Comparative Integrated Omics Analysis of the Hfq Regulon in Bordetella pertussis

 ,
,
Abstract
:1. Introduction
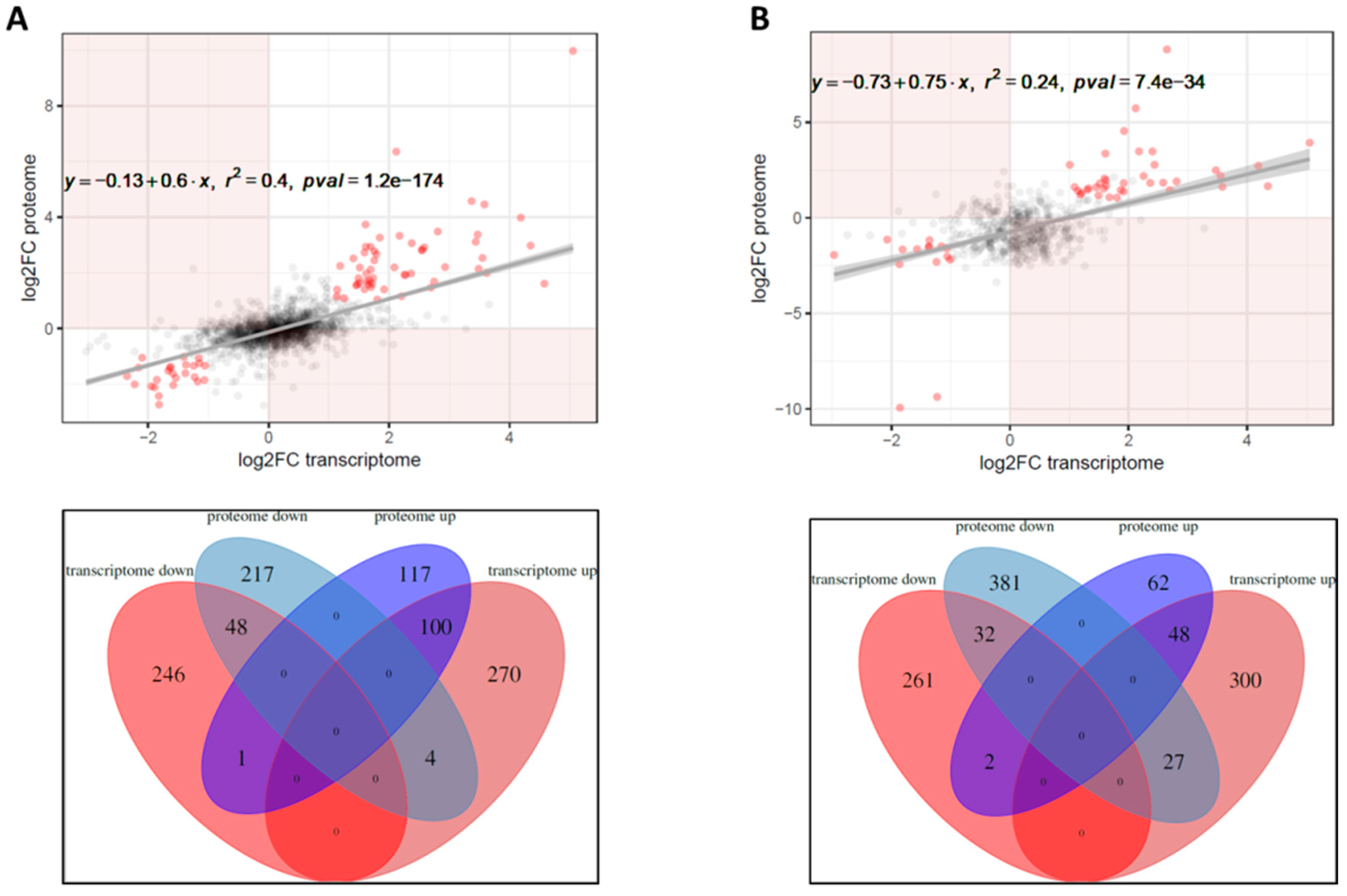
2. Results
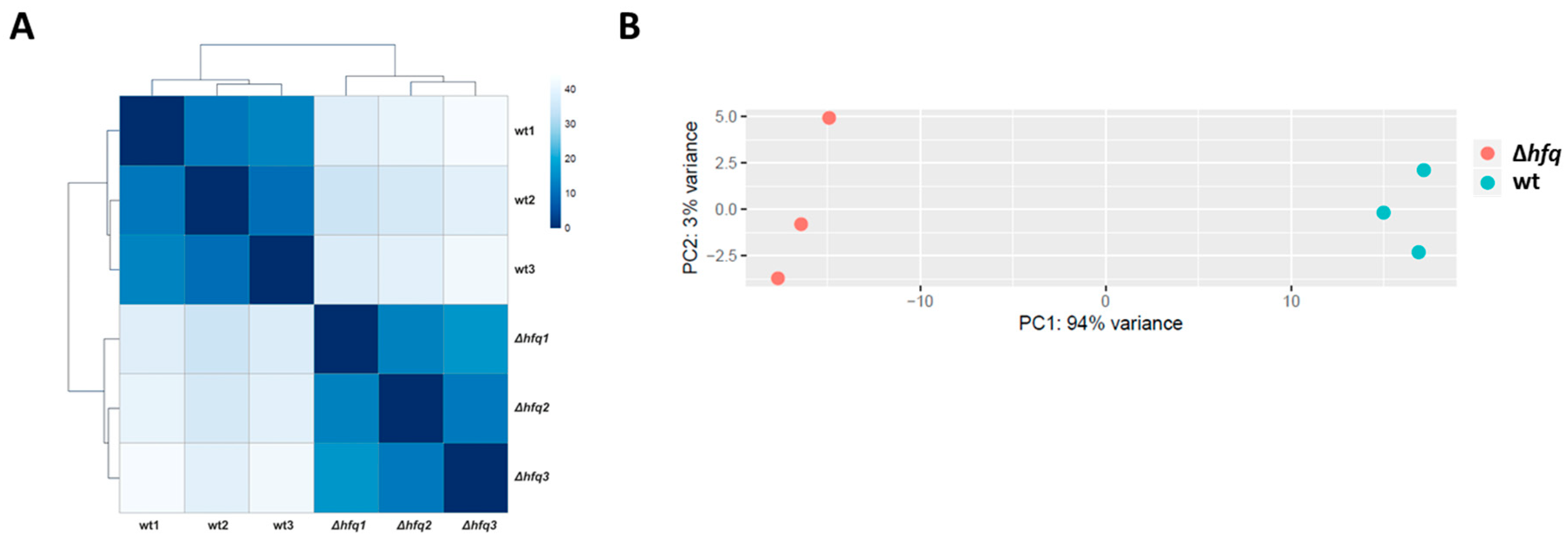
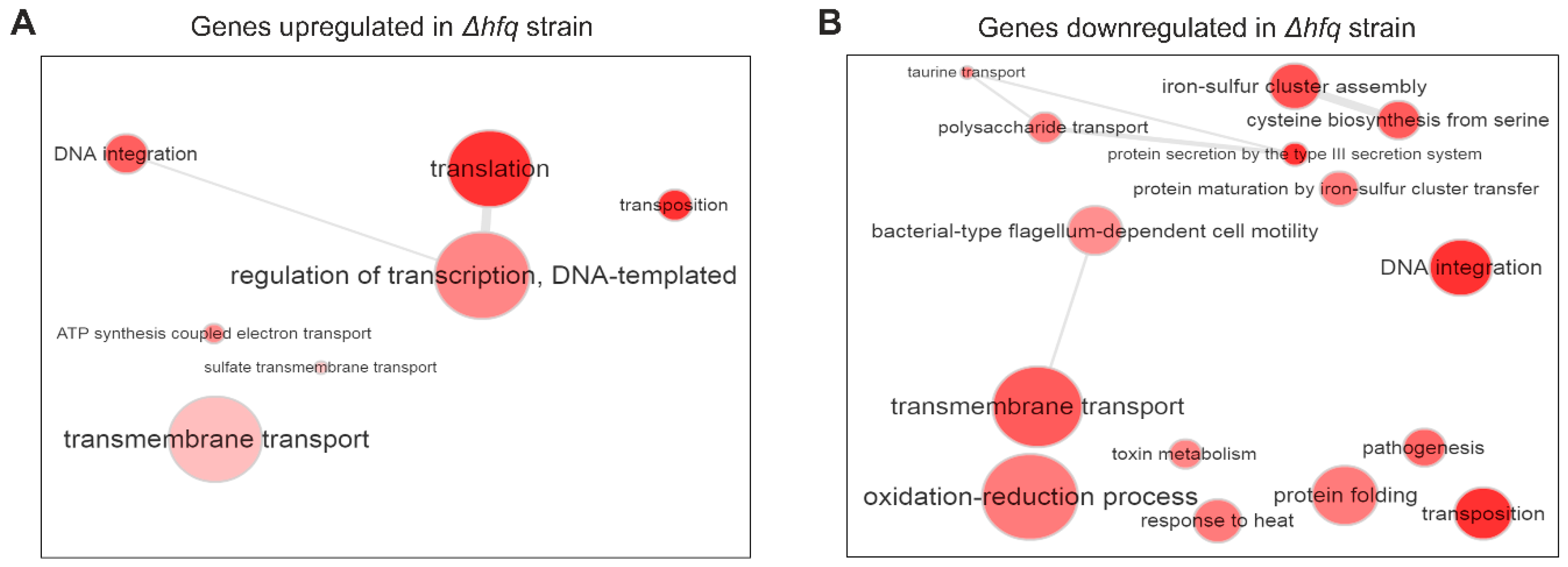
2.1. Identification of the Hfq Regulon by RNA-seq
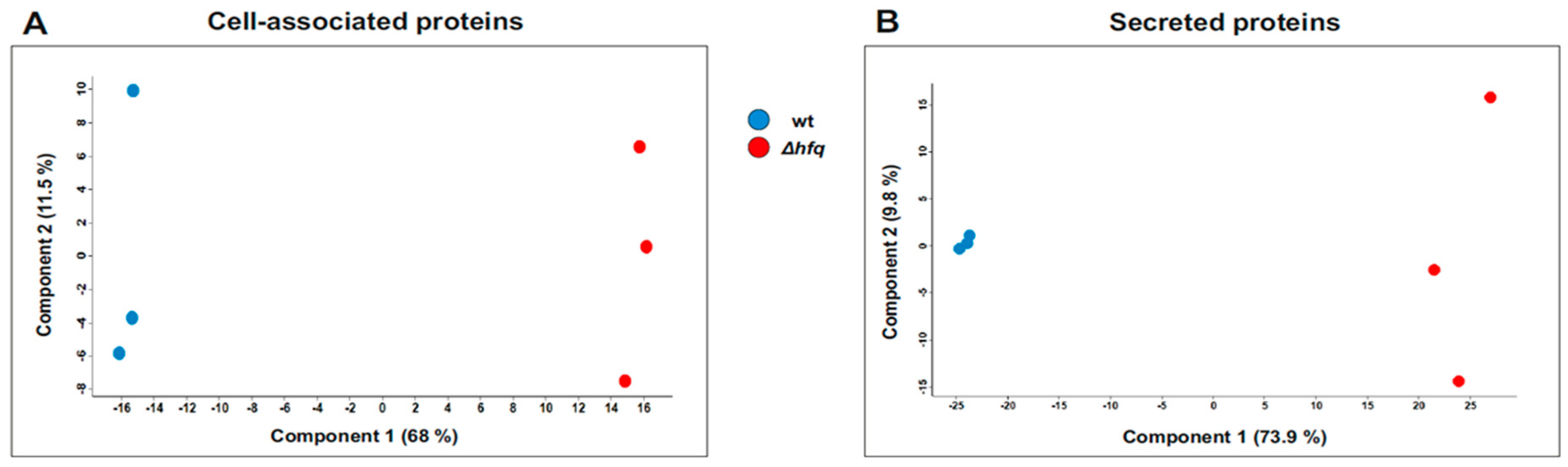
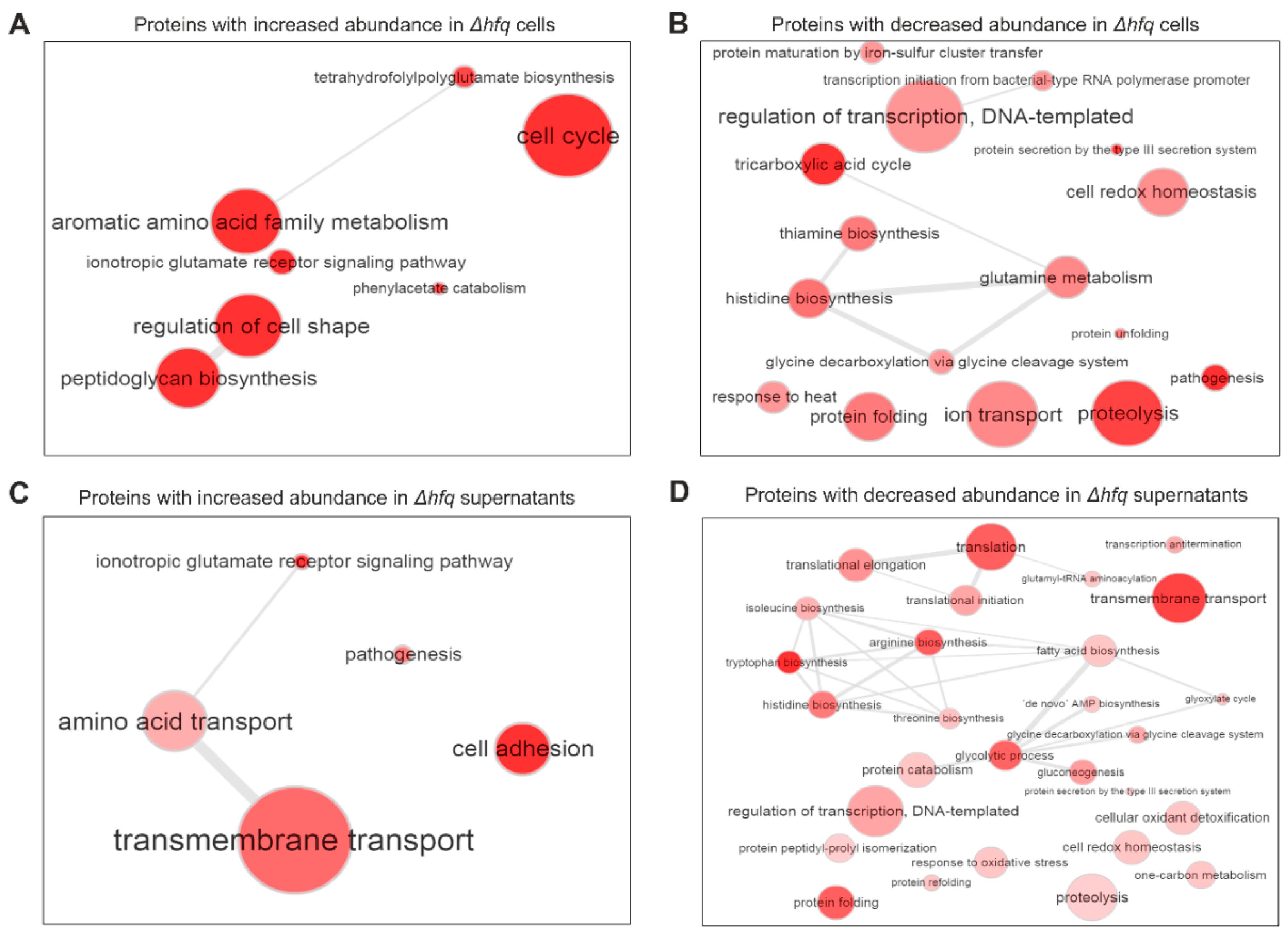
2.2. The Effect of Hfq on Proteome and Secretome Composition of B. pertussis
2.3. Correlation between Transcriptome, Proteome and Secretome Datasets
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. RNA Isolation
4.3. Library Preparation and Deep Sequencing
4.4. Protein Isolation and Sample Preparation for Proteomics
4.5. Label-Free Proteomic Analysis by LC-MS/MS
4.6. GO Term Enrichment Analysis
4.7. Transcriptome–Proteome Correlation Analyses
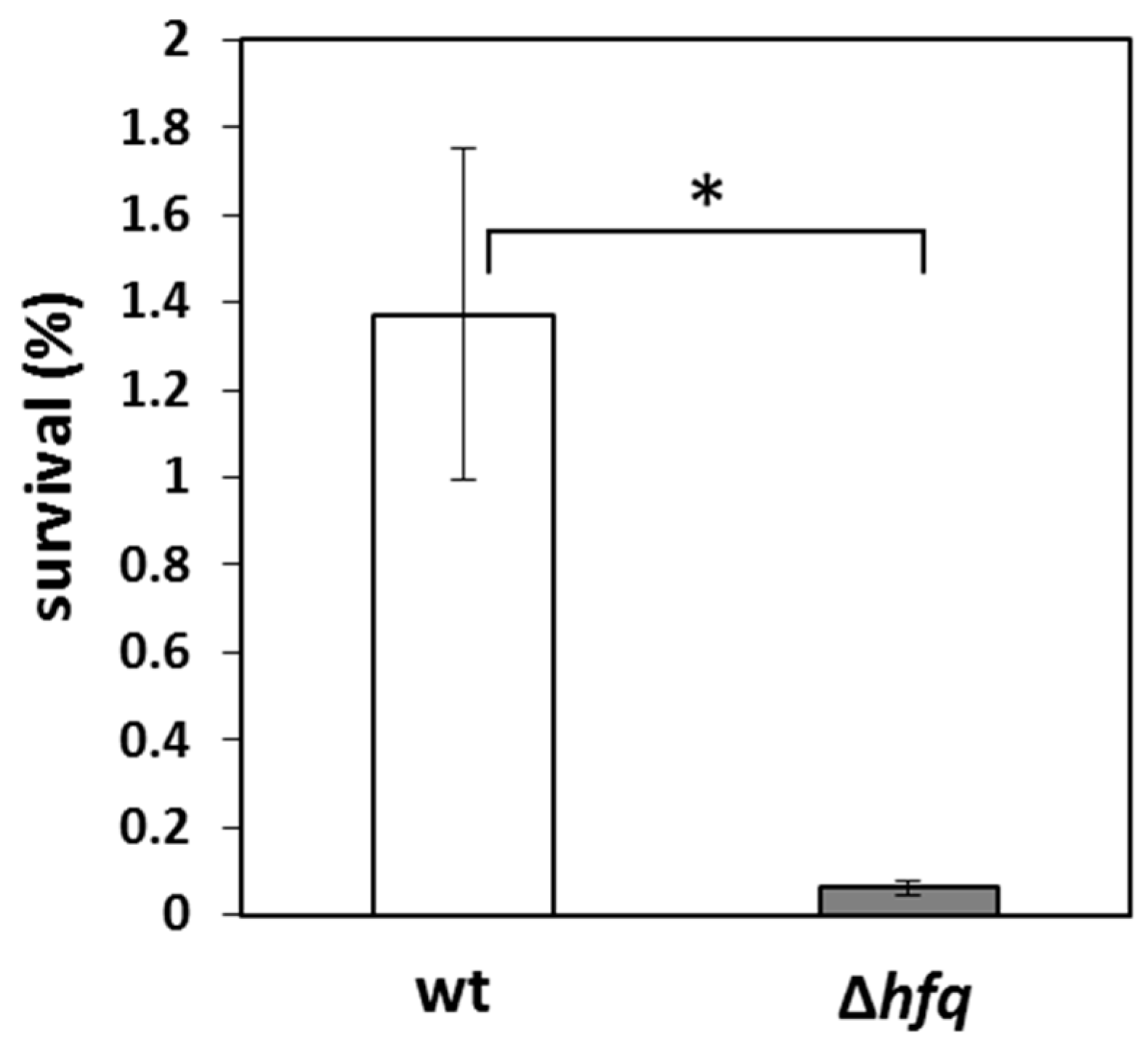
4.8. Serum Killing Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| BGA | Bordet-Gengou agar |
| FDR | False discovery rate |
| TEAB | Triethylammonium bicarbonate |
| EDTA | Ethylenediamine tetra-acetic acid |
| CFU | Colony forming unit |
| DE | Differential expression |
| TTT | Tripartite tricarboxylate transporters |
| TRAP | Tripartite ATP-independent periplasmic transporters |
| PCA | Principal component analysis |
| GO | Gene ontology |
| LFQ | Label-free quantification |
| T3SS | Type 3 secretion system |
| FC | Fold change |
References
- Mattoo, S.; Cherry, J.D. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to bordetella pertussis and other bordetella subspecies. Clin. Microbiol. Rev. 2005, 18, 326–382. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Vaccine preventable deaths and the global immunization vision and strategy, 2006–2015. MMWR Morb. Mortal. Wkly. Rep. 2006, 55, 511–515. [Google Scholar]
- Raguckas, S.E.; VandenBussche, H.L.; Jacobs, C.; Klepser, M.E. Pertussis resurgence: Diagnosis, treatment, prevention, and beyond. Pharmacotherapy 2007, 27, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D. The present and future control of pertussis. Clin. Infect. Dis. 2010, 51, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D. Pertussis: Challenges today and for the future. PLoS Pathog. 2013, 9, e1003418. [Google Scholar] [CrossRef] [PubMed]
