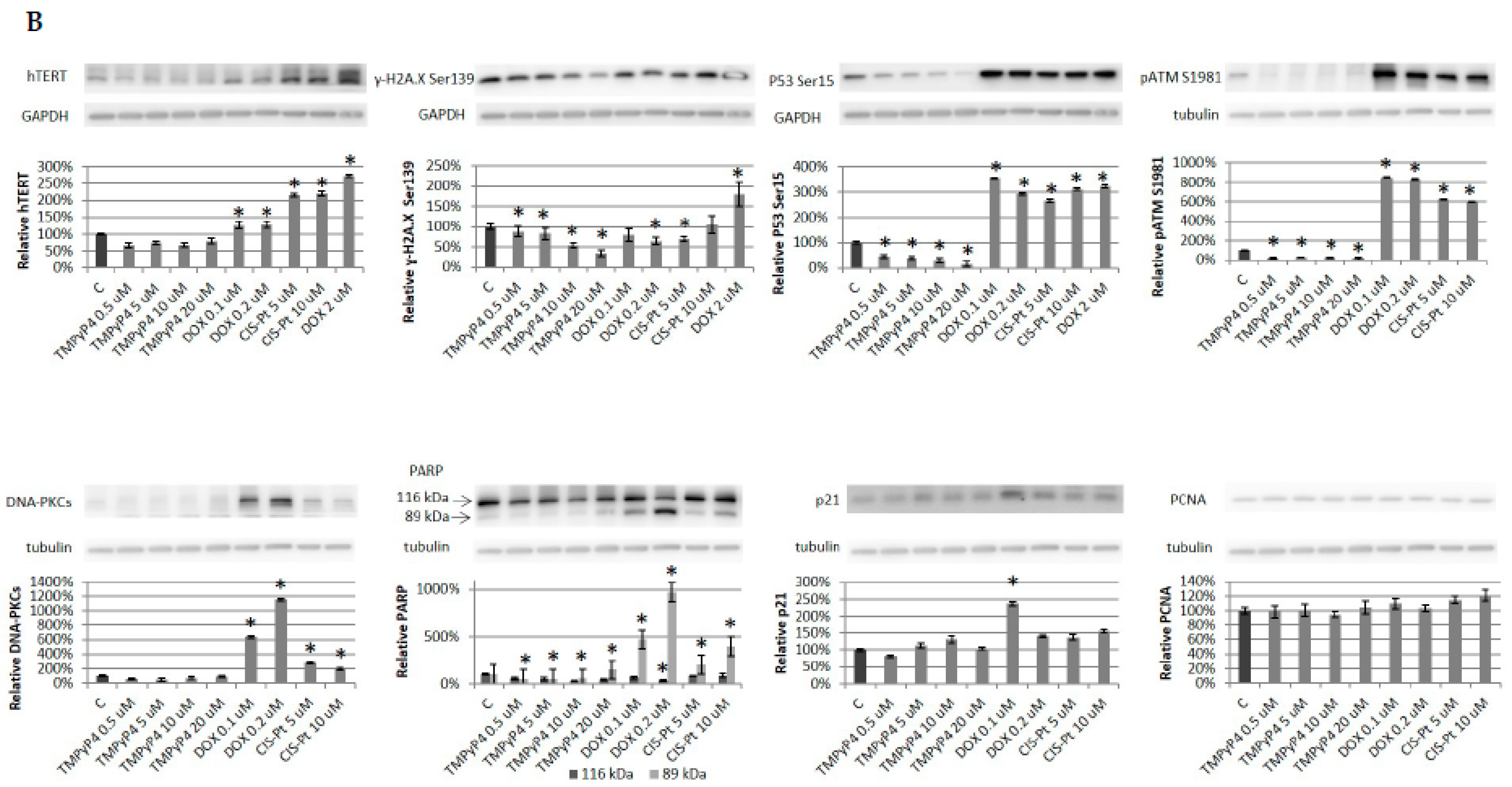
2. Results
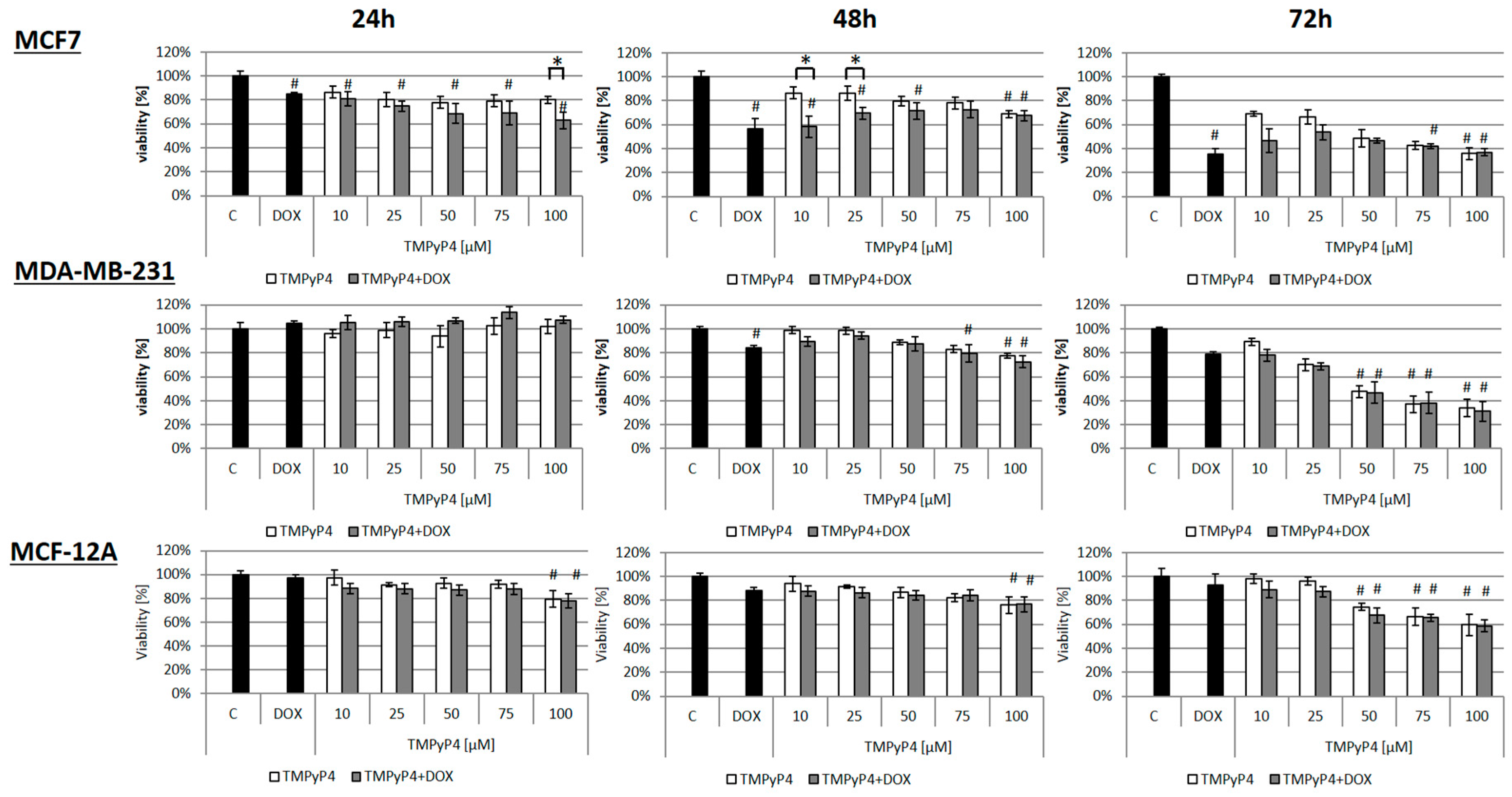
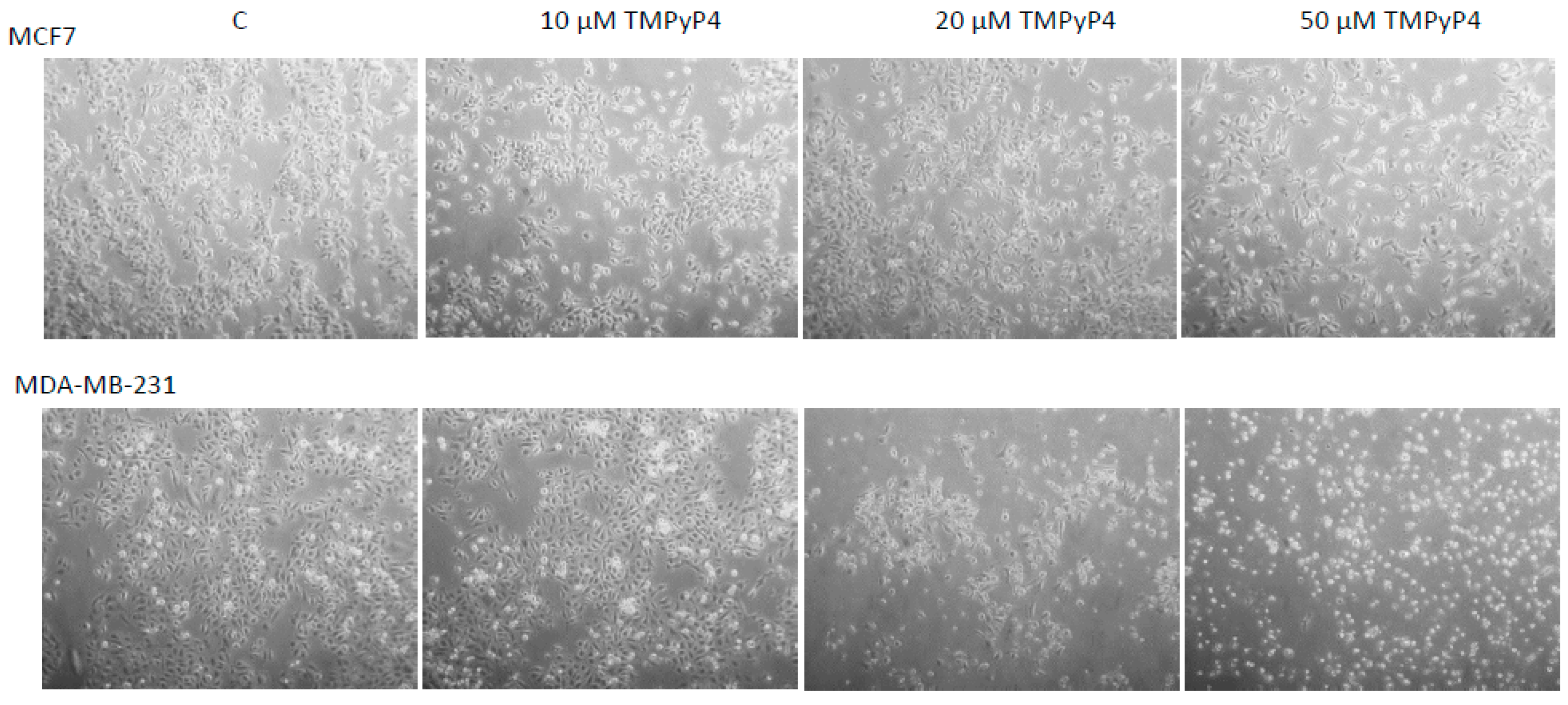
2.1. Cellular Toxicity of TMPyP4
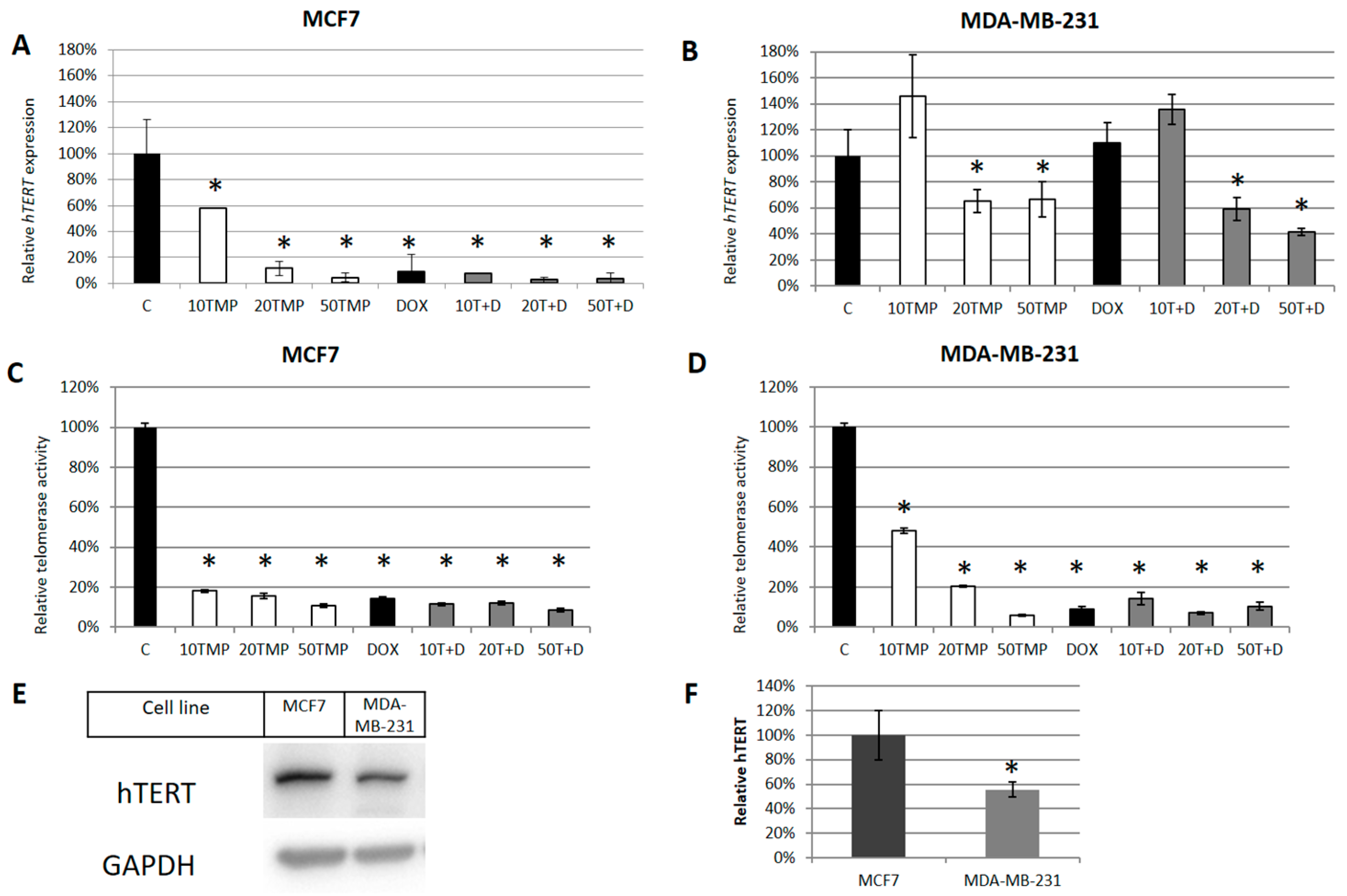
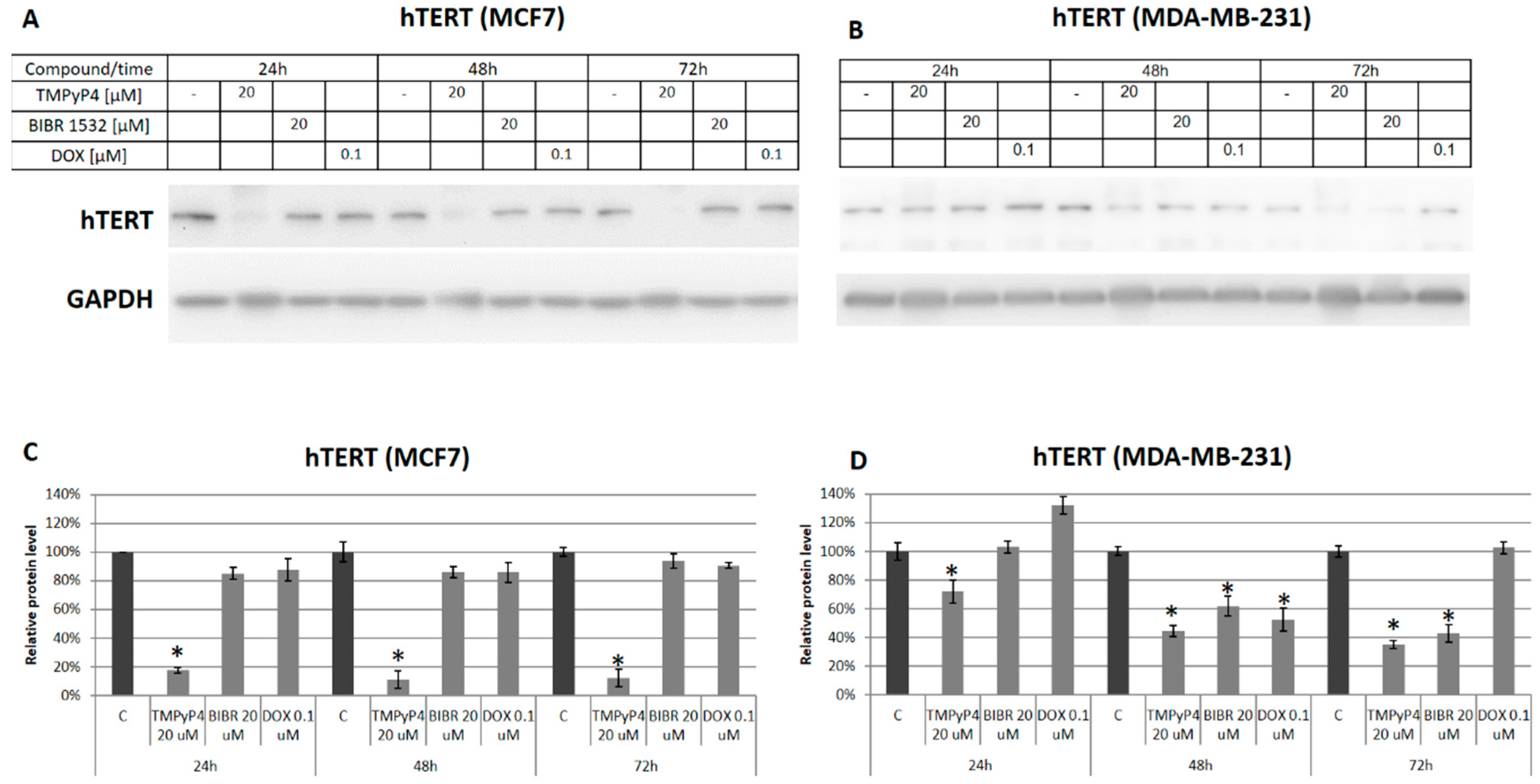
2.2. TMPyP4 Alters Telomerase Expression and Activity
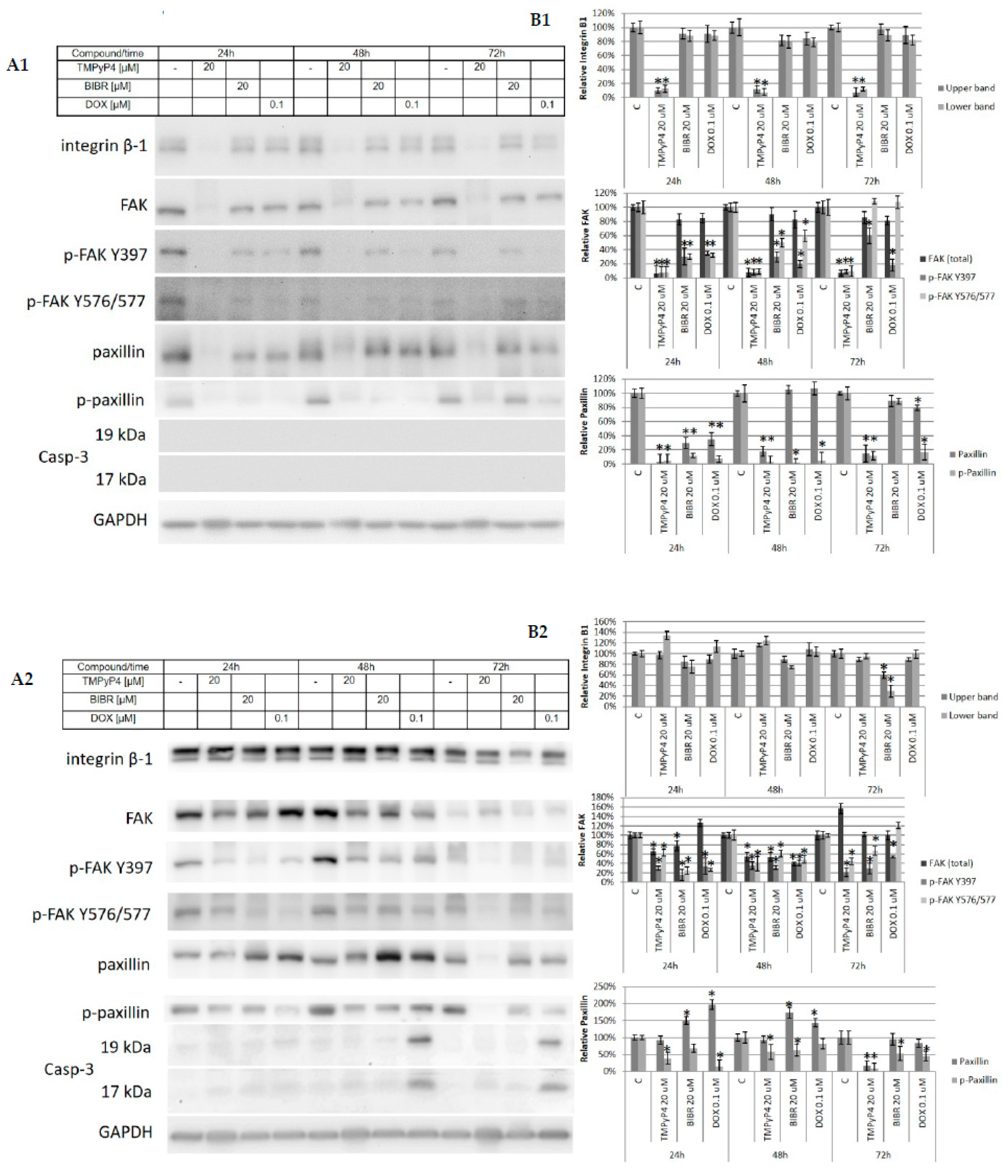
2.3. TMPyP4—Mechanism of Action
2.4. Effect of TMPyP4 on DNA Damage and Repair Signaling
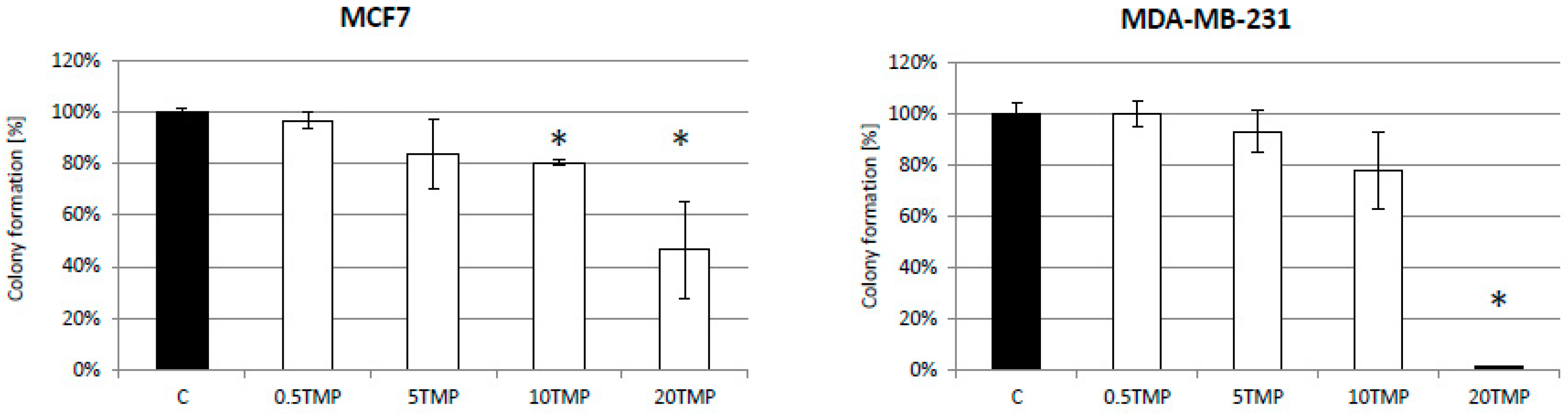
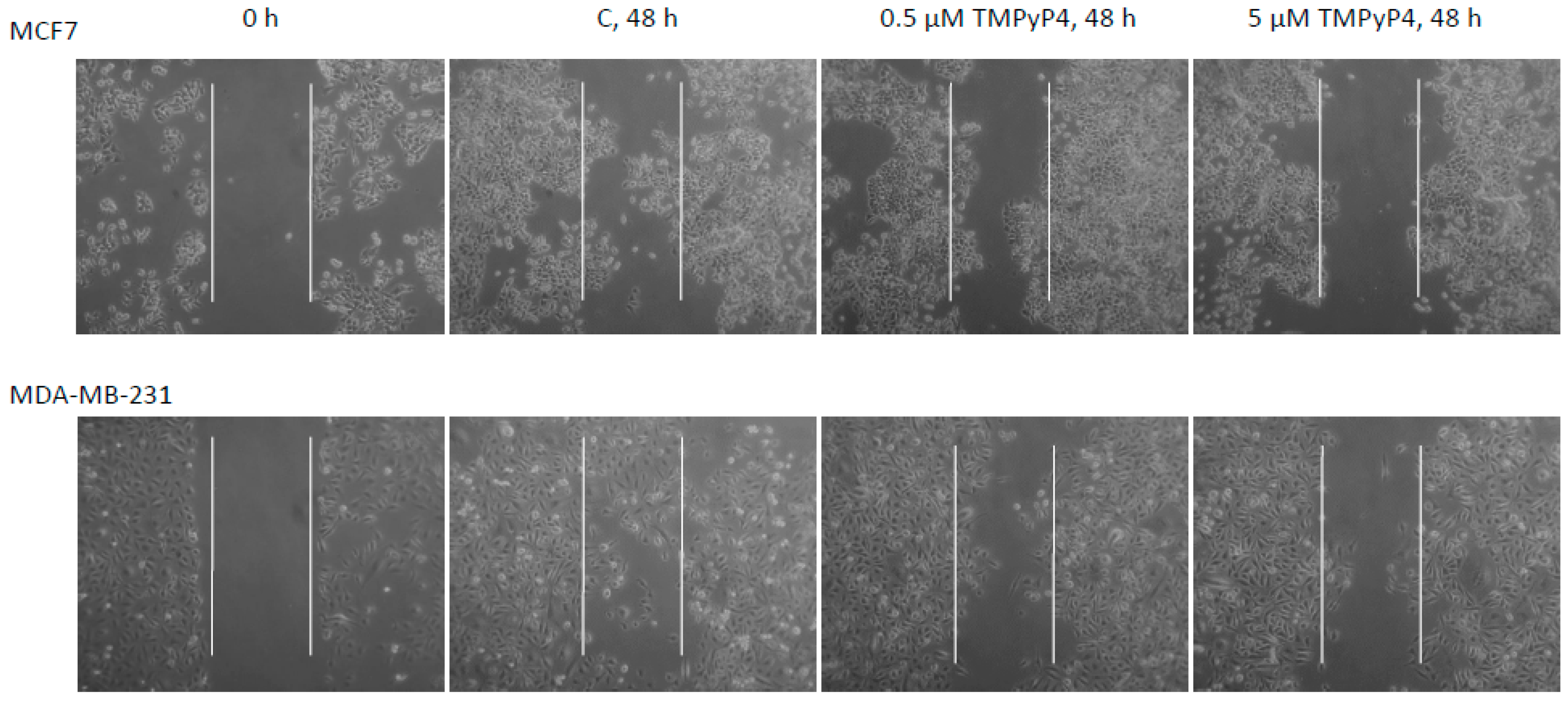
2.5. TMPyP4 Affects Adhesion and Migration Processes
3. Discussion
3.1. TMPyP4 as a Tool in Targeting Telomerase in Breast-Cancer Therapy
3.2. Contribution of TMPyP4 to Cell-Cycle Modulation
3.3. TMPyP4 in DNA Damage/Repair Pathway
3.4. Contribution of TMPyP4 to Functional Impairment of Migration and Adhesion
4. Materials and Methods
4.1. Cell Lines and Study Compounds
4.2. Cytotoxicity Assay
4.3. Quantitative Assessment of hTERT Expression
4.4. Telomerase Activity
4.5. Western Blot Analysis
4.6. Colony Formation Assay
4.7. Cell-Cycle Analysis
4.8. Scratch Assay
4.9. Cell Adhesion Assessment
