Phenformin as an Anticancer Agent: Challenges and Prospects

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cancer and Diabetes Type 2: Common Risk Factors
2.1. Hyperinsulinemia and Insulin Resistance
2.2. Metabolic Syndrome
2.3. Diabetes Type 2 Increases Risk of Cancer
2.4. Biguanides Have a Protective Function on Cancer in Diabetic Patients
3. Phenformin and Cancer
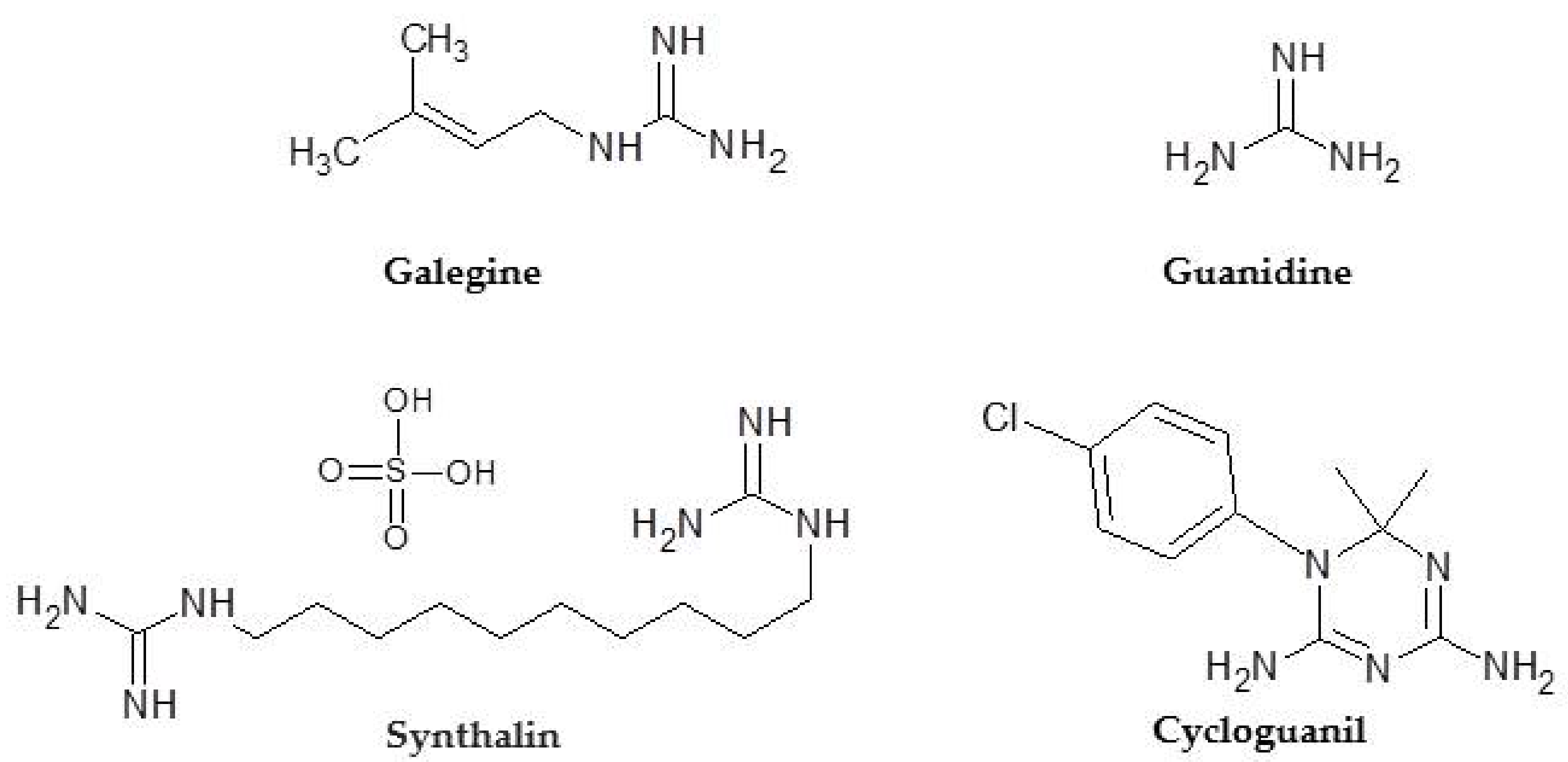
3.1. Phenformin: the Origin
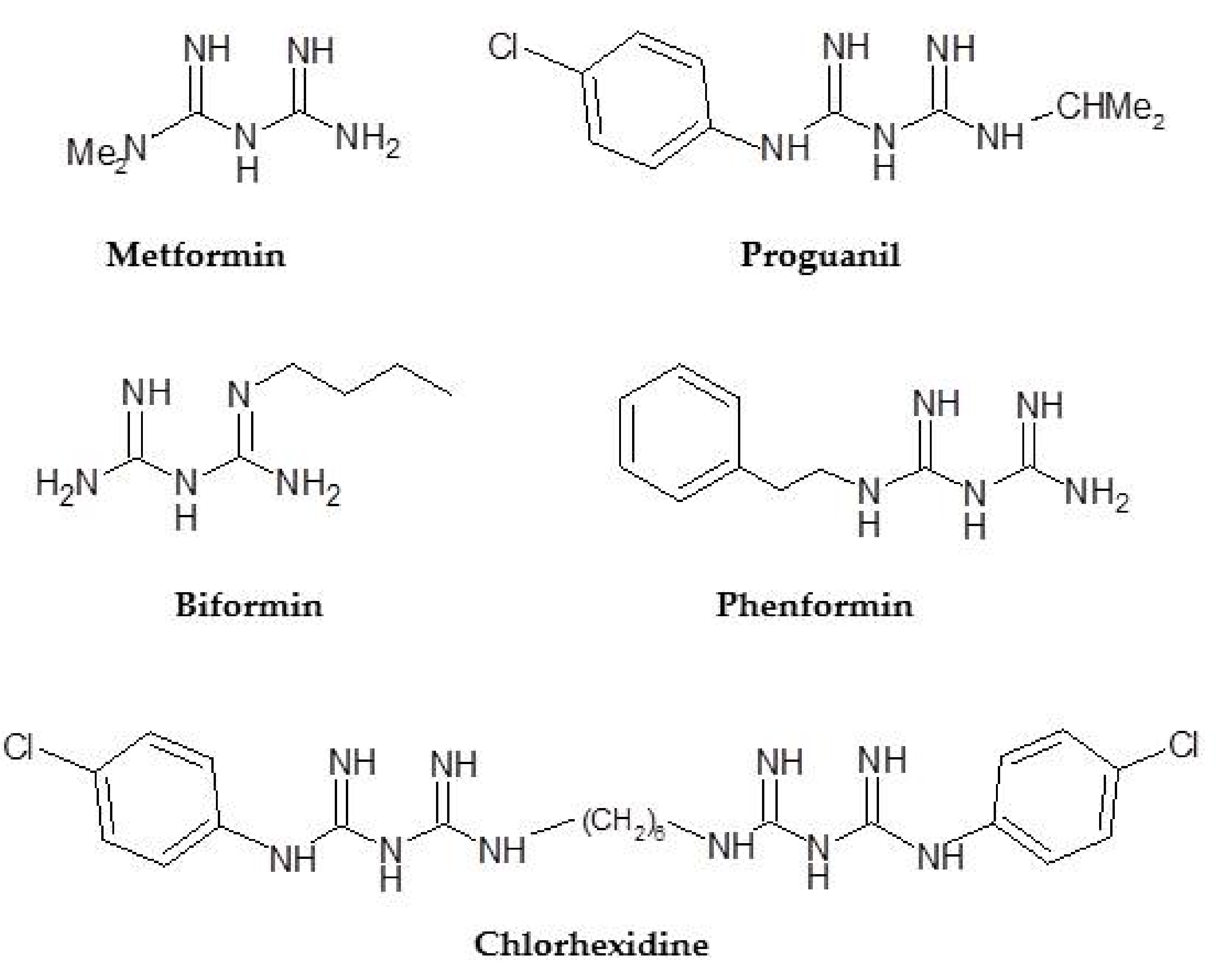
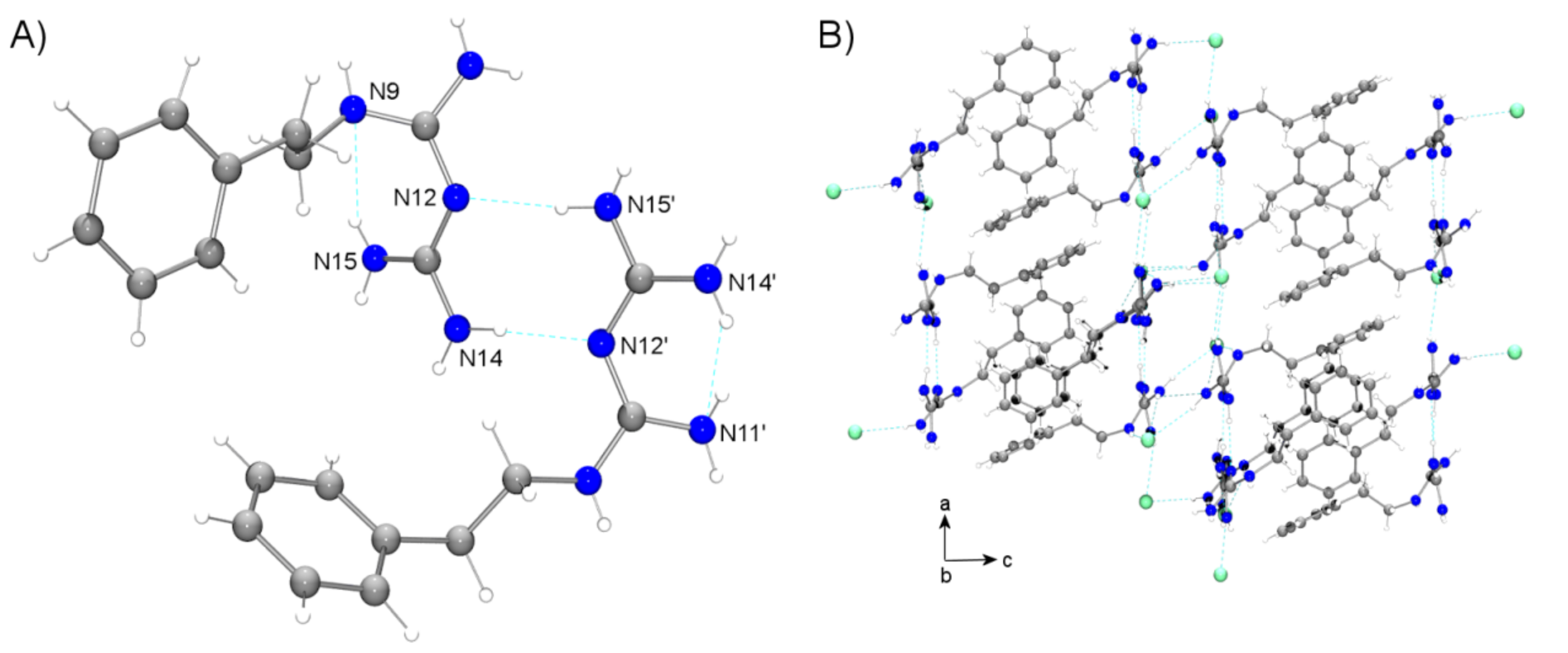
3.2. Phenformin: Chemistry
3.3. Phenformin as Anticancer Agent
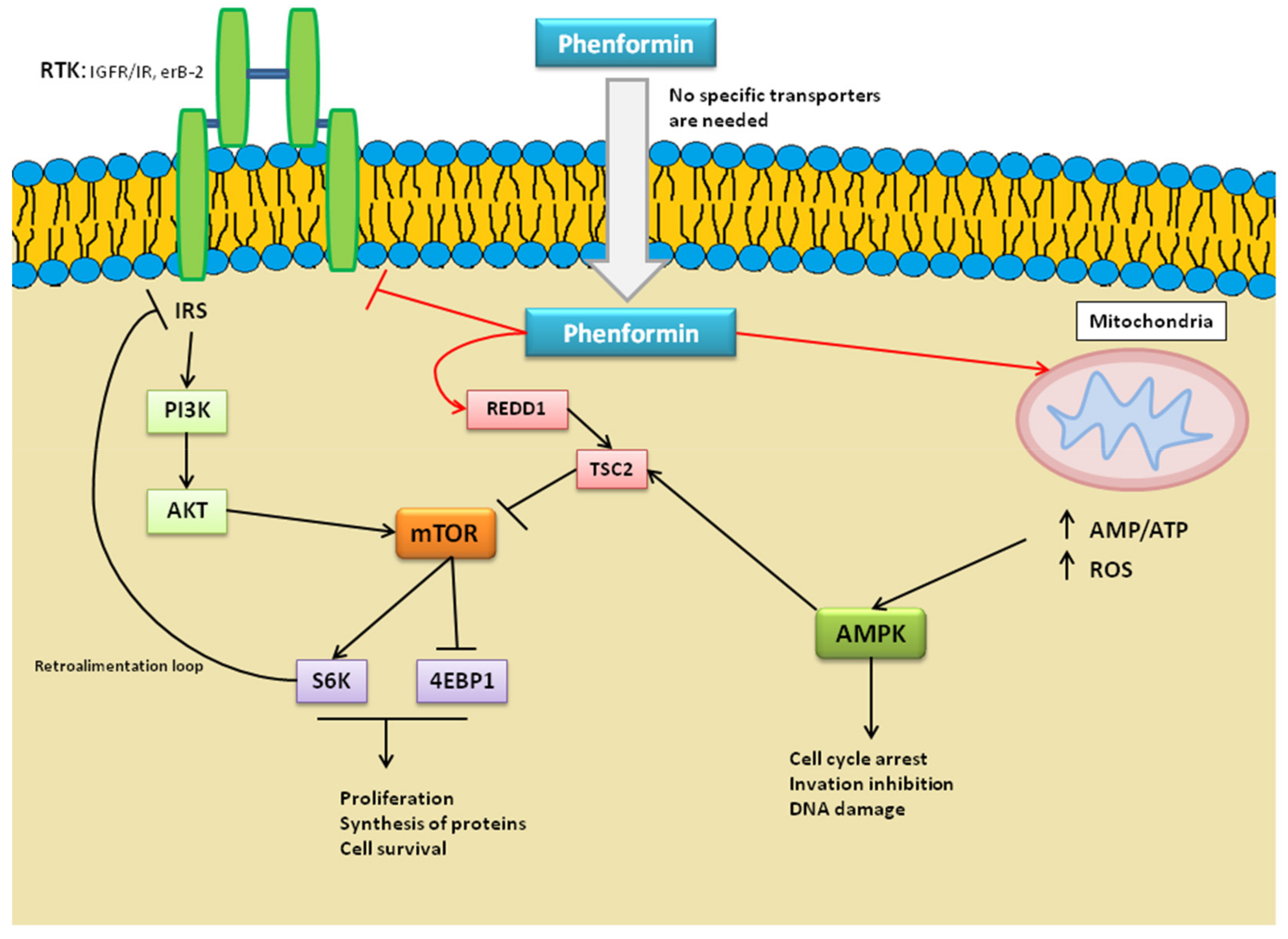
3.3.1. Mechanism of Action of Phenformin
3.3.2. Phenformin in Cancer Treatment
Phenformin Effect on Differentiated Cancer Cells
Phenformin’s Effect on Cancer Stem Cells
Combined Therapy Using Phenformin
4. Current Challenges and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hruby, A.; Hu, F.B. The epidemiology of obesity: A big picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Sami, W.; Ansari, T.; Butt, N.S.; Hamid, M.R.A. Effect of diet on type 2 diabetes mellitus: A review. Int. J. Health Sci. 2017, 11, 65–71. [Google Scholar]
- Guariguata, L.; Whiting, D.R.; Hambleton, I.; Beagley, J.; Linnenkamp, U.; Shaw, J.E. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 2014, 103, 103137–103149. [Google Scholar] [CrossRef] [PubMed]
- Scappaticcio, L.; Maiorino, M.I.; Bellastella, G.; Giugliano, D.; Esposito, K. Insights into the relationships between diabetes, prediabetes, and cancer. Endocrine 2017, 56, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, T.A. Diabetes sand cancer. QJM. 2010, 103, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. CA: Cancer J. Clin. 2010, 60, 207–221. [Google Scholar]
- Leighton, E.; Sainsbury, C.A.; Jones, G.C. A Practical Review of C-Peptide Testing in Diabetes. Diabetes Ther. 2017, 8, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef]
- Handelsman, Y.; Leroith, D.; Bloomgarden, Z.T.; Dagogo-Jack, S.; Einhorn, D.; Garber, A.J.; Grunberger, G.; Harrell, R.M.; Gagel, R.F.; Lebovitz, H.E.; et al. Diabetes and cancer an AACE/ACE consensusstatement. Endocr. Pract. 2013, 19, 675–693. [Google Scholar] [CrossRef]