- Mooi, F.R.; Van Der Maas, N.A.; De Melker, H.E. Pertussis resurgence: Waning immunity and pathogen adaptation—Two sides of the same coin. Epidemiol. Infect. 2014, 142, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Sealey, K.L.; Belcher, T.; Preston, A. Bordetella pertussis epidemiology and evolution in the light of pertussis resurgence. Infect. Genet. Evol. 2016, 40, 136–143. [Google Scholar] [CrossRef]
- Burdin, N.; Handy, L.K.; Plotkin, S.A. What is wrong with pertussis vaccine immunity? The problem of waning effectiveness of pertussis vaccines. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Hewlett, E.L.; Burns, D.L.; Cotter, P.A.; Harvill, E.T.; Merkel, T.J.; Quinn, C.P.; Stibitz, E.S. Pertussis pathogenesis—What we know and what we don’t know. J. Infect. Dis. 2014, 209, 982–985. [Google Scholar] [CrossRef]
- The Periscope Consortium. Periscope: Road towards effective control of pertussis. Lancet Infect. Dis. 2019, 19, 179–186. [Google Scholar] [CrossRef]
- Chao, Y.; Vogel, J. The role of hfq in bacterial pathogens. Curr. Opin. Microbiol. 2010, 13, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, J.R.; Grilo, A.M.; Guerreiro, S.I.; Sousa, S.A.; Leitao, J.H. Hfq: A multifaceted rna chaperone involved in virulence. Fut. Microbiol. 2016, 11, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Vogel, J. Regulatory rna in bacterial pathogens. Cell Host Microbe 2010, 8, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Luisi, B.F. Hfq and its constellation of rna. Nat. Rev. Microbiol. 2011, 9, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Vecerek, B.; Rajkowitsch, L.; Sonnleitner, E.; Schroeder, R.; Blasi, U. The c-terminal domain of escherichia coli hfq is required for regulation. Nucleic Acids Res. 2008, 36, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Updegrove, T.B.; Zhang, A.; Storz, G. Hfq: The flexible rna matchmaker. Curr. Opin. Microbiol. 2016, 30, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Brennan, R.G.; Link, T.M. Hfq structure, function and ligand binding. Curr. Opin. Microbiol. 2007, 10, 125–133. [Google Scholar] [CrossRef]
