Cystathionine-Gamma-Lyase-Derived Hydrogen Sulfide-Regulated Substance P Modulates Liver Sieve Fenestrations in Caecal Ligation and Puncture-Induced Sepsis


 and
and 
Abstract
:1. Introduction
2. Results
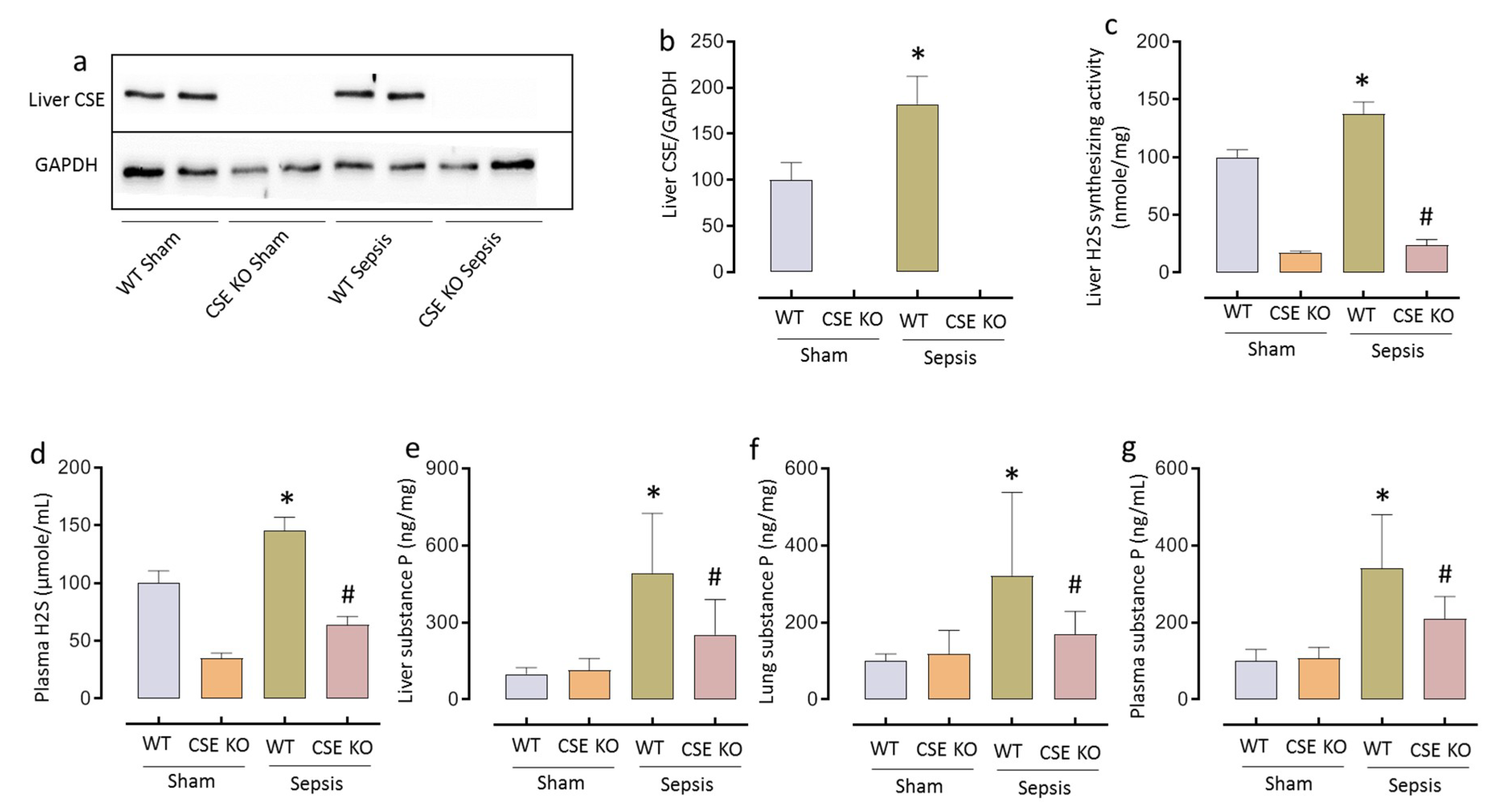
2.1. CSE-derived H2S Regulates SP Following CLP-Induced Sepsis
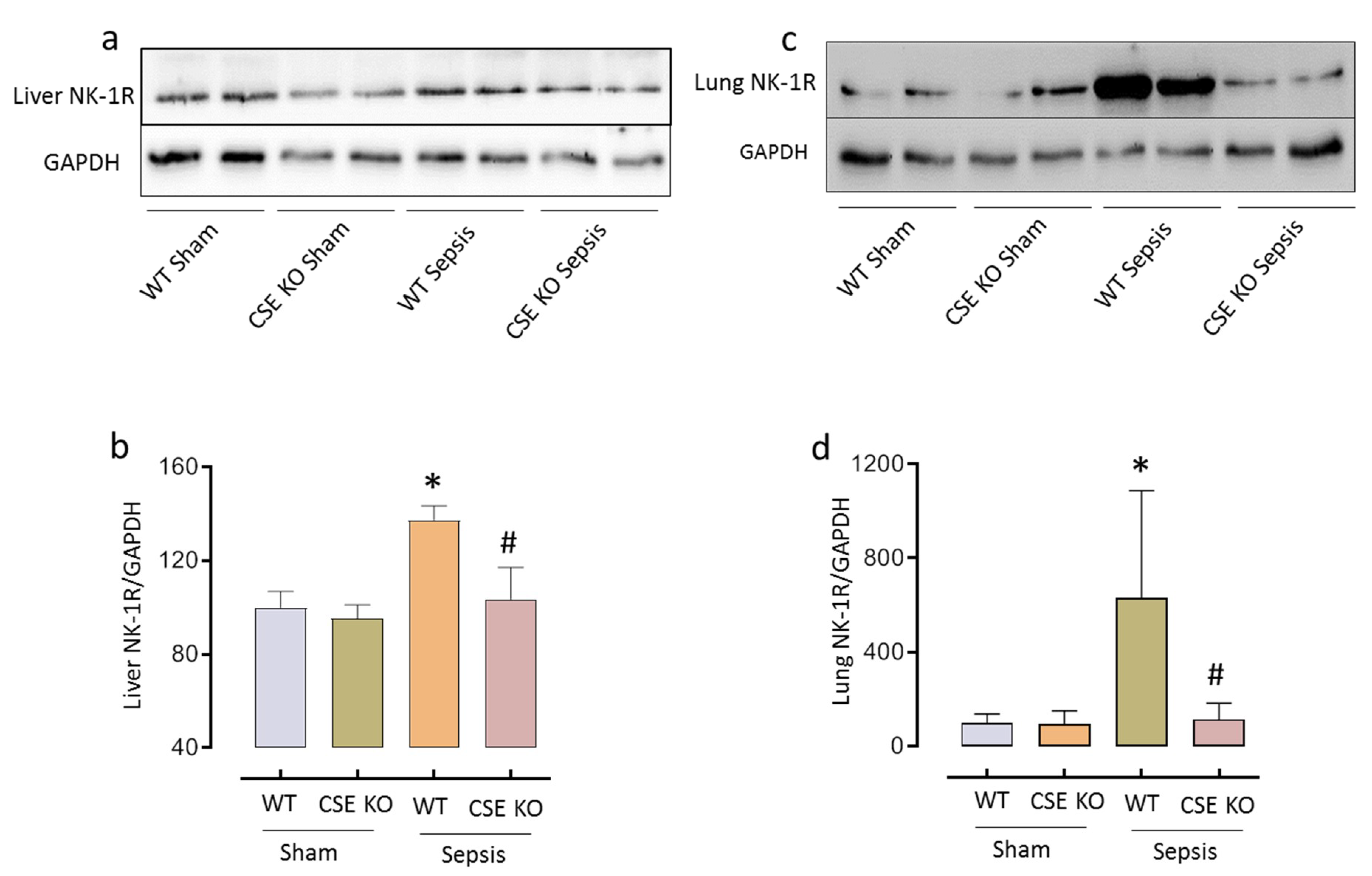
2.2. CSE-Derived H2S Regulates NK-1R Expression in Sepsis
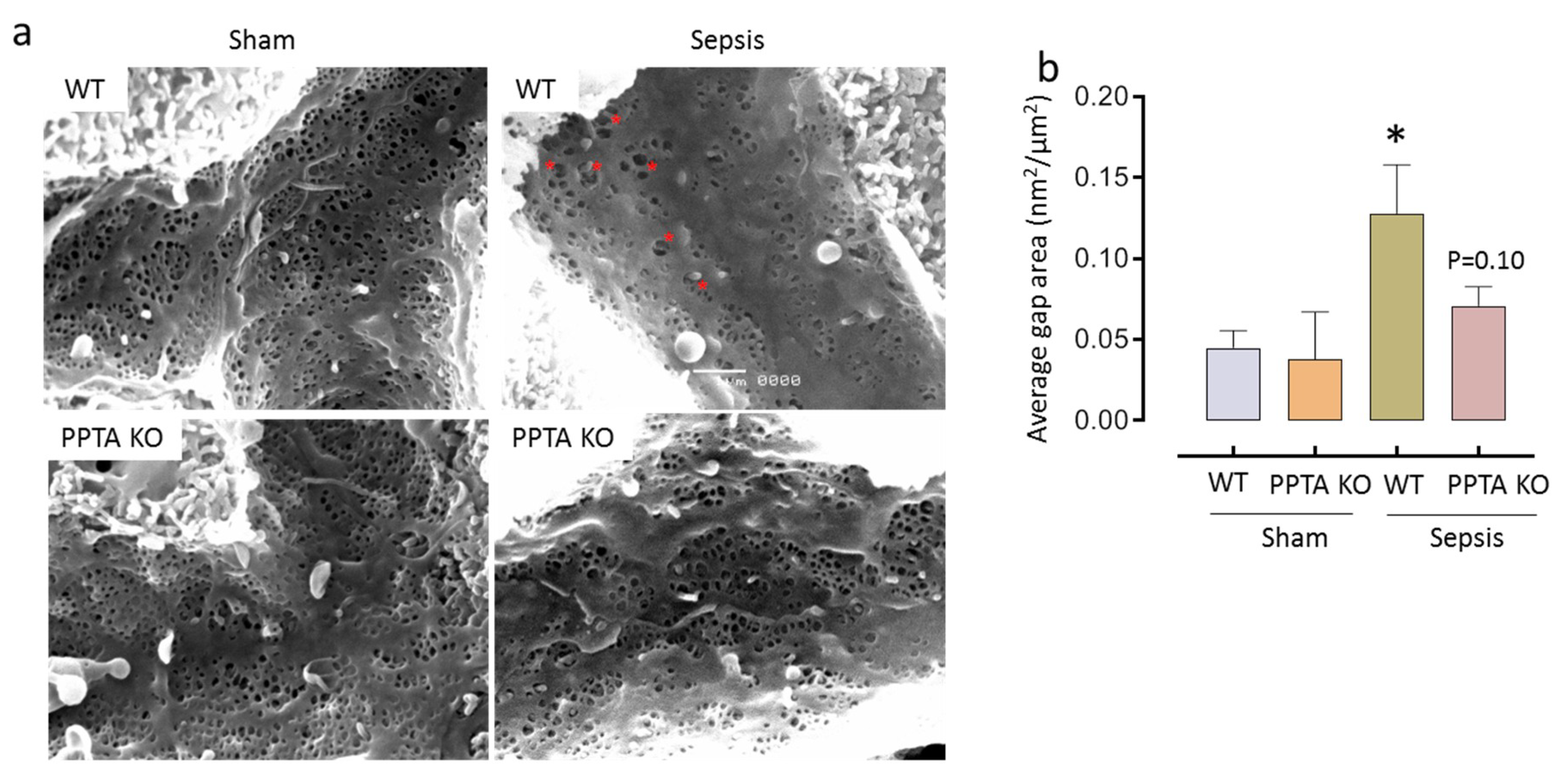
2.3. PPTA Gene Deletion Protects Against Sepsis-Induced Damage to LSECs
3. Discussion
4. Materials and Methods
4.1. Induction of Sepsis
4.2. H2S-Synthesizing Activity Assay
4.3. Measurement of H2S Levels in Plasma
4.4. Measurement of Substance P Levels
4.5. Western Blotting
4.6. Scanning Electron Microscopy (SEM)
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, K.; Li, H.; Li, S.; Yang, G. Regulation of cystathionine gamma-lyase/H₂S system and its pathological implication. Front. Biosci. (Landmark edition) 2013, 19, 1355–1369. [Google Scholar] [CrossRef]
- Zhang, H.; Zhi, L.; Moochhala, S.M.; Moore, P.K.; Bhatia, M. Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. J. Leukoc. Biol. 2007, 82, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhi, L.; Moore, P.K.; Bhatia, M. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1193–L1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badiei, A.; Chambers, S.; Gaddam, R.; Bhatia, M. Cystathionine-γ-lyase gene silencing with siRNA in monocytes/macrophages attenuates inflammation in cecal ligation and puncture-induced sepsis in the mouse. J. Biosci. 2016, 41, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Moochhala, S.M.; Bhatia, M. Endogenous hydrogen sulfide regulates inflammatory response by activating the ERK pathway in polymicrobial sepsis. J. Immunol. 2008, 181, 4320–4331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhi, L.; Moochhala, S.; Moore, P.K.; Bhatia, M. Hydrogen sulfide acts as an inflammatory mediator in cecal ligation and puncture-induced sepsis in mice by upregulating the production of cytokines and chemokines via NF-κB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L960–L971. [Google Scholar] [CrossRef] [PubMed]