- Salani, B.; Del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, cancer and glucose metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471. [Google Scholar] [CrossRef] [Green Version]
- Drzewoski, J.; Drozdowska, A.; Sliwińska, A. Do we have enough data to confirm the link between antidiabetic drug use and cancer development? Pol. Arch. Med. Wewn. 2011, 121, 81–87. [Google Scholar] [PubMed]
- Anisimov, V.N. Metformin for aging and cancer prevention. Aging (Albany NY) 2010, 2, 760–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.J.; Zhen, Z.J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar] [CrossRef] [PubMed]
- Del Barco, S.; Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagosklonny, M.V.; Campisi, J. Cancer and aging: More puzzles, more promises? Cell Cycle 2008, 7, 2615–2618. [Google Scholar] [CrossRef] [PubMed]
- Mercken, E.M.; Carboneau, B.A.; Krzysik-Walker, S.M.; de Cabo, R. Of mice and men: The benefits of caloric restriction, exercise, and mimetics. Ageing Res. Rev. 2012, 11, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.H.; LeRoith, D. Obesity, type 2 diabetes, and cancer: The insulin and IGF connection. Endocr Relat Cancer 2012, 19, F27–F45. [Google Scholar] [CrossRef]
- Gómez-Hernández, A.; Beneit Redondo, N.; Perdomo Loaiza, L.; Escribano Illanes, O.; Díaz-Castroverde, S.; Benito de las Heras, M. Role of insulin receptor A isoform anf IGF-1R in the development of atherosclerotic plaque. Anales de la Real Academia Nacional de Farmacia 2016, 82, 129–142. [Google Scholar]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Andres, S.F.; Simmons, J.G.; Mah, A.T.; Santoro, M.A.; Van Landeghem, L.; Lund, P.K. Insulin receptor isoform switching in intestinal stem cells, progenitors, differentiated lineages and tumors: Evidence that IR-B limits proliferation. J. Cell Sci. 2013, 15, 5645–5656. [Google Scholar] [CrossRef]
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Sakaguchi, M.; Kleinridders, A.; Gonzalez-Del Pino, G.; Dreyfuss, J.M.; O’Neill, B.T.; Ramirez, A.K.; Pan, H.; Winnay, J.N.; Boucher, J.; et al. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat. Commun. 2017, 8, 14892. [Google Scholar] [CrossRef] [PubMed]