- Bibova, I.; Skopova, K.; Masin, J.; Cerny, O.; Hot, D.; Sebo, P.; Vecerek, B. The rna chaperone hfq is required for virulence of bordetella pertussis. Infect. Immun. 2013, 81, 4081–4090. [Google Scholar] [CrossRef]
- Bibova, I.; Hot, D.; Keidel, K.; Amman, F.; Slupek, S.; Cerny, O.; Gross, R.; Vecerek, B. Transcriptional profiling of bordetella pertussis reveals requirement of rna chaperone hfq for type iii secretion system functionality. RNA Biol. 2015, 12, 175–185. [Google Scholar] [CrossRef]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mrna expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Kumar, D.; Bansal, G.; Narang, A.; Basak, T.; Abbas, T.; Dash, D. Integrating transcriptome and proteome profiling: Strategies and applications. Proteomics 2016, 16, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between protein and mrna abundance in yeast. Mol. Cell Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mrna expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Ghazalpour, A.; Bennett, B.; Petyuk, V.A.; Orozco, L.; Hagopian, R.; Mungrue, I.N.; Farber, C.R.; Sinsheimer, J.; Kang, H.M.; Furlotte, N.; et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 2011, 7, e1001393. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mrna and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Nie, L.; Wu, G.; Zhang, W. Correlation of mrna expression and protein abundance affected by multiple sequence features related to translational efficiency in desulfovibrio vulgaris: A quantitative analysis. Genetics 2006, 174, 2229–2243. [Google Scholar] [CrossRef]
- Arraiano, C.M.; Andrade, J.M.; Domingues, S.; Guinote, I.B.; Malecki, M.; Matos, R.G.; Moreira, R.N.; Pobre, V.; Reis, F.P.; Saramago, M.; et al. The critical role of rna processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 2010, 34, 883–923. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef]
- Grenga, L.; Chandra, G.; Saalbach, G.; Galmozzi, C.V.; Kramer, G.; Malone, J.G. Analyzing the complex regulatory landscape of hfq—An integrative, multi-omics approach. Front. Microbiol. 2017, 8, 1784. [Google Scholar] [CrossRef]