- Gaddam, R.R.; Fraser, R.; Badiei, A.; Chambers, S.; Cogger, V.C.; Le Couteur, D.G.; Ishii, I.; Bhatia, M. Cystathionine-Gamma-Lyase Gene Deletion Protects Mice against Inflammation and Liver Sieve Injury following Polymicrobial Sepsis. PLoS ONE 2016, 11, e0160521. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.M.; O’Connell, J.; O’Brien, D.I.; Goode, T.; Bredin, C.P.; Shanahan, F. The role of substance P in inflammatory disease. J. Cell. Physiol. 2004, 201, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.; Koh, Y.H.; Moochhala, S.M.; Bhatia, M. Neurokinin-1 receptor antagonist treatment in polymicrobial sepsis: Molecular insights. Int. J. Inflamm. 2010, 20, 601098. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.; Tamizhselvi, R.; Manikandan, J.; Melendez, A.J.; Moochhala, S.M.; Bhatia, M. Substance P in polymicrobial sepsis: Molecular fingerprint of lung injury in preprotachykinin-A-/-mice. Mol. Med. 2010, 16, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Gaddam, R.R.; Chambers, S.; Murdoch, D.; Shaw, G.; Bhatia, M. Circulating levels of hydrogen sulfide and substance P in patients with sepsis. J. Infect. 2017, 4, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Zhi, L.; Zhang, H.; Ng, S.-W.; Moore, P.K. Role of substance P in hydrogen sulfide-induced pulmonary inflammation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L896–L904. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.-F.; Moochhala, S.M.; Bhatia, M. Hydrogen sulfide promotes transient receptor potential vanilloid 1-mediated neurogenic inflammation in polymicrobial sepsis*. Crit. Care Med. 2010, 38, 619–628. [Google Scholar] [CrossRef]