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hahn, W.C.; Meyerson, M. Telomerase activation, cellular immortalization and cancer. Ann. Med. 2001, 33, 123–129. [Google Scholar] [CrossRef]
- Romaniuk, A.; Paszel-Jaworska, A.; Totoń, E.; Lisiak, N.; Hołysz, H.; Królak, A.; Grodecka-Gazdecka, S.; Rubiś, B. The non-canonical functions of telomerase: To turn off or not to turn off. Mol. Biol. Rep. 2019, 46, 1401. [Google Scholar] [CrossRef]
- Hornsby, P.J. Cellular aging and cancer. Crit. Rev. Oncol. Hematol. 2011, 79, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubis, B.; Holysz, H.; Gladych, M.; Toton, E.; Paszel, A.; Lisiak, N.; Kaczmarek, M.; Hofmann, J.; Rybczynska, M. Telomerase downregulation induces proapoptotic genes expression and initializes breast cancer cells apoptosis followed by DNA fragmentation in a cell type dependent manner. Mol. Biol. Rep. 2013, 40, 4995–5004. [Google Scholar] [CrossRef] [Green Version]
- Hockemeyer, D.; Sfeir, A.J.; Shay, J.W.; Wright, W.E.; de Lange, T. POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 2005, 24, 2667–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Podell, E.R.; Zaug, A.J.; Yang, Y.; Baciu, P.; Cech, T.R.; Lei, M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 2007, 445, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-Quadruplexes in Gene Promoters: A Novel Anticancer Strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Maziveyi, M.; Alahari, S.K. Cell matrix adhesions in cancer: The proteins that form the glue. Oncotarget 2017, 8, 48471–48487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickreuter, E.; Cordes, N. The cancer cell adhesion resistome: Mechanisms, targeting and translational approaches. Biol. Chem. 2017, 398, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, Y.; Zhao, M.; Wu, C.; Zhang, P.; Chen, X.; Yang, Y.; Liu, G. miRNA-29c Suppresses Lung Cancer Cell Adhesion to Extracellular Matrix and Metastasis by Targeting Integrin β1 and Matrix Metalloproteinase2 (MMP2). PLoS ONE 2013, 8, e70192. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. Mechanisms of Motility in Metastasizing Cells. Mol. Cancer Res. 2010, 8, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Liu, Q.; Ge, Y.; Zhao, Q.; Zheng, X.; Zhao, Y. hTERT promotes cell adhesion and migration independent of telomerase activity. Sci. Rep. 2016, 6, 22886. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.Y.; Yao, Y.G. Mitochondrial dysfunction and nuclear-mitochondrial shuttling of TERT are involved in cell proliferation arrest induced by G-quadruplex ligands. FEBS Lett. 2013, 587, 1656–1662. [Google Scholar] [CrossRef]