- Delgado-Ortega, M.; Marc, D.; Dupont, J.; Trapp, S.; Berri, M.; Meurens, F. Socs proteins in infectious diseases of mammals. Vet. Immunol. Immunopathol. 2013, 151, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Marr, N.; Shah, N.R.; Lee, R.; Kim, E.J.; Fernandez, R.C. Bordetella pertussis autotransporter vag8 binds human c1 esterase inhibitor and confers serum resistance. PLoS ONE 2011, 6, e20585. [Google Scholar] [CrossRef] [PubMed]
- Hovingh, E.S.; van den Broek, B.; Kuipers, B.; Pinelli, E.; Rooijakkers, S.H.M.; Jongerius, I. Acquisition of c1 inhibitor by bordetella pertussis virulence associated gene 8 results in c2 and c4 consumption away from the bacterial surface. PLoS Pathog. 2017, 13, e1006531. [Google Scholar] [CrossRef] [PubMed]
- Brookes, C.; Freire-Martin, I.; Cavell, B.; Alexander, F.; Taylor, S.; Persaud, R.; Fry, N.; Preston, A.; Diavatopoulos, D.; Gorringe, A. Bordetella pertussis isolates vary in their interactions with human complement components. Emerg. Microbes Infect. 2018, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. Rna-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of rna-seq and microarray in transcriptome profiling of activated t cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Linhartova, I.; Halada, P.; Novotna, J.; Bezouskova, S.; Osicka, R.; Weiser, J.; Vohradsky, J.; Sebo, P. The iron-regulated transcriptome and proteome of neisseria meningitidis serogroup c. Proteomics 2006, 6, 6194–6206. [Google Scholar] [CrossRef]
- Yuk, M.H.; Harvill, E.T.; Miller, J.F. The bvgas virulence control system regulates type iii secretion in bordetella bronchiseptica. Mol. Microbiol. 1998, 28, 945–959. [Google Scholar] [CrossRef]
- Ahuja, U.; Shokeen, B.; Cheng, N.; Cho, Y.; Blum, C.; Coppola, G.; Miller, J.F. Differential regulation of type iii secretion and virulence genes in bordetella pertussis and bordetella bronchiseptica by a secreted anti-sigma factor. Proc. Natl. Acad. Sci. USA 2016, 113, 2341–2348. [Google Scholar] [CrossRef]