- Ang, S.-F.; Moochhala, S.M.; MacAry, P.A.; Bhatia, M. Hydrogen sulfide and neurogenic inflammation in polymicrobial sepsis: Involvement of substance P and ERK-NF-kappaB signaling. PLoS ONE 2011, 6, e24535. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Zhang, H.; Hegde, A.; Ng, S.W.; Adhikari, S.; Moochhala, S.M.; Bhatia, M. Hydrogen sulfide up-regulates substance P in polymicrobial sepsis-associated lung injury. J. Immunol. 2007, 179, 4153–4160. [Google Scholar] [CrossRef]
- Svistounov, D.; Warren, A.; McNerney, G.P.; Owen, D.M.; Zencak, D.; Zykova, S.N.; Crane, H.; Huser, T.; Quinn, R.J.; Smedsrod, B.; et al. The Relationship between fenestrations, sieve plates and rafts in liver sinusoidal endothelial cells. PLoS ONE 2012, 7, e46134. [Google Scholar] [CrossRef]
- Cogger, V.C.; Roessner, U.; Warren, A.; Fraser, R.; Le Couteur, D.G. A Sieve-Raft Hypothesis for the regulation of endothelial fenestrations. Comput. Struct. Biotechnol. J. 2013, 8, 1–9. [Google Scholar] [CrossRef]
- Deaciuc, I.V.; Bagby, G.J.; Niesman, M.R.; Skrepnik, N.; Spitzer, J.J. Modulation of hepatic sinusoidal endothelial cell function by Kupffer cells: An example of intercellular communication in the liver. Hepatology 1994, 19, 464–470. [Google Scholar] [CrossRef]
- Knolle, P.A.; Löser, E.; Protzer, U.; Duchmann, R.; Schmitt, E.; Zum Büschenfelde, K.H.; Rose-John, S.; Gerken, G. Regulation of endotoxin-induced IL-6 production in liver sinusoidal endothelial cells and Kupffer cells by IL-10. Clin. Exp. Immunol. 1997, 107, 555–561. [Google Scholar] [CrossRef]
- Kuo, H.-p.; Lin, H.-c.; Hwang, K.-h.; Wang, C.-h.; Lu, L.-c. Lipopolysaccharide enhances substance P-mediated neutrophil adherence to epithelial cells and cytokine release. Am. J. Respir. Crit. Care Med. 2000, 162, 1891–1897. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.; Zhang, H.; Moochhala, S.M.; Bhatia, M. Neurokinin-1 receptor antagonist treatment protects mice against lung injury in polymicrobial sepsis. J. Leukoc. Biol. 2007, 82, 678–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorente, L.; Martín, M.M.; Almeida, T.; Hernández, M.; Ferreres, J.; Solé-Violán, J.; Labarta, L.; Díaz, C.; Jiménez, A. Association