- López de la Torre Casares, M. Diabetes mellitus y cáncer: Una visión global. MGF 2011, 137, 144–147. [Google Scholar]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 5, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Bellastella, G.; Scappaticcio, L.; Esposito, K.; Giugliano, D.; Maiorino, M.I. Metabolic syndrome and cancer: “The common soil hypothesis”. Diabetes Res. Clin. Pract. 2018, 143, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Bortolotti, M.; Polito, L.; Bolognesi, A. Metabolic syndrome and cancer risk: The role of xanthine oxidoreductase. Redox Biol. 2019, 21, 101070. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 12, 27–47. [Google Scholar] [CrossRef]
- Chakraborti, C.K. Role of adiponectin and some other factors linking type 2 diabetes mellitus and obesity. World J. Diabetes 2015, 6, 1296–1308. [Google Scholar] [CrossRef]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef]
- García, E.Y. Flumamine, a new synthetic analgesic and anti-flu drug. J. Philipp. Med. Assoc. 1950, 26, 287–293. [Google Scholar]
- Sterne, J. Report on 5-years’ experience with dimethylbiguanide (metformin, glucophage) in diabetic therapy. Wien. Med. Wochenschr. 1963, 3, 599–602. [Google Scholar]
- Sterne, J.; Hirsch, C. Experimental basis for combined treatment of diabetes with the biguanide-sulfonamide association. Diabetes 1964, 12, 171–175. [Google Scholar]
- Bailey, C.J. Biguanides and NIDDM. Diabetes Care 1992, 15, 755–772. [Google Scholar] [CrossRef] [PubMed]
- Nattrass, M.; Alberti, K.G. Biguanides. Diabetologia 1978, 14, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geoghegan, F.; Chadderton, N.; Farrar, G.J.; Zisterer, D.M.; Porter, R.K. Direct effects of phenformin on metabolism/bioenergetics and viability of SH-SY5Y neuroblastoma cells. Oncol. Lett. 2017, 14, 6298–6306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrnstadt, C.; Mootz, D.; Wunderlich, H. Protonation sides of organic bases with several nitrogen function: Crystal structure of salts of chlordiazepoxide, dihydralazine and phenformin. J. Chem. Soc. Perkin Trans. 1979, 2, 735–740. [Google Scholar] [CrossRef]