- Grand, C.L.; Han, H.; Muñoz, R.M.; Weitman, S.; Von Hoff, D.D.; Hurley, L.H.; Bearss, D.J. The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol. Cancer Ther. 2002, 1, 565–573. [Google Scholar] [PubMed]
- Cheng, M.J.; Cao, Y.G. TMPYP4 exerted antitumor effects in human cervical cancer cells through activation of p38 mitogen-activated protein kinase. Biol. Res. 2017, 50, 24. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhou, Q.; Yang, X. Caspase-3 status is a determinant of the differential responses to genistein between MDA-MB-231 and MCF-7 breast cancer cells. Biochim. Biophys. Acta 2007, 1773, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, T.W.; Naylor, M.J. Breast cancer stem cells. Front. Physiol. 2013, 4, 225. [Google Scholar] [CrossRef] [Green Version]
- Georgoulis, A.; Vorgias, C.E.; Chrousos, G.P.; Rogakou, E.P. Genome Instability and γH2AX. Int. J. Mol. Sci. 2017, 18, 1979. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.H.; Nie, X.; Liu, H.Y.; Fang, Y.M.; Zhao, Y.; Xia, L.X. TMPyP4 promotes cancer cell migration at low doses but induces cell death at high doses. Sci. Rep. 2016, 6, 26592. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef]