- Mattoo, S.; Yuk, M.H.; Huang, L.L.; Miller, J.F. Regulation of type iii secretion in bordetella. Mol. Microbiol. 2004, 52, 1201–1214. [Google Scholar] [CrossRef]
- Kurushima, J.; Kuwae, A.; Abe, A. The type iii secreted protein bspr regulates the virulence genes in bordetella bronchiseptica. PLoS ONE 2012, 7, e38925. [Google Scholar] [CrossRef] [PubMed]
- Fennelly, N.K.; Sisti, F.; Higgins, S.C.; Ross, P.J.; van der Heide, H.; Mooi, F.R.; Boyd, A.; Mills, K.H. Bordetella pertussis expresses a functional type iii secretion system that subverts protective innate and adaptive immune responses. Infect. Immun. 2008, 76, 1257–1266. [Google Scholar] [CrossRef]
- Gaillard, M.E.; Bottero, D.; Castuma, C.E.; Basile, L.A.; Hozbor, D. Laboratory adaptation of bordetella pertussis is associated with the loss of type three secretion system functionality. Infect. Immun. 2011, 79, 3677–3682. [Google Scholar] [CrossRef] [PubMed]
- Brickman, T.J.; Cummings, C.A.; Liew, S.Y.; Relman, D.A.; Armstrong, S.K. Transcriptional profiling of the iron starvation response in bordetella pertussis provides new insights into siderophore utilization and virulence gene expression. J. Bacteriol. 2011, 193, 4798–4812. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, T.; Kamachi, K.; Yonezawa, H.; Fukutomi, T.; Kawakami, H.; Kamiya, S. Glutamate limitation, bvgas activation, and (p)ppgpp regulate the expression of the bordetella pertussis type 3 secretion system. J. Bacteriol. 2016, 198, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Fantappie, L.; Metruccio, M.M.; Seib, K.L.; Oriente, F.; Cartocci, E.; Ferlicca, F.; Giuliani, M.M.; Scarlato, V.; Delany, I. The rna chaperone hfq is involved in stress response and virulence in neisseria meningitidis and is a pleiotropic regulator of protein expression. Infect. Immun. 2009, 77, 1842–1853. [Google Scholar] [CrossRef]
- Barnes, M.G.; Weiss, A.A. Brka protein of bordetella pertussis inhibits the classical pathway of complement after c1 deposition. Infect. Immun. 2001, 69, 3067–3072. [Google Scholar] [CrossRef]
- Berggard, K.; Johnsson, E.; Mooi, F.R.; Lindahl, G. Bordetella pertussis binds the human complement regulator c4bp: Role of filamentous hemagglutinin. Infect. Immun. 1997, 65, 3638–3643. [Google Scholar]