- Ernst, S.R.; Cagle, Jr. F.W. Biguanide hydrochloride. Acta Cryst Allogr. 1997, B33, 235–237. [Google Scholar] [CrossRef]
- Klepser, T.B.; Kelly, M.W. Metformin hydrochloride: An antihyperglycemic agent. Am. J. Health Syst. Pharm. 1997, 54, 893–903. [Google Scholar] [CrossRef]
- Soriano-Lesh, M.A.; Holt, E.M.; Aguirre-García, F.; Soriano-García, M. 1-Phenethylguanidinium Chloride. Acta Crystallogr. 1998, C54, 1187–1189. [Google Scholar] [CrossRef]
- Guo, D.S.; Zhang, H.Q.; Ding, F.; Liu, Y. Thermodynamic origins of selective binding affinity between p-sulfonatocalix (4,5) arenes with biguanidiniums. Org. Biomol. Chem. 2012, 10, 1527–1536. [Google Scholar] [CrossRef]
- Safe, S.; Nair, V.; Karki, K. Metformin-induced anticancer activities: Recent insights. Biol. Chem. 2018, 28, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Sun, H.; Qi, Y.; Zhao, X.; Feng, M.; Wu, X. Insulin-like growth factor 1/mammalian target of rapamycin and amp-activated protein kinase signaling involved in the effects of metformin in the human endometrial cancer. Int. J. Gynecol. Cancer 2016, 26, 1667–1672. [Google Scholar] [CrossRef]
- Soliman, P.T.; Zhang, Q.; Broaddus, R.R.; Westin, S.N.; Iglesias, D.; Munsell, M.F.; Schmandt, R.; Yates, M.; Ramondetta, L.; Lu, K.H. Prospective evaluation of the molecular effects of metformin on the endometrium in women with newly diagnosed endometrial cancer: A window of opportunity study. Gynecol Oncol. 2016, 143, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Pusceddu, S.; de Braud, F.; Concas, L.; Bregant, C.; Leuzzi, L.; Formisano, B.; Buzzoni, R. Rationale and protocol of the MetNET-1 trial, a prospective, single center, phase II study to evaluate the activity and safety of everolimus in combination with octreotide LAR and metformin in patients with advanced pancreatic neuroendocrine tumors. Tumori 2014, 100, e286–e289. [Google Scholar] [PubMed]
- Zhuang, Y.; Miskimins, W.K. Metformin induces both caspase-dependent and poly(ADP-ribose) polymerase-dependent cell death in breast cancer cells. Mol. Cancer Res. 2011, 9, 603–615. [Google Scholar] [CrossRef]