- Jäger, K.; Walter, M. Therapeutic Targeting of Telomerase. Genes 2016, 7, 39. [Google Scholar] [CrossRef]
- Romaniuk, A.; Kopczynski, P.; Ksiazek, K.; Rubis, B. Telomerase modulation in therapeutic approach. Curr. Pharm. Des. 2014, 20, 6438–6451. [Google Scholar] [CrossRef]
- Xu, B.; Lefringhouse, J.; Liu, Z.; West, D.; Baldwin, L.A.; Ou, C.; Chen, L.; Napier, D.; Chaiswing, L.; Brewer, L.D.; et al. Inhibition of the integrin/FAK signaling axis and c-Myc synergistically disrupts ovarian cancer malignancy. Oncogenesis 2017, 6, e295. [Google Scholar] [CrossRef]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Shi, Y.; Sun, L.; Chen, G.; Zheng, D.; Li, L.; Wei, W. A combination of the telomerase inhibitor, BIBR1532, and paclitaxel synergistically inhibit cell proliferation in breast cancer cell lines. Target. Oncol. 2015, 10, 565. [Google Scholar] [CrossRef]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef]
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N.; et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008, 4, e10. [Google Scholar] [CrossRef] [PubMed]
- Singhapol, C.; Pal, D.; Czapiewski, R.; Porika, M.; Nelson, G.; Saretzki, G.C. Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS ONE 2013, 8, e52989. [Google Scholar] [CrossRef]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, J.; Matsuo, T.; Shimose, S.; Kubo, T.; Ishikawa, M.; Yasunaga, Y.; Ochi, M. Antitumor effects of telomerase inhibitor TMPyP4 in osteosarcoma cell lines. J. Orthophaedic Res. 2011, 29, 1707–1711. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, O.; Shapira, M.; Hershko, D.D. Differential effects of doxorubicin treatment on cell cycle arrest and Skp2 expression in breast cancer cells. Anticancer. Drugs 2007, 18, 1113–1121. [Google Scholar] [CrossRef]
- Tauchi, T.; Shin-Ya, K.; Sashida, G.; Sumi, M.; Nakajima, A.; Shimamoto, T.; Ohyashiki, J.H.; Ohyashiki, K. Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells: Involvement of ATM-dependent DNA damage response pathways. Oncogene 2003, 22, 5338–5347. [Google Scholar] [CrossRef]
- Rapozzi, V.; Zorzet, S.; Zacchigna, M.; Della Pietra, E.; Cogoi, S.; Xodo, L.E. Anticancer Activity of Cationic Porphyrins in Melanoma Tumour-Bearing Mice and Mechanistic in Vitro