between serum substance P levels and mortality in patients with severe sepsis. J. Crit. Care 2015, 30, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Puneet, P.; Hegde, A.; Ng, S.W.; Lau, H.Y.; Lu, J.; Moochhala, S.M.; Bhatia, M. Preprotachykinin-A gene products are key mediators of lung injury in polymicrobial sepsis. J. Immunol. 2006, 176, 3813–3820. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; Mross, P.E.; Hosie, M.J.; Ansselin, A.D.; McLean, A.J. The effect of acute oxidative stress on the ultrastructure of the perfused rat liver. Pharmacol. Toxicol. 2001, 89, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; Muller, M.; Fraser, R.; McLean, A.J.; Khan, J.; Le Couteur, D.G. The effects of oxidative stress on the liver sieve. J. Hepatol. 2004, 41, 370–376. [Google Scholar] [CrossRef]
- Dobbs, B.; Rogers, G.; Xing, H.; Fraser, R. Endotoxin-induced defenestration of the hepatic sinusoidal endothelium: A factor in the pathogenesis of cirrhosis? Liver 1994, 14, 230–233. [Google Scholar] [CrossRef]
- Koo, D.J.; Chaudry, I.H.; Wang, P. Kupffer cells are responsible for producing inflammatory cytokines and hepatocellular dysfunction during early sepsis. J. Surg. Res. 1999, 83, 151–157. [Google Scholar] [CrossRef]
- Cheluvappa, R.; Cogger, V.C.; Kwun, S.Y.; O’Reilly, J.N.; Le Couteur, D.G.; Hilmer, S.N. Liver sinusoidal endothelial cells and acute non-oxidative hepatic injury induced by Pseudomonas aeruginosa pyocyanin. Int. J. Exp. Pathol. 2008, 89, 410–418. [Google Scholar] [CrossRef]
- Sarphie, T.G.; D’Souza, N.B.; Deaciuc, I.V. Kupffer cell inactivation prevents lipopolysaccharide-induced structural changes in the rat liver sinusoid: An electron-microscopic study. Hepatology 1996, 23, 788–796. [Google Scholar] [CrossRef]
- Cheluvappa, R.; Jamieson, H.A.; Hilmer, S.N.; Muller, M.; Le Couteur, D.G. The effect of Pseudomonas aeruginosa virulence factor, pyocyanin, on the liver sinusoidal endothelial cell. J. Gastroenterol. Hepatol. 2007, 22, 1350–1351. [Google Scholar] [CrossRef]