- Wu, C.; Qiu, S.; Zhu, X.; Lin, H.; Li, L. OCT1-mediated metformin uptake regulates pancreatic stellate cell activity. Cell Physiol. Biochem. 2018, 47, 1711–1720. [Google Scholar] [CrossRef]
- Veiga, S.R.C.; Ge, X.C.; Mercer, C.A.; Hernández-Álvarez, M.I.; Thomas, H.E.; Hernandez-Losa, J.; Ramón, Y.; Cajal, S.; Zorzano, A.; Thomas, G.; et al. Phenformin-induced mitochondrial dysfunction sensitizes hepatocellular carcinoma for dual inhibition of mTOR. Clin. Cancer Res. 2018, 1, 3767–3780. [Google Scholar] [CrossRef]
- Jackson, A.L.; Sun, W.; Kilgore, J.; Guo, H.; Fang, Z.; Yin, Y.; Jones, H.M.; Gilliam, T.P.; Zhou, C.; Bae-Jump, V.L. Phenformin has anti-tumorigenic effects in human ovarian cancer cells and in an orthotopic mouse model of serous ovarian cancer. Oncotarget 2017, 21, 100113–100127. [Google Scholar] [CrossRef]
- Jossé, L.; Xie, J.; Proud, C.G.; Smales, C.M. mTORC1 signalling and eIF4E/4E-BP1 translation initiation factor stoichiometry influence recombinant protein productivity from GS-CHOK1 cells. Biochem. J. 2016, 15, 4651–4664. [Google Scholar] [CrossRef]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR Signaling in protein translation regulation: Implications in cancer genesis and therapeutic interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhao, M.; Howard, E.W.; Zhao, Q.; Parris, A.B.; Ma, Z.; Yang, X. Phenformin inhibits growth and epithelial-mesenchymal transition of ErbB2-overexpressing breast cancer cells through targeting the IGF1R pathway. Oncotarget 2017, 1, 60342–60357. [Google Scholar] [CrossRef] [PubMed]
- Sivendran, S.; Agarwal, N.; Gartrell, B.; Ying, J.; Boucher, K.M.; Choueiri, TK.; Sonpavde, G.; Oh, W.K.; Galsky, M.D. Metabolic complications with the use of mTOR inhibitors for cancer therapy. Cancer Treat. Rev. 2014, 40, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Petrachi, T.; Romagnani, A.; Albini, A.; Longo, C.; Argenziano, G.; Grisendi, G.; Dominici, M.; Ciarrocchi, A.; Dallaglio, K. Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget 2017, 24, 6914–6928. [Google Scholar]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; Pasca di Magliano, M.; Swanson, K.D.; Zheng, B. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J. Invest. Dermatol. 2017, 137, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Tokajuk, A.; Krzyżanowska-Grycel, E.; Tokajuk, A.; Grycel, S.; Sadowska, A.; Car, H. Antidiabetic drugs and risk of cancer. Pharmacol. Rep. 2015, 67, 1240–1250. [Google Scholar]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 22, 10574–10579. [Google Scholar] [CrossRef]
- Appleyard, M.V.; Murray, K.E.; Coates, P.J.; Wullschleger, S.; Bray, S.E.; Kernohan, N.M.; Fleming, S.; Alessi, D.R.; Thompson, A.M. Phe henformin as prophylaxis and therapy in breast cancer xenografts. Br. J. Cancer 2012, 13, 1117–1122. [Google Scholar] [CrossRef]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.S.; Choi, J.Y. Synergistic anti-cancer effect of phenformin and oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef]
- Wandee, J.; Prawan, A.; Senggunprai, L.; Kongpetch, S.; Tusskorn, O.; Kukongviriyapan, V. Metformin enhances cisplatin induced inhibition of cholangiocarcinoma cells via AMPK-mTOR pathway. Life Sci. 2018, 15, 172–183. [Google Scholar] [CrossRef]
- Pollak, M. Metformin and other biguanides in oncology: Advancing the research agenda. Cancer Prev. Res. (Phila). 2010, 3, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; Shaw, R.J. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 11, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.E.; Bae-Jump, V. Metformin and gynecologic cancers. Obstet. Gynecol. Surv. 2014, 69, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Biglia, N.; Tana, R.; Cosio, S.; Gallo, M. Metformin use and gynecological cancers: A novel treatment option emerging from drug repositioning. Crit. Rev. Oncol. Hematol. 2016, 105, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Dabrafenib plus trametinib: A review in advanced melanoma with a BRAF (V600) mutation. Target. Oncol. 2016, 11, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Kangwan, N.; Park, J.M.; Kim, E.H.; Hahm, K.B. Chemoquiescence for ideal cancer treatment and prevention: Where are we now? J. Cancer Prev. 2014, 19, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.X.; Yang, S.W.; Li, P.A.; Luo, X.; Li, Z.Y.; Hao, Y.X.; Yu, P.W. The promotion of the transformation of quiescent gastric cancer stem cells by IL-17 and the underlying mechanisms. Oncogene 2017, 36, 1256–1264. [Google Scholar] [CrossRef]