- Noofeli, M.; Bokhari, H.; Blackburn, P.; Roberts, M.; Coote, J.G.; Parton, R. Bapc autotransporter protein is a virulence determinant of bordetella pertussis. Microb. Pathog. 2011, 51, 169–177. [Google Scholar] [CrossRef]
- Rosa, L.T.; Bianconi, M.E.; Thomas, G.H.; Kelly, D.J. Tripartite atp-independent periplasmic (trap) transporters and tripartite tricarboxylate transporters (ttt): From uptake to pathogenicity. Front. Cell Infect. Microbiol. 2018, 8, 33. [Google Scholar] [CrossRef]
- Antoine, R.; Jacob-Dubuisson, F.; Drobecq, H.; Willery, E.; Lesjean, S.; Locht, C. Overrepresentation of a gene family encoding extracytoplasmic solute receptors in bordetella. J. Bacteriol. 2003, 185, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Huvent, I.; Belrhali, H.; Antoine, R.; Bompard, C.; Locht, C.; Jacob-Dubuisson, F.; Villeret, V. Crystal structure of bordetella pertussis bugd solute receptor unveils the basis of ligand binding in a new family of periplasmic binding proteins. J. Mol. Biol. 2006, 356, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Huvent, I.; Belrhali, H.; Antoine, R.; Bompard, C.; Locht, C.; Jacob-Dubuisson, F.; Villeret, V. Structural analysis of bordetella pertussis buge solute receptor in a bound conformation. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H. Contribution of pertussis toxin to the pathogenesis of pertussis disease. Pathog. Dis. 2015, 73, ftv073. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Elliott, T. Efficient translation of the rpos sigma factor in salmonella typhimurium requires host factor i, an rna-binding protein encoded by the hfq gene. J. Bacteriol. 1996, 178, 3763–3770. [Google Scholar] [CrossRef] [PubMed]
- Muffler, A.; Fischer, D.; Hengge-Aronis, R. The rna-binding protein hf-i, known as a host factor for phage qbeta rna replication, is essential for rpos translation in escherichia coli. Genes Dev. 1996, 10, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Davis, B.M.; Waldor, M.K. Hfq is essential for vibrio cholerae virulence and downregulates sigma expression. Mol. Microbiol. 