- Yan, J.; Li, S.; Li, S. The role of the liver in sepsis. Int. Rev. Immunol. 2014, 6, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Gaddam, R.R.; Fraser, R.; Badiei, A.; Chambers, S.; Cogger, V.C.; Le Couteur, D.G.; Bhatia, M. Differential Effects of Kupffer Cell Inactivation on Inflammation and The Liver Sieve Following Caecal-Ligation and Puncture-Induced Sepsis in Mice. Shock 2017, 4, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, N.A.; Chung, C.S.; Borgerding, J.N.; Ayala, C.A.; Ayala, A. Kupffer cells protect liver sinusoidal endothelial cells from Fas-dependent apoptosis in sepsis by down-regulating gp130. Am. J. Pathol. 2013, 182, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Ishii, I.; Akahoshi, N.; Yamada, H.; Nakano, S.; Izumi, T.; Suematsu, M. Cystathionine γ-lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem. 2010, 285, 26358–26368. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.G.; Ganea, D.; Gamero, A.M. Cecal ligation puncture procedure. J. Vis. Exp. JoVE 2011. [Google Scholar] [CrossRef]
- Siempos, I.I.; Lam, H.C.; Ding, Y.; Choi, M.E.; Choi, A.M.; Ryter, S.W. Cecal Ligation and Puncture-induced Sepsis as a Model To Study Autophagy in Mice. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; O’Reilly, J.N.; Warren, A.; Le Couteur, D.G. A standardized method for the analysis of liver sinusoidal endothelial cells and their fenestrations by scanning electron microscopy. J. Vis. Exp. 2015. [Google Scholar] [CrossRef]
- Chunyu, Z.; Junbao, D.; Dingfang, B.; Hui, Y.; Xiuying, T.; Chaoshu, T. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem. Biophys. Res. Commun. 2003, 302, 810–816. [Google Scholar] [CrossRef]