Studies. Mol. Cancer 2014, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gérard, C.; Goldbeter, A. The balance between cell cycle arrest and cell proliferation: Control by the extracellular matrix and by contact inhibition. Interface Focus 2014, 4, 20130075. [Google Scholar] [CrossRef]
- Wardi, L.; Alaaeddine, N.; Raad, I.; Sarkis, R.; Serhal, R.; Khalil, C.; Hilal, G. Glucose restriction decreases telomerase activity and enhances its inhibitor response on breast cancer cells: Possible extra-telomerase role of BIBR 1532. Cancer Cell Int. 2014, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Rubis, B.; Kaczmarek, M.; Szymanowska, N.; Galezowska, E.; Czyrski, A.; Juskowiak, B.; Hermann, T.; Rybczynska, M. The biological activity of G-quadruplex DNA binding papaverine-derived ligand in breast cancer cells. Investig. New Drugs 2009, 27, 289–296. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Duensing, T.D.; Watson, S.R. Assessment of Apoptosis (Programmed Cell Death) by Flow Cytometry. Cold Spring Harb. Protoc. 2018, 2, 38–40. [Google Scholar] [CrossRef] [PubMed]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konieczna, N.; Romaniuk-Drapała, A.; Lisiak, N.; Totoń, E.; Paszel-Jaworska, A.; Kaczmarek, M.; Rubiś, B. Telomerase Inhibitor TMPyP4 Alters Adhesion and Migration of Breast-Cancer Cells MCF7 and MDA-MB-231. Int. J. Mol. Sci. 2019, 20, 2670. https://doi.org/10.3390/ijms20112670
Konieczna N, Romaniuk-Drapała A, Lisiak N, Totoń E, Paszel-Jaworska A, Kaczmarek M, Rubiś B. Telomerase Inhibitor TMPyP4 Alters Adhesion and Migration of Breast-Cancer Cells MCF7 and MDA-MB-231. International Journal of Molecular Sciences. 2019; 20(11):2670. https://doi.org/10.3390/ijms20112670
Chicago/Turabian StyleKonieczna, Natalia, Aleksandra Romaniuk-Drapała, Natalia Lisiak, Ewa Totoń, Anna Paszel-Jaworska, Mariusz Kaczmarek, and Błażej Rubiś. 2019. "Telomerase Inhibitor TMPyP4 Alters Adhesion and Migration of Breast-Cancer Cells MCF7 and MDA-MB-231" International Journal of Molecular Sciences 20, no. 11: 2670. https://doi.org/10.3390/ijms20112670
APA StyleKonieczna, N., Romaniuk-Drapała, A., Lisiak, N., Totoń, E., Paszel-Jaworska, A., Kaczmarek, M., & Rubiś, B. (2019). Telomerase Inhibitor TMPyP4 Alters Adhesion and Migration of Breast-Cancer Cells MCF7 and MDA-MB-231. International Journal of Molecular Sciences, 20(11), 2670. https://doi.org/10.3390/ijms20112670