- Landman, G.W.; Kleefstra, N.; van Hateren, K.J.; Groenier, K.H.; Gans, R.O.; Bilo, H.J. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 2010, 33, 322–326. [Google Scholar] [CrossRef]
- Schöckel, L.; Glasauer, A.; Basit, F.; Bitschar, K.; Truong, H.; Erdmann, G.; Algire, C.; Hägebarth, A.; Willems, P.H.; Kopitz, C.; et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015, 3, 11. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 30, 4305–4319. [Google Scholar]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 2016, 30, 56456–56470. [Google Scholar]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 1, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Ng, V.W.; Gao, S.; Tan, M.H.; Yang, Y.Y. Phenformin-loaded polymeric micelles for targeting both cancer cells and cancer stem cells in vitro and in vivo. Biomaterials 2014, 35, 9177–9186. [Google Scholar] [CrossRef] [PubMed]
- Bayat, S.; Shekari Khaniani, M.; Choupani, J.; Alivand, M.R.; Mansoori Derakhshan, S. HDACis (class I), cancer stem cell, and phytochemicals: Cancer therapy and prevention implications. Biomed. Pharmacother. 2018, 97, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, J.A.; Oliveras-Ferraros, C.; Cufí, S.; Corominas-Faja, B.; Joven, J.; Martin-Castillo, B.; Vazquez-Martin, A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle 2012, 11, 2782–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 10, 37281–37299. [Google Scholar] [CrossRef]
- Ravnan, M.C.; Matalka, M.S. Vemurafenib in patients with BRAF V600E mutation-positive advanced melanoma. Clin. Ther. 2012, 34, 1474–1486. [Google Scholar] [CrossRef]
- Dasgupta, T.; Olow, A.K.; Yang, X.; Hashizume, R.; Nicolaides, T.P.; Tom, M.; Aoki, Y.; Berger, M.S.; Weiss, W.A.; Stalpers, L.J.; et al. Survival advantage combining a BRAF inhibitor and radiation in BRAF V600E-mutant glioma. J. Neurooncol. 2016, 126, 385–393. [Google Scholar] [CrossRef]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc. Natl. Acad. Sci. USA 2013, 5, 18226–18231. [Google Scholar] [CrossRef]