- Bhatia, M.; Saluja, A.K.; Hofbauer, B.; Frossard, J.-L.; Lee, H.S.; Castagliuolo, I.; Wang, C.-C.; Gerard, N.; Pothoulakis, C.; Steer, M.L. Role of substance P and the neurokinin 1 receptor in acute pancreatitis and pancreatitis-associated lung injury. Proc. Natl. Acad. Sci. 1998, 95, 4760–4765. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Group | Diameter of Fenestrae (nm) | Number of Fenestrae/µm2 | Porosity (%) |
|---|---|---|---|
| WT Sham | 149.39 ± 12.40 | 10.63 ± 1.54 | 18.44 ± 2.18 |
| PPTA KO Sham | 135.83 ± 7.83 | 11.60 ± 2.56 | 17.64 ± 3.28 |
| WT Sepsis | 143.91 ± 9.04 | 6.40 ± 2.49 * | 11.05 ± 3.23 * |
| PPTA KO Sepsis | 144.68 ± 8.39 | 9.05 ± 0.94 | 15.71 ± 2.19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaddam, R.R.; Chambers, S.; Fraser, R.; Cogger, V.C.; Le Couteur, D.G.; Ishii, I.; Bhatia, M. Cystathionine-Gamma-Lyase-Derived Hydrogen Sulfide-Regulated Substance P Modulates Liver Sieve Fenestrations in Caecal Ligation and Puncture-Induced Sepsis. Int. J. Mol. Sci. 2019, 20, 3191. https://doi.org/10.3390/ijms20133191
Gaddam RR, Chambers S, Fraser R, Cogger VC, Le Couteur DG, Ishii I, Bhatia M. Cystathionine-Gamma-Lyase-Derived Hydrogen Sulfide-Regulated Substance P Modulates Liver Sieve Fenestrations in Caecal Ligation and Puncture-Induced Sepsis. International Journal of Molecular Sciences. 2019; 20(13):3191. https://doi.org/10.3390/ijms20133191
Chicago/Turabian StyleGaddam, Ravinder R, Stephen Chambers, Robin Fraser, Victoria C Cogger, David G Le Couteur, Isao Ishii, and Madhav Bhatia. 2019. "Cystathionine-Gamma-Lyase-Derived Hydrogen Sulfide-Regulated Substance P Modulates Liver Sieve Fenestrations in Caecal Ligation and Puncture-Induced Sepsis" International Journal of Molecular Sciences 20, no. 13: 3191. https://doi.org/10.3390/ijms20133191
APA StyleGaddam, R. R., Chambers, S., Fraser, R., Cogger, V. C., Le Couteur, D. G., Ishii, I., & Bhatia, M. (2019). Cystathionine-Gamma-Lyase-Derived Hydrogen Sulfide-Regulated Substance P Modulates Liver Sieve Fenestrations in Caecal Ligation and Puncture-Induced Sepsis. International Journal of Molecular Sciences, 20(13), 3191. https://doi.org/10.3390/ijms20133191