2004, 53, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Bossi, N.; Lemire, S.; Maloriol, D.; Balbontin, R.; Casadesus, J.; Bossi, L. Loss of hfq activates the sigmae-dependent envelope stress response in salmonella enterica. Mol. Microbiol. 2006, 62, 838–852. [Google Scholar] [CrossRef] [PubMed]
- Keidel, K.; Amman, F.; Bibova, I.; Drzmisek, J.; Benes, V.; Hot, D.; Vecerek, B. Signal transduction-dependent small regulatory rna is involved in glutamate metabolism of the human pathogen bordetella pertussis. RNA 2018, 24, 1530–1541. [Google Scholar] [CrossRef]
- Amman, F.; D’Halluin, A.; Antoine, R.; Huot, L.; Bibova, I.; Keidel, K.; Slupek, S.; Bouquet, P.; Coutte, L.; Caboche, S.; et al. Primary transcriptome analysis reveals importance of is elements for the shaping of the transcriptional landscape of bordetella pertussis. RNA Biol. 2018, 15, 967–975. [Google Scholar] [CrossRef]
- Kasuga, T.; Nakase, Y.; Ukishima, K.; Takatsu, K. Studies on haemophilis pertussis. Iii. Some properties of each phase of h. Pertussis. Kitasato Arch. Exp. Med. 1954, 27, 37–47. [Google Scholar] [PubMed]
- Stainer, D.W.; Scholte, M.J. A simple chemically defined medium for the production of phase i bordetella pertussis. J. Gen. Microbiol. 1970, 63, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermuller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. Htseq—A python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The one hour yeast proteome. Mol. Cell Proteom. 2014, 13, 339–347. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. Maxquant enables high peptide identification rates, individualized p.P.B.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the maxquant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed maxlfq. Mol. Cell Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The pride database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | RNA-seq1 | Proteome1 | Secretome1 | Function |
|---|---|---|---|---|
| BP1358 | 4.18 | 3.93 | 2.78 | TTT transporter |
| BP1487 | 4.34 | 2.92 | 1.65 | TRAP transporter |
| BP1489 | 3.48 | 2.06 | ND | TRAP transporter |