- Wong, D.J.; Robert, L.; Atefi, M.S.; Lassen, A.; Avarappatt, G.; Cerniglia, M.; Avramis, E.; Tsoi, J.; Foulad, D.; Graeber, T.G.; et al. Antitumor activity of the ERK inhibitor SCH772984 [corrected] against BRAF mutant, NRAS mutant and wild-type melanoma. Mol. Cancer 2014, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Trousil, S.; Chen, S.; Mu, C.; Shaw, F.M.; Yao, Z.; Ran, Y.; Shakuntala, T.; Merghoub, T.; Manstein, D.; Rosen, N.; et al. Phenformin enhances the efficacy of erk inhibition in NF1-mutant melanoma. J. Invest. Dermatol. 2017, 137, 1135–1143. [Google Scholar] [CrossRef]
- Gopal, Y.N.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1α and oxidative phosphorylation in melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 6, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Campoy, J.M. Use of metformin in clinical endocrinology. Endocr. Pract. 2016, 22, 1024–1026. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García Rubiño, M.E.; Carrillo, E.; Ruiz Alcalá, G.; Domínguez-Martín, A.; A. Marchal, J.; Boulaiz, H. Phenformin as an Anticancer Agent: Challenges and Prospects. Int. J. Mol. Sci. 2019, 20, 3316. https://doi.org/10.3390/ijms20133316
García Rubiño ME, Carrillo E, Ruiz Alcalá G, Domínguez-Martín A, A. Marchal J, Boulaiz H. Phenformin as an Anticancer Agent: Challenges and Prospects. International Journal of Molecular Sciences. 2019; 20(13):3316. https://doi.org/10.3390/ijms20133316
Chicago/Turabian StyleGarcía Rubiño, Mª Eugenia, Esmeralda Carrillo, Gloria Ruiz Alcalá, Alicia Domínguez-Martín, Juan A. Marchal, and Houria Boulaiz. 2019. "Phenformin as an Anticancer Agent: Challenges and Prospects" International Journal of Molecular Sciences 20, no. 13: 3316. https://doi.org/10.3390/ijms20133316
APA StyleGarcía Rubiño, M. E., Carrillo, E., Ruiz Alcalá, G., Domínguez-Martín, A., A. Marchal, J., & Boulaiz, H. (2019). Phenformin as an Anticancer Agent: Challenges and Prospects. International Journal of Molecular Sciences, 20(13), 3316. https://doi.org/10.3390/ijms20133316