| BP2066 | 2.64 | ON | 8.80 | TTT transporter |
| BP2090 | 1.92 | ON | 4.40 | ABC transporter |
| BP2271 | 5.05 | 9.75 | 3.98 | lipoprotein |
| BP2352 | 3.58 | 4.50 | 1.90 | TRAP transporter |
| BP2667 | 2.11 | 6.47 | 5.72 | adhesin FhaS |
| BP2692 | 1.51 | 2.10 | 1.90 | ABC transporter |
| BP3501 | 3.36 | 4.86 | ON | TTT transporter |
| BP3783 | 1.50 | 1.62 | 1.05 | pertussis toxin subunit A |
| BP3784 | 1.80 | 1.14 | 1.04 | pertussis toxin subunit B |
| BP3785 | 1.47 | 1.57 | 0.81 | pertussis toxin subunit D |
| BP3786 | 1.14 | ND | 1.60 | pertussis toxin subunit E |
| BP3787 | 1.49 | 1.76 | 1.26 | pertussis toxin subunit C |
| Gene | Name | RNA-seq1 | Proteome1 | Secretome1 |
|---|---|---|---|---|
| BP0500 | bopC | −0.69 | −1.19 | OFF |
| BP2248 | bscJ | −1.58 | −2.16 | ND |
| BP2250 | bcr4 | −1.81 | −2.75 | OFF |
| BP2251 | bcrH2 | −1.58 | −1.44 | ND |
| BP2252 | bopB | −1.89 | −2.23 | OFF |
| BP2253 | bopD | −1.85 | −1.83 | −9.95 |
| BP2254 | bcrH1 | −1.26 | −0.96 | OFF |
| BP2256 | bsp22 | −1.22 | −1.76 | -9.13 |
| BP2257 | bopN | −0.85 | −1.71 | OFF |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dienstbier, A.; Amman, F.; Štipl, D.; Petráčková, D.; Večerek, B. Comparative Integrated Omics Analysis of the Hfq Regulon in Bordetella pertussis. Int. J. Mol. Sci. 2019, 20, 3073. https://doi.org/10.3390/ijms20123073
Dienstbier A, Amman F, Štipl D, Petráčková D, Večerek B. Comparative Integrated Omics Analysis of the Hfq Regulon in Bordetella pertussis. International Journal of Molecular Sciences. 2019; 20(12):3073. https://doi.org/10.3390/ijms20123073
Chicago/Turabian StyleDienstbier, Ana, Fabian Amman, Daniel Štipl, Denisa Petráčková, and Branislav Večerek. 2019. "Comparative Integrated Omics Analysis of the Hfq Regulon in Bordetella pertussis" International Journal of Molecular Sciences 20, no. 12: 3073. https://doi.org/10.3390/ijms20123073
APA StyleDienstbier, A., Amman, F., Štipl, D., Petráčková, D., & Večerek, B. (2019). Comparative Integrated Omics Analysis of the Hfq Regulon in Bordetella pertussis. International Journal of Molecular Sciences, 20(12), 3073. https://doi.org/10.3390/